

Abstract
A suite of pharmacokinetic and pharmacological studies show that bromophycolide A (1), an inhibitor of drug-sensitive and drug-resistant Plasmodium falciparum, displays a typical small molecule profile with low toxicity and good bioavailability. Despite susceptibility to liver metabolism and a short in vivo half-life, 1 significantly decreased parasitemia in a malaria mouse model. Combining these data with prior SAR analyses, we demonstrate the potential for future development of 1 and its bioactive ester analogs.
Keywords: Bromophycolide, Malaria
The human malaria parasite, Plasmodium falciparum, is responsible for more than 90 % of malarial infections worldwide.1 Despite a 17 % decrease in reported malarial cases and 25 % reduction in malaria related deaths since 2000, there were 216 million cases of malaria reported worldwide in 2010.1 Recently a shift in the treatment paradigm has been implemented due to rapidly increasing resistance to traditional therapies such as chloroquine, amodiaquine, mefloquine, sulfadoxine-pyrimethamine, and artemisinin.2, 3 This paradigm shift focuses on combining an artemisinin derivative with an agent possessing a different mode of action in order to reduce the further expansion of drug resistant strains.2 Unfortunately, resistance to at least one of the approved partner drugs already exists in multiple countries including Thailand and Cambodia.4 This fast evolution of resistance not only offers clinical challenges, but has also generated an immediate need for new pharmacophores that act on resistant strains.
In 2005, the discovery of a novel class of meroditerpenes from the marine red alga Callophycus serratus led to their exploration as potential antimalarial leads.5–8 Bromophycolide A (BrA, 1; Figure 1), the most abundant member of this compound class, exhibited submicromolar inhibition of both chloroquine-sensitive and chloroquine-resistant P. falciparum.9 Mechanistic studies revealed that 1 prevents heme detoxification, a vital process for the parasite during its bloodborne stages.9 Drugs such as chloroquine that target heme detoxification have had great success in malaria treatment, despite emergence of drug resistance.10 Although both chloroquine and 1 prevent heme detoxification in the parasite, 1 is unaffected by the resistance mechanism P. falciparum has evolved for chloroquine, providing support for its further development.9
Figure 1.
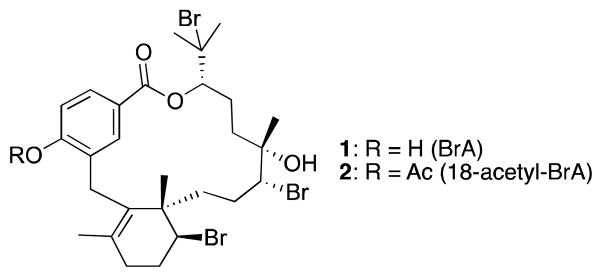
Structures of bromophycolide A (1) and its semi-synthetic derivative 18-acetyl-BrA (2).
Lead optimization requires identification of biochemical and physiochemical properties such as clearance from the blood stream, distribution between blood and other tissues, and understanding of metabolic fate in order to focus drug modification efforts. Here we report on these parameters, including in vivo efficacy, which ascertain key properties of 1 towards deriving a clinical drug candidate.
In mice infected with Plasmodium yoelii dosed once daily with 1 at 10 mg/kg, after four days the parasite load was 47 ± 12 % lower than in infected mice injected with vehicle only (Figure 2). Fluorescence Activated Cell Sorter (FACS) data revealed a final parasitemia of 32 ± 2 % for control animals compared to 17 ± 4 % for those dosed at 10 mg/kg and 20 ± 1 % dosed at 5 mg/kg. Twenty-four hours after the first dose of 10 mg/kg, parasitemia was 27 % lower for treated than control mice. After the second dose, parasite load was 55 % lower, reflected in 10.0 ± 0.5 % vs. 4.5 ± 1.5 % parasitemia in treated vs. control mice, respectively. BrA (1) was non-toxic to mice with this dosing regime. At the termination of efficacy trials, the concentration of 1 in blood and plasma of infected mice was below the limit of detection, indicating rapid elimination of 1 from the blood.
Figure 2.
