

Abstract
Masked acyl cyanide (MAC) reagents are shown to be effective umpolung synthons for enantioselective Michael addition to substituted enones. The reactions are catalyzed by chiral squaramides and afford adducts in high yields (90–99%) and with excellent enantioselectivities (85–98%). The addition products are unmasked to produce dicyanohydrins that, upon treatment with a variety of nucleophiles, provide γ-keto acids, esters, and amides. The use of this umpolung synthon has enabled, in enantiomerically enriched form, the first total synthesis of the prenylated polyphenol (+)-fornicin C.
The importance of umpolung chemistry stems from its capacity to provide access to functional group arrangements that are difficult to realize through normal chemical reactivity considerations.1 For example, whereas 1,3-dicarbonyl compounds are readily synthesized through classical reactivity (nucleophilic enolate plus an electrophilic carbonyl reactant), 1,4-dicarbonyl compounds, having dissonant connectivity,2 can prove challenging. An efficient route to such compounds is through an umpolung strategy, the reaction of an enone with a masked acyl anion. Given their value in synthesis, many acyl anion equivalents have been developed, and utilized for a variety of synthesis problems.3 Among useful and versatile umpolung synthons are protected hydroxyl malononitriles, known as MAC reagents (masked acyl cyanide), developed by Yamamoto and Nemoto.4 Treatment of a MAC reagent with base generates a nucleophilic acyl anion equivalent that will react with a variety of electrophilic units. Subsequent unmasking of the MAC unit produces an acyl cyanide, which can be intercepted to afford carboxylic acids, esters, or amides.5 Thus, unlike typical acyl anion equivalents, MAC reagents also harbor masked acid chloride-like reactivity. This dual capability, found in few acyl anion equivalents, allows MAC reagents to function as carbon monoxide equivalents.6 The usefulness of these reagents is demonstrated by their use in the total syntheses of (–)-bestatin, cyclotheonamide C, and oseltamivir (Tamiflu).7,8,9 Despite their importance in synthesis, there appears to be no report of a catalytic enantioselective reaction of a MAC reagent.10 The ease of deprotonation of MAC reagents suggested that their reactions could be rendered enantioselective using a bifunctional chiral base. In this report, we disclose the first enantioselective reactions of the MAC family of umpolung synthons.11,12 Specifically, we show that Michael additions of MAC reagents to enones are efficiently catalyzed by chiral squaramides, affording adducts in high yields and excellent enantioselectivities, and, upon unmasking, provide ready access to chiral γ-keto-carboxylic acids, -esters and -amides (Figure 1). Our initial efforts focused on the identification of effective catalysts and conditions for the enantioselective Michael reaction between trans-chalcone (1a) and MOM-MAC (2) to afford adduct 3a (Fig. 2).13,14 Although quinine (I) promoted this reaction, it afforded the adduct with poor enantioselectivity. The cinchonidine-derived thiourea catalyst (II) gave better enantioselectivity, but low conversion. On the other hand, the corresponding squaramide (III) provided both superior conversion and enantioselectivity. As anticipated, the pseudo-enantiomeric, quinidine-derived squaramide (IV) provided the product with comparable selectivity and conversion, but enriched in the opposite enantiomer. Among the other squaramide catalysts examined, those based on 1,2-diaminocyclohexane proved most effective, particularly the pyrrolidine catalyst VIc, which formed 3a at a high rate and with 90% ee.15,16 The enantiomeric excess improved significantly, to 97%, when the reaction was carried out at –30 °C. It is noteworthy that comparable enantioselectivity was observed even at 1 mol % catalyst loading, albeit with lower conversion. Moreover, increasing the catalyst loading to 10 mol % did not significantly increase the conversion over the 5 mol % reaction, presumably due to the limited solubility of the catalyst.17 Additional solvents were also investigated for the reaction, but they did not lead to an improved outcome.
Figure 1.

Synthesis of 1,4-dicarbonyls via addition of MAC reagents
Figure 2.

