

Abstract
Members of the seven transmembrane receptor (7TMR) superfamily are sequestered from the plasma membrane following stimulation both to limit cellular responses as well as to initiate novel G protein-independent signaling pathways. The best studied mechanism for 7TMR internalization is via clathrin-coated pits, where clathrin and adaptor protein complex 2 (AP-2) nucleate and polymerize upon encountering the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to form the outer layer of the clathrin-coated vesicle. Activated receptors are recruited to clathrin-coated pits by β-arrestins, scaffolding proteins which interact with agonist-occupied 7TMRs as well as AP-2 and clathrin. We report here that following stimulation of the β2-adrenergic receptor (β2-AR), a