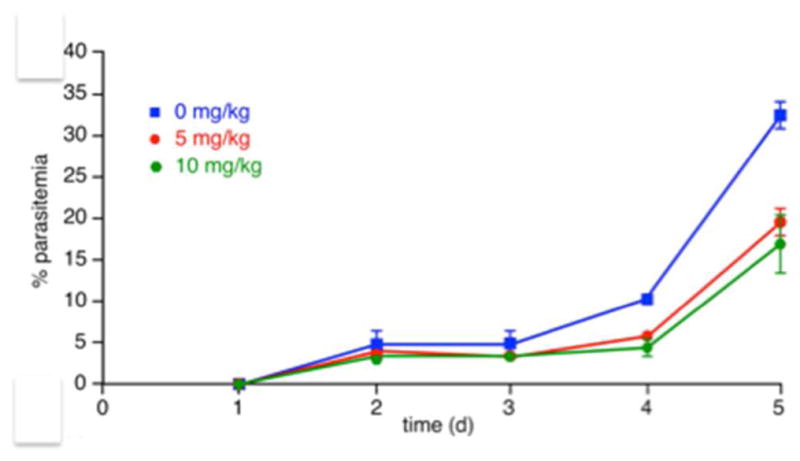
Bromophycolide A (1) decreases parasitemia in vivo. BrA (1) was administered to Swiss webster female mice on day 2 after confirmation of infection with Plasmodium yoelii. Experiment was terminated on day 5 and daily parasite load determined by FACS analysis by staining the parasites with Hoechst 33342 (n=3; ± standard deviation [SD]).
BrA (1) has previously been shown to potently inhibit the malaria parasite P. falciparum in vitro, having an IC50 of 499 nM against chloroquine-sensitive parasites and an IC50 of 377 nM against chloroquine-resistant parasites.9 In comparison to the few reported in vivo efficacies of other non-artemisinin derived antimalarial natural products, the activity of 1 is promising but not exceptional. For instance, gomphostenin A, which displayed an in vitro IC50 of 8.8 μM against P. falciparum, only suppressed parasitemia in vivo by 45% when dosed orally at 50 mg/kg,11 which is less efficacious than 1. Other leads displayed improved in vivo activity. For instance, spirotetrahydro-β-carboline NITD609, which displayed in vitro IC50 value of 0.9 nM against P. falciparum, was significantly more active in vivo than 1, reducing mouse parasitemia by 99% when dosed orally at 10 mg/kg.12 Similarly, simalikalactone E, with an in vitro IC50 of 24–68 nM, reduced parasitemia in vivo by 50% at 0.5 mg/kg IP dosing and 1 mg/kg oral dosing.13
The encouraging decrease in mouse parasitemia associated with BrA treatment (Figure 2) was observed despite the rapid elimination of 1 from systemic circulation. The pharmacokinetic profile of 1, determined following single injections by intravenous (IV) or intraperitoneal (IP) dosing to healthly mice, is characterized by rapid, but short-lived, pharmacological effects in vivo, as shown by a first order elimination process, short half-life, high clearance, fast absorption, and good bioavailability (Table 1; Figure 3). The preliminary pharmacokinetics displayed a high plasma clearance (Cl) of 16 ± 3 mL/min/kg in mice following IV administration. This is similar to the clearance rate for chloroquine (17 mL/min/kg),14 but higher than the spiroindolone NITD609, whose clearance rate in mice was shown to be 9.8 mL/min/kg.12, 15
Table 1.
Pharmacokinetic parameters following either intravenous bolus (IV) or intraperitoneal (IP) injection of BrA (1) in mice (n=3; mean ± SD).
| Parameter | Units | IV | IP |
|---|---|---|---|
| Dose | mg/kg | 23 | 23 |
| Kel | h−1 | 0.93 ± 0.13 | - |
| Kab | h−1 | - | 0.24 ± 0.03 |
| t1/2 | h | 0.75 ± 0.11 | - |
| Cmax | μg/mL | 5.8 ± 3.9 | |
| Tmax | h | - | 1.5 |
| AUC | μg*h/mL | 26 ± 5 | 20 |
| Cp0 | μg/mL | 24 ± 6 | - |
| Vd | L/kg | 0.95 ± 0.27 | - |
| Cl | mL/min/kg | 16 ± 3 | - |
Figure 3.

In vivo pharmacokinetic study of BrA (1) by a single intravenous (IV) or intraperitoneal (IP) injection. Data points are mean mouse plasma concentration (n=3, ±SD). Initial dose was 23 mg/kg.