A selection of chiral catalysts examined
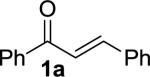
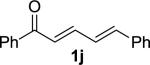
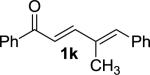
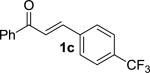
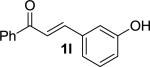
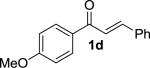
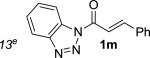
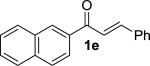
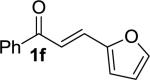
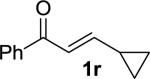
We next examined the practical utility and substrate scope of squaramide-catalyzed enantioselective Michael reactions of MAC reagents (Table 1). In general, high yields and enantioselectivities were observed for a broad range of enones. The isolated yield for the parent trans-chalcone reaction exceeded that determined through NMR during optimization studies (entry 1). Importantly, the related TBS-MAC reagent (4)4b was also found to be effective for the enantioselective Michael reaction. Although it displays lower reactivity compared to 2, requiring higher reaction temperatures and/or longer times for complete reaction, the TBS-MAC provides comparable enantioselectivity while offering an alternative deprotection protocol (vide infra). Enones having electron-deficient aryl groups at the β-position gave excellent results (entries 2–3). The more electron-rich substrate, p-methoxyphenyl enone 1d, required higher reaction temperatures and provided reduced enantioselection (entry 4). The Michael addition was also effective with naphthyl enone 1e and various heteroaryl-substituted enones (entries 5–9). Conjugated enone 1j reacted selectively at the β-position to afford the addition products in excellent yields and enantioselectivities (entry 10). The related substituted dienone 1k delivered the addition product with a notable drop in enantioselectivity, coinciding with its reduced reactivity (entry 11). The reaction of enone 1l, which contains a free phenol, is noteworthy as it demonstrates the tolerance for acidic hydrogen bond donor groups in the substrate (entry 12). The benzotriazole-containing enone 1m (entry 13) was also an effective substrate.
Table 1.
Substrate scope of squaramide catalyzed MAC addition reactionsa
|
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | enone | R3 | product | temp (°c) | time (h) | yieldc (%) | eed (%) | entry | enone | R3 | product | temp (°C) | time (h) | yieldc (%) | eed (%) |
| 1 |
|
MOM TBS |
3a
5a |
–30 –20 |
24 36 |
98 96 |
97 96 |
10 |
|
MOM TBS |
3j
5j |
–10 0 |
28 48 |
98 96 |
95 94 |
| 2 |
|
MOM | 3b | –30 | 20 | 98 | 97 | 11 |
|
MOM | 3k | 23 | 24 | 97 | 88 |
| 3 |
|
MOM | 3c | –30 | 24 | 98 | 97 | 12 |
|
MOM | 3l | 23 | 24 | 96 | 92 |
| 4 |
|
MOM TBS |
3d
5d |
0 0 |
28 72 |
99 96 |
92 90 |
13e |
|
MOM | 3m | –10 | 72 | 96 | 89 |
| 5 |
|
MOM TBS |
3e
5e |
–30 –20 |
24 36 |
99 95 |
98 97 |
14 |
|
MOM TBS |
3n
5n |
–30 –20 |
20 36 |
96 98 |
96 94 |
| 6 |
|
TBS | 5f | 0 | 24 | 93 | 90 | 15 |
|
MOM | 3o | –30 | 28 | 96 | 97 |
| 7 |
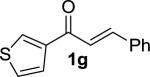
|
TBS | 5g | –10 | 48 | 94 | 90 | 16 |
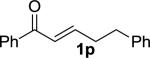
|
MOM TBS |
3p
5p |
–30 –20 |
24 36 |
98 92 |
96 94 |
| 8 |
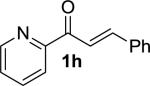
|
MOM | 3h | –30 | 30 | 98 | 91 | 17 |
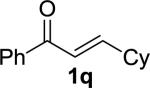
|
MOM | 3q | 23 | 24 | 94 | 89 |
| 18 |
|
MOM | 3r | –30 | 24 | 98 | 97 | ||||||||
| 9 |
|
MOM TBS |
3i
5i |
0 0 |
36 72 |
97 90 |
92 89 |
19 |
|
MOM TBS |
3s
5s |
–30 –20 |
24 36 |
99 93 |
94 85 |
Conditions: Reactions performed with 1 (0.36 mmol), 2 or 4 (0.30 mmol), VIc (5 mol %), CH2Cl2 (0.6 mL).
Isolated yield.
Enantiomeric excess determined by chiral stationary phase HPLC.
4Å MS were used as an additive to avoid hydrolysis.
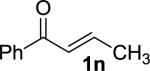
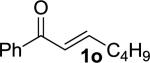
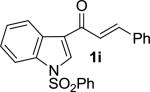
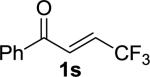
Enones having β-alkyl-substituents were also found to be effective substrates for the MAC-Michael reaction.18 The reaction of both MAC reagents with enones having primary alkyl substitutents proceeded well, affording the addition products in near-quantitative yields and with high enantioselectivities (entries 14–16). The more sterically hindered cyclohexyl-enone 1q (entry 17) required room temperature for the reaction to proceed, but still gave the product with 89% ee. Cyclopropyl enone 1r, on the other hand, was more reactive and gave the addition product in excellent yield and selectivity (entry 18). The Michael reaction of MOM-MAC and trifluoromethyl-substituted enone 1s proceeded nicely, providing straightforward access to chiral units containing the trifluoromethyl group. Curiously, for reasons that are unclear, the same reaction with TBS-MAC gave significantly lower selectivity with this substrate.
The optimized conditions are suitable for the preparation of gram quantities of the Michael adducts. For example, the reaction with chalcone was scaled up to produce one gram of product, formed in the same yield and enantioselectivity as noted in entry 1. Additionally, in large-scale reactions the low solubility of the catalyst makes it convenient to recover most of it (84%) from the ether extracts by simple filtration. Such recovered catalyst can be used multiple times without measurable loss in efficacy.
We recognized that the above reactions would be of value only if the MAC group in the Michael adducts could be transformed into useful functional groups without eroding enantioselectivity. A careful examination of hydrolysis conditions allowed us to identify mild, highly effective conditions for unmasking the MAC moiety. For the MOM-MAC adduct excellent results were obtained if the intermediate acyl cyanide, which is prone to racemization, were intercepted at a relatively low temperature (ca. –40 °C).19 Thus, treatment of the labile dicyanohydrin (6), formed upon removal of the MOM group, at –40 °C with MeOH/Et3N or morpholine/Et3N, gave ketoester 7 or ketoamide 9, respectively, in high yields and with little or no ee loss (Scheme 1). The corresponding ketoacid (8) can be formed directly by treating the MOM-adduct with CSA in aqueous acetic acid. Conditions were also developed for the efficient transformation of the TBSMAC adduct 5a to methyl ester 7, Weinreb amide 10, glycine-derived amide 11, as well as for its direct transformation to ketoacid 8.20
Scheme 1.

Functionalization of MAC adducts
The absolute stereochemistry induced during the Michael reaction was determined by taking advantage of the unique reactivity of acyl benzotriazoles.21 The MAC addition product of cinnamylbenzotriazole (1m) catalyzed by (R,R)-VIc was first converted to methyl ester 12, which upon unmasking generated the known succinate ester 13, enriched in the S enantiomer (Scheme 2). The observed asymmetric induction is consistent with a pre-transition state assembly shown in Figure 3, wherein the ammonium salt directs nucleophilic addition of MAC anion to chalcone, which in turn is activated through hydrogen bonding. Analogous models have been posited for thiourea and squaramide catalyzed asymmetric reactions.14b,h
Scheme 2.

Determination of absolute stereochemistry
Figure 3.