BrA (1) has a short in vivo residence as displayed by an IV half-life of 0.75 ± 0.11 h. However, this value is not limiting as the marketed antimalarial drug, artesunate, exhibits a half-life of less than 10 min by IV dosing.16 Additionally a short plasma half-life suggests a decreased probability of the evolution of drug-resistance in the parasite.17 The apparent volume of distribution, 0.95 ± 0.27 L/kg of body weight, following IV dosing, is greater than the body of water volume suggesting that 1 is moderately distributed to the tissues, with a portion remaining in systemic circulation to be transferred throughout the body. In contrast, chloroquine has a much larger apparent volume of distribution of 200 L/kg, indicating primary localization in non-blood tissues, with very little of the drug remaining in the plasma circulating throughout the body.14
The rapid removal of 1 from systemic circulation as indicated by the preliminary pharmacokinetic study would suggest a much lower in vivo efficacy than achieved by 1 in the mouse model (Figure 2). Therefore, in the context of pharmacokinetic modeling, the in vivo efficacy of 1 is better than expected.
Despite its rapid clearance rate and low half-life, 1 did not suffer decomposition or loss of solubility in serum. Blood contains many enzymes that can lead to rapid degradation and erroneous pharmacokinetic parameters.18 While no significant loss of 1 was observed after incubation in mouse serum at 37 °C for 24 h (r2 = 0.20, p=0.27), a 27 ± 9 % loss was observed after 48 h. These data indicate that 1 is stable in mouse serum over 24 h and is not subject to rapid degradation by blood enzymes.
In contrast to its stability in serum, we found that human liver enzymes efficiently metabolized 1. In order to establish the most active routes to BrA metabolism in a model relevant for drug development, we used human liver enzymes to model the metabolic profile of BrA. As negligible metabolism of BrA was observed after 4 h, experiments with the human liver sub-cellular fractions were extended to 24 h. Incubation in the presence of the S9 liver fraction significantly decreased the concentration of 1 in the reaction mixture by 55 ± 4 % over 24 h (Figure 4); no such metabolism was noted in the absence of enzyme or NADPH (data not shown). Calculated apparent intrinsic clearance (CLint) for 1 exposed to the S9 fraction was 0.52 ± 0.06 mL/min/kg with an in vitro t1/2 of 20 ± 2 h. Upon addition of the glucuronidation co-factor UDPGA, 1 was subject to an additional 21 % metabolism when exposed to the S9 liver fraction (Figure 5). The calculated CLint for 1 exposed to the S9 fraction with UDPGA was 1.1 ± 0.1 mL/min/kg with an in vitro t1/2 of 9.5 ± 0.8 h, almost double the CLint without UDPGA, indicating that glucuronidation is a major pathway in the metabolism of 1.
Figure 4.

BrA (1) is metabolized by human liver subcellular fractions in vitro (n=3; ±SD)
Figure 5.

Glucuronidation is a significant pathway for the metabolism of BrA (1), as exhibited by the addition of UDPGA (n=3; ±SD).
In order to identify the liver enzymes and products involved in its metabolism, we exposed 1 to both microsomal and cytosolic subcellular fractions in addition to the S9 liver enzymes, resulting in dramatically different profiles (p ≤ 0.020 comparing all three treatments at t = 6 and 24 h). Incubation of 1 with the microsomal subcellular fraction resulted in a comparable result as that obtained with exposure to S9 enzymes (Figures 4 and 5). Without the addition of UDPGA, the concentration of 1 was reduced by 61 ± 1% in the presence of human liver microsomes. The calculated CLint of the microsomal fraction was 0.72 ± 0.09 mL/mg/kg with a t1/2 of 14 ± 4 h. In the presence of UDPGA, only 15.3 ± 3 % of 1 remained after 24 h with a calculated CLint of 1.6 ± 0.5 mL/min/kg with a t1/2 of 6.6 ± 1.9 h. The same distinction seen with the S9 subcellular fraction between incubations with UDPGA and without is noted, further supporting the role of glucuronidation as a major pathway in the metabolism of 1. However, 1 was stable in the presence of the human liver cytosolic fraction (r2 = 0.24, p = 0.10, Figure 3).
LC-MS analysis of the reaction products of 1 treated with S9 or microsomal enzymes supported the production of hydroxylated (m/z 677) and doubly hydroxylated (m/z of 693) metabolites such as M1-M4 (Scheme 1) in the absence of UDPGA. Both microsomal and cytosolic fractions contain methyl transferases, glutathione-S-transferases, and reductases.19 Unique to the microsomal fraction are cytochrome P450 enzymes and UDP-glucuronosyltransferases.19 Detection of hydroxylated derivatives of bromophycolide A with the S9 and microsomal fraction in the presence of NADPH suggested that cytochrome P450 monooxygenases are involved in liver metabolism of 1. In the presence of UDPGA, a glucuronidated metabolite, potentially M5, was detected (m/z 837) (Scheme 1), providing evidence for the role of UDP-glucuronosyltransferases in the metabolism of 1.
Scheme 1.

Possible phase I and II liver metabolites of BrA(1) based on LC-MS analysis. Shaded regions denote the position of modification.
The macrocyclic lactone of 1 was surprisingly tolerant to both blood and liver which contain a diversity of hydrolytic enzymes that can rapidly hydrolyze esters, lactones, amides, and lactams.18 The fact that products of hydrolysis of the macrolactone core of 1 were not detected in either the liver metabolism or mouse serum experiments provides strong support that the core motif of 1 is a stable entity.
Phase I hydroxylation of 1 may occur on either the aromatic ring or on the aliphatic methyl group (Scheme 1), although we were unsuccessful at definitively placing these functional groups by MS/MS analysis or via isolation and NMR characterization of metabolic products. Successful modifications to prevent hydroxylation at metabolically reactive sites on the aromatic ring have been performed by halogenation of the spiroindolone NITD609. Incorporation of either a fluorine or chlorine in both the positions meta and para to the pyrrole significantly increased the metabolic half-life and in vivo efficacy of spiroindolone NITD609.20 Comparable methods have also been seen for other leads such as triazolopyrimidine where replacement of an aromatic hydrogen with a trifluoromethyl group dramatically enhanced metabolic stability and in vivo efficacy.21
Hydroxyl groups make 1 susceptible to glucuronidation, which is expected to primarily occur at the aromatic hydroxyl group.22 The phenol of 1 has been previously shown to be vital to biological activity as the methylation of this functional group led to complete loss of in vitro activity.9 Thus, protecting the phenol could stabilize 1 in the presence of liver enzymes. The addition of electron withdrawing halogens to the aromatic ring could result in a reduction in the glucuronidation of the phenol of 1.23 Alternatively, esterification of the phenol may also reduce phase II glucuronidation, creating a pro-drug motif cleaved in the presence of hydrolytic enzymes found in the blood, releasing 1.24 This route is promising as semi-synthetic 18-acetyl-BrA (2) exhibits an IC50 value of 241 nM in vitro against P. falciparum.9
BrA (1) exhibits poor solubility in aqueous buffers (clog P = 8.8) relative to desirable drug candidates with clog P under 5.25 To date, we have been able to solubilize 1 starting from a solution in DMSO using serial dilutions with solutol HS-15 in saline solution. Attempts using other common materials such as Tween-80, cyclodextrins, and polyethylene glycol 400 failed. Expansion of next generation ester analogs could include the incorporation of alkoxy amines26, 27 or the addition of a phosphate group as a further modified prodrug-motif.28
Overall, in comparison to current early lead criteria published by the Medicines for Malaria Venture (MMV)29, 1 represents a relatively strong candidate for further development. BrA (1) has an in vitro potency against both drug-sensitive and drug-resistant parasites less than the required 1 μM with an established mode of action. BrA (1) is stable in mouse serum and moderately stable in the presence of human microsomes. The lack of mouse toxicity (in either the PK study or the in vivo efficacy study) and significant reduction in parasitemia in vivo despite a short half-life suggests promise. Previously published data indicate that 1 displays 10 fold differential selectivity among eukaryotic cell lines, having an IC50 above 6 uM against human cancer cells.5 Although 1 is not “rule-of-5” compliant and is poorly soluble in buffer, this study identifies key metabolic issues and provides a first step towards developing a strong structure pharmacologic relationship (SPR) map for indicating the next step to deliver a viable clinical lead.