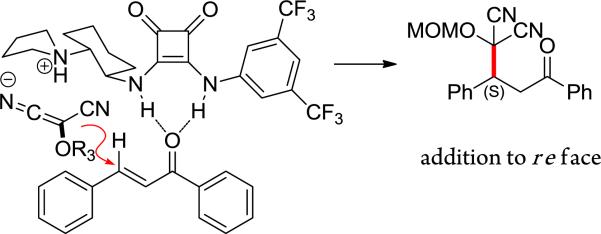
Model for asymmetric induction
The utility of the above enantioselective methodology is demonstrated through the total synthesis of prenylated polyphenol (+)-fornicin C. Isolated from Ganoderma fornicatum, extracts of which are reported in Chinese medicine to have health promoting properties,22 fornicin C displays moderate in vitro cytotoxic activity against the human larynx carcinoma cell line Hep-2 (23 μg/mL). Triol 17 can be prepared directly from the aldol reaction of the trianion of 2,5-dihydroxyacetophenone (14) with geraniol-derived aldehyde 16 in ca. 50% yield,23 or in a higher yield via the Mukaiyama aldol reaction of silyl enol ether 15 with 16. Dehydration to the enone was an unforeseen challenge since basic conditions promoted cyclization to a chromanone, while strongly acidic conditions introduced complications through reactions with the geranyl side-chain. Furthermore, with dehydrating agents such as Martin's sulfurane or Burgess’ reagent, the phenols competed with the secondary alcohol. We found that sulfur trioxide-pyridine complex was effective in achieving the dehydration of triol 17 to enone 18.24 Addition of TBS-MAC (4) to enone 18 provided adduct 19 with 92% enantioselectivity.25 When subjected to AcOH-buffered TBAF, 19 was successfully unmasked to reveal (+)-fornicin C in 51% overall yield, with complete retention of enantiomeric excess.
To summarize, we have developed the first catalytic, enantioselective reaction of the MAC family of umpolung synthons. Specifically, we have shown that chiral squaramide VIc catalyzes the conjugate addition reactions of MAC reagents 2 and 4 to a collection of electronically and structurally diverse enones to afford adducts in high yields and with excellent enantioselectivities. The addition products can be unmasked to afford γ-keto-carboxylic acids, -esters and –amides, all formed in excellent yields and with near complete retention of enantiomeric excesses. We have utilized this methodology for the total synthesis of (+)-fornicin C, prepared in five steps from known alde-hyde 16, in 51% overall yield and 92% ee. The development of other enantioselective reactions of MAC and related umpolung reagents are expected to be of great value in organic synthesis.
Supplementary Material
Scheme 3.

Total synthesis of Fornicin C
ACKNOWLEDGMENT
Financial support from the National Institutes of Health (R01GM069990) is gratefully acknowledged.
Footnotes
Supporting Information
Experimental procedures and detailed characterization data of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- 1.Wittig G, Davis P, Koenig G. Chem. Ber. 1951:627. Seebach D, Kolb M. Chem. Ind. 1974:687. The phrase “charge affinity inversion” was introduced independently: Evans DA, Andrews GC. Acc. Chem. Res. 1974;147, and references cited therein.
- 2.The concept of dissonant and consonant connectivity was presented by Professor David A. Evans at a UCLA Physical Organic Chemistry Seminar, May 6, 1971. See SI for information on an unpublished manuscript on the subject by Evans Zweifel GS, Nantz MH. Modern Organic Synthesis: An Introduction. W.H. Freeman; New York: 2007. Synthetic Design. pp. 1–30., and references cited therein.
- 3.a Grobel BT, Seebach D. Synthesis. 1977:357. [Google Scholar]; b Seebach D. Angew. Chem. Int. Ed. 1979;18:239. [Google Scholar]; c Hase TA. Umpoled Synthons: A Survey of Sources and Uses in Synthesis. Wiley; New York: 1987. [Google Scholar]
- 4.a Nemoto H, Kubota Y, Yamamoto Y. J. Org. Chem. 1990;55:4515. [Google Scholar]; b Nemoto H, Li X, Ma R, Suzuki I. Tetrahedron Lett. 2003;44:73. [Google Scholar]; c Nemoto H, Kawamura T, Miyoshi N. J. Am. Chem. Soc. 2005;127:14546. doi: 10.1021/ja054010h. [DOI] [PubMed] [Google Scholar]; d Nemoto H, Ma R, Kawamura T, Kamiya M, Shibuya M. J. Org. Chem. 2006;71:6038. doi: 10.1021/jo0607515. [DOI] [PubMed] [Google Scholar]; e Nemoto H, Kawamura T, Kitasaki K, Yatsuzuka K, Kamiya M, Yoshioka Y. Synthesis. 2009:1694. and references cited therein. [Google Scholar]
- 5.For a review on acyl cyanides, see: Hunig S, Schaller R. Angew. Chem. Int. Ed. 1982;21:36.