Supplementary Material
Acknowledgments
The authors thank the Government of Fiji and the customary fishing right owners for permission to perform research in their territorial waters. We also thank M. Hay and C. Clements for collection of algal samples, E. Platzer for providing the P. yoelii strain and B. Walter for performing FACS analysis.
Funding Sources
This work was supported by ICBG grant U01-TW007401 from the Fogarty International Center of the U.S. National Institutes of Health and National Science Foundation.
ABBREVIATIONS
- BrA
bromophycolide A
- DMSO
dimethylsulfoxide
- MeOH
methanol
- CH3CN
acetonitrile
- UDPGA
uridine 5′-diphosphoglucuronic acid tri-sodium salt
- FACS
Fluorescence-activated cell sorting
- IV
intravenous
- and IP
intraperitoneal
- Kel
first order rate constant for the elimination phase
- Kab
first order rate constant for the absorption phase
Footnotes
SUPPORTING INFORMATION AVAILABLE
Detailed materials, methods and procedures are located in the supporting information. This material is free of charge via the Internet at http://pubs.acs.org.
References
- 1.WHO. World Malaria Report. 2011. pp. 1–121. [Google Scholar]
- 2.WHO. Global report on antimalarial drug efficacy and drug resistance 2000–2010. 2010. [Google Scholar]
- 3.Dondorp AM, Yeung S, White L, Nguon C, Day NPJ, Socheat D, von Seidlein L. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8:272–275. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 4.Wongsrichanalai C, Sirichaisinthop J, Karwacki JJ, Congpuong K, Miller RS, Pang L, Thimasarn K. Drug resistant malaria on the Thai-Myanmar and Thai-Cambodian borders. SE Asian J Trop Med Pub Health. 2001;32:41–49. [PubMed] [Google Scholar]
- 5.Kubanek J, Prusak AC, Snell TW, Giese RA, Hardcastle KI, Fairchild CR, Aalbersberg W, Raventos-Suarez C, Hay ME. Antineoplastic diterpene-benzoate macrolides from the Fijian red alga. Callophycus serratus Org Lett. 2005;7:5261–5264. doi: 10.1021/ol052121f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubanek J, Prusak AC, Snell TW, Giese RA, Fairchild CR, Aalbersberg W, Hay ME. Bromophycolides C-I from the Fijian red alga Callophycus serratus. J Nat Prod. 2006;69:731–735. doi: 10.1021/np050463o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane AL, Stout EP, Lin AS, Prudhomme J, Le Roch K, Fairchild CR, Franzblau SG, Hay ME, Aalbersberg W, Kubanek J. Antimalarial bromophycolides J-Q from the Fijian red algaCallophycus serratus. J Org Chem. 2009;74:2736–2742. doi: 10.1021/jo900008w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin AS, Stout EP, Prudhomme J, Le Roch K, Fairchild CR, Franzblau SG, Aalbersberg W, Hay ME, Kubanek J. Bioactive bromophycolides R-U from the Fijian red alga Callophycus serratus. J Nat Prod. 2010;73:275–278. doi: 10.1021/np900686w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stout EP, Cervantes S, Prudhomme J, France S, La Clair JJ, Le Roch K, Kubanek J. Bromophycolide A targets heme crystallization in the human malaria parasite Plasmodium falciparum. Chem Med Chem. 2011;6:1572–1577. doi: 10.1002/cmdc.201100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Na-Bangchang K, Karbwang J. Current status of malaria chemotherapy and the role of pharmacology in antimalarial drug research and development. Fundam Clin Pharmacol. 2009;23:387–409. doi: 10.1111/j.1472-8206.2009.00709.x. [DOI] [PubMed] [Google Scholar]
- 11.Sathe M, Kaushik MP. Gomphostenins: two new antimalarial compounds from the leaves of Gomphostemma niveum. Bioorg Med Chem Lett. 2010;20:1312–1314. doi: 10.1016/j.bmcl.2009.02.120. [DOI] [PubMed] [Google Scholar]
- 12.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cachet N, Hoakwie F, Bertani S, Bourdy G, Deharo E, Stien D, Houel E, Gornitzka H, Fillaux J, Chevalley S, Valentin A, Jullian V. Antimalarial activity of simalikalactone E, a new quassinoid from Quassia amara L. (Simaroubaceae) Antimicrob Agents Chemother. 2009;53:4393–4398. doi: 10.1128/AAC.00951-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore BR, Page-Sharp M, Stoney JR, Ilett KF, Jago JD, Batty KT. Pharmacokinetics, pharmacodynamics, and allometric scaling of chloroquine in a murine malaria model. Antimicrob Agents Chemother. 2011;55:3899–3907. doi: 10.1128/AAC.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friskholmberg M, Bergqvist Y, Termond E, Domeijnyberg B. The single dose kinetics of chloroquine and its major metabolite desethylchloroquine in healthy-subjects. Eur J Clin Pharmacol. 1984;26:521–530. doi: 10.1007/BF00542151. [DOI] [PubMed] [Google Scholar]
- 16.Morris CA, Duparc S, Borghini-Fuhrer I, Jung D, Shin CS, Fleckenstein L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar J. 2011;10:263–280. doi: 10.1186/1475-2875-10-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MMV Stategy for the selection and early development of combination drugs for the treatment of uncomplicated P. falciparum malaria. (5 July )
- 18.Di L, Kerns EH. Solution stability--plasma, gastrointestinal, bioassay. Curr Drug Metab. 2008;9:860–868. doi: 10.2174/138920008786485218. [DOI] [PubMed] [Google Scholar]
- 19.Kerns E, Di L. Drug-like properties: concepts, structure design and methods: from ADME to toxicity optimization. Academic Press; 2008. [Google Scholar]
- 20.Chatterjee AK, Yeung BK. Back to the future: lessons learned in modern target-based and whole-cell lead optimization of antimalarials. Curr Top Med Chem. 2012;12:473–483. doi: 10.2174/156802612799362977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coteron JM, Marco M, Esquivias J, Deng XY, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu BJ, Basu S, Meng SN, Wang XQ, Hu M. Regioselective sulfation and glucuronidation of phenolics: insights into the structural basis. Curr Drug Metab. 2011;12:900–916. doi: 10.2174/138920011797470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madsen P, Ling A, Plewe M, Sams CK, Knudsen LB, Sidelmann UG, Ynddal L, Brand CL, Andersen B, Murphy D, Teng M, Truesdale L, Kiel D, May J, Kuki A, Shi SH, Johnson MD, Teston KA, Feng J, Lakis J, Anderes K, Gregor V, Lau J. Optimization of alkylidene hydrazide based human glucagon receptor antagonists. Discovery of the highly potent and orally available 3-cyano-4-hydroxybenzoic acid[1-(2,3,5,6-tetramethylbenzyl)-1H-indol-4-ylmethylene]hydrazide. J Med Chem. 2002;45:5755–5775. doi: 10.1021/jm0208572. [DOI] [PubMed] [Google Scholar]
- 24.Hussain MA, Shefter E. Naltrexone-3-salicylate (a prodrug of naltrexone) - synthesis and pharmacokinetics in dogs. Pharm Res. 1988;5:113–115. doi: 10.1023/a:1015944302567. [DOI] [PubMed] [Google Scholar]
- 25.Hill AP, Young RJ. Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity. Drug Discov Today. 2010;15:648–655. doi: 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 26.Arrowsmith JE, Campbell SF, Cross PE, Stubbs JK, Burges RA, Gardiner DG, Blackburn KJ. Long-acting dihydropyridine calcium-antagonists. 1.2-Alkoxymethyl derivatives Incorporating basic substituents. J Med Chem. 1986;29:1696–1702. doi: 10.1021/jm00159a022. [DOI] [PubMed] [Google Scholar]
- 27.Lundquist JT, Harnish DC, Kim CY, Mehlmann JF, Unwalla RJ, Phipps KM, Crawley ML, Commons T, Green DM, Xu WX, Hum WT, Eta JE, Feingold I, Patel V, Evans MJ, Lai K, Borges-Marcucci L, Mahaney PE, Wrobel JE. Improvement of physiochemical properties of the tetrahydroazepinoindole series of farnesoid X receptor (FXR) agonists: beneficial modulation of lipids in primates. J Med Chem. 2010;53:1774–1787. doi: 10.1021/jm901650u. [DOI] [PubMed] [Google Scholar]
- 28.Stella VJ, Nti-Addae KW. Prodrug strategies to overcome poor water solubility. Adv Drug Deliv Rev. 2007;59:677–694. doi: 10.1016/j.addr.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 29.MMV Compound progression criteria. 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.