- 6.Selected carboxyl anion equivalents: Utimoto K, Wakabayashi Y, Shishiyama Y, Inoue M, Nozaki H. Tetrahedron Lett. 1981;22:4279. Bates GS;, Ramaswamy S. Can. J. Chem. 1983;61:2006. Trost BM, Quayle P. J. Am. Chem. Soc. 1984;106:2469. Satoh T, Onda K-I, Yamakawa K. Tetrahedron Lett. 1990;31:3567. Aggarwal VK, Thomas A, Schade S. Tetrahedron. 1997;53:16213.
- 7.Nemoto H, Ma R, Suzuki I, Shibuya M. Org. Lett. 2000;2:4245. doi: 10.1021/ol006816m. [DOI] [PubMed] [Google Scholar]
- 8.Roche SP, Faure S, Aitken DJ. Angew. Chem. Int. Ed. 2008;47:6840. doi: 10.1002/anie.200802005. [DOI] [PubMed] [Google Scholar]
- 9.Yamatsugu K, Yin L, Kamijo S, Kimura Y, Kanai M, Shibasaki M. Angew. Chem. Int. Ed. 2009;48:1070. doi: 10.1002/anie.200804777. [DOI] [PubMed] [Google Scholar]
- 10.For auxiliary-based asymmetric syntheses using MAC reagents see: Nemoto H, Ma RJ, Moriguchi H, Suzuki I, Shibuya M. J. Organomet. Chem. 2000;611:445. Nemoto H, Ma RJ, Kawamura T, Kamiya M, Shibuya M. J. Org. Chem. 2006;71:6038. doi: 10.1021/jo0607515. Nemoto H, Moriguchi H, Ma R, Kawamura T, Kamiya M. Shibuya, M. Tetrahedron: Asymmetry. 2007;18:383.
- 11.For selected examples of acyl anion equivalents in enantioselective reactions, see: Yet L. Angew. Chem. Int. Ed. 2001;40:875. Gröger H. Chem. Rev. 2003;103:2795. doi: 10.1021/cr020038p. Boruwa J, Gogoi N, Saikia PP, Barua NC. Tetrahedron: Asymmetry. 2006;17:3315. Palomo C, Oiarbide M, Laso A. Eur. J. Org. Chem. 2007:2561. Bugaut X, Glorius F. Chem. Soc. Rev. 2012;41:3511. doi: 10.1039/c2cs15333e.
- 12.For recent examples of catalytic enantioselective addition of carboxyl synthons, see: Herrera RP, Monge D, Martin-Zamora E, Fernandez R, Lassaletta JM. Org. Lett. 2007;9:3303. doi: 10.1021/ol071292c. Uraguchi D, Ueki Y, Ooi T. Science. 2009;326:120. doi: 10.1126/science.1176758. Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027. doi: 10.1038/nature09125. Prakash GKS, Wang F, Zhang Z, Ni C, Haiges R, Olah GA. Org. Lett. 2012;14:3260. doi: 10.1021/ol301112m. Fernandez M, Uria U, Vicario JL, Reyes E, Carrillo L. J. Am. Chem. Soc. 2012;134:11872. doi: 10.1021/ja3041042. Uraguchi D, Ueki Y, Ooi T. Chem. Sci. 2012;3:842. Leighty M;, Shen B, Johnston JN. J. Am. Chem. Soc. 2012;134:15233. doi: 10.1021/ja306225u.
- 13.Please refer to SI for a table listing some of the conditions examined.
- 14.For examples of enantioselective Michael addition of malononitrile, see: Taylor MS, Zalatan DN, Lerchner AM, Jacobsen EN. J. Am. Chem. Soc. 2005;127:1313–1317. doi: 10.1021/ja044999s. Hoashi Y, Okino T, Takemoto Y. Angew. Chem. Int. Ed. 2005;44:4032. doi: 10.1002/anie.200500459. Wang J, Li H, Zu L, Jiang W, Xie H, Duan W, Wang W. J. Am. Chem. Soc. 2006;128:12652. doi: 10.1021/ja065187u. Li X, Cun L, Lian C, Zhong L, Chen Y, Liao J, Zhu J, Deng J. Org. & Bio. Chem. 2008;6:349. doi: 10.1039/b713129a. Shi J, Wang M, He L, Zheng K, Liu X, Lin L, Feng X. Chem. Commun. 2009:4711. doi: 10.1039/b908632c. Russo A, Perfetto A, Lattanzi A. Adv. Syn. & Cat. 2009;351:3067. Molleti N, Rana NK, Singh VK. Org. Lett. 2012;14:4322. doi: 10.1021/ol3015607. Yang W, Jia Y, Du DM. Org. Biomol. Chem. 2012;10:332. doi: 10.1039/c1ob06302b.
- 15.For earlier applications of chiral squaramides from this laboratory, see: Malerich JP, Hagihara K, Rawal VH. J. Am. Chem. Soc. 2008;130:14416. doi: 10.1021/ja805693p. Konishi H, Lam TY, Malerich JP, Rawal VH. Org. Lett. 2010;12:2028. doi: 10.1021/ol1005104. Zhu Y, Malerich JP, Rawal VH. Angew. Chem. Int. Ed. 2010;49:153. doi: 10.1002/anie.200904779.
- 16.For general reviews on hydrogen bond catalysis, see: Doyle AG, Jacobsen EN. Chem. Rev. 2007;107:5713. doi: 10.1021/cr068373r. Türkmen YE, Zhu Y, Rawal VH. In: Brønsted Acids. In Comprehensive Enantioselective Organocatalysis. Dalko PI, editor. Vol. 2. Wiley-VCH; Weinheim: 2013. Chapt. 10.
- 17.The catalyst does not dissolve fully even at 5 mol % loading.
- 18.Benzylideneacetone reacted very slowly under the standard conditions, giving the product in low yield (<10%).
- 19.Standard deprotection conditions (e.g., TsOH, TFA, LiBF4, TMSBr, ZnBr2, BF3OEt2, MgBr2, TMSOTf, Amberlyst 15, PPTS, Sc(OTf)3, Bi(OTf)3, Y(OTf)3, and ZrCl4) led to low yields and racemization. DME was used since THF was found to be unstable under these conditions.
- 20.Erosion of enantioselectivity was observed for some substrates when using the reported conditions for unmasking (TBAF, 3 equiv. MeOH; see Ref. 4d).
- 21.Katritzky AR, Lan XF, Yang JZ, Denisko OV. Chem. Rev. 1998;98:409. doi: 10.1021/cr941170v. [DOI] [PubMed] [Google Scholar]
- 22.Niu X-M, Li S-H, Sun H-D, Che C-TJ. Nat. Prod. 2006;69:1364. doi: 10.1021/np060218k. [DOI] [PubMed] [Google Scholar]
- 23.a Tsuda Y, Hosoi HS, Goto Y. Chem. Pharm. Bull. 1991;39:18. [Google Scholar]; b Hosoi S, Kiuchi F, Nakamura N, Imasho M, Ahad Ali M, Sakaki Y. Chem. Pharm. Bull. 1999;47:37. doi: 10.1248/cpb.47.37. [DOI] [PubMed] [Google Scholar]
- 24.Sulfur trioxide was discovered while screening solvents with Burgess re-agent; DMF reacted with Burgess reagent to form sulfur trioxide-triethylamine complex and (E)-methyl ((dimethylamino)methylene)carbamate:
- 25.The MOM-MAC reagent also reacted well with the free dihydroxyenone 18 (>90% ee), but the adduct gave side products during unmasking, due to acid incompatibility of the side chain.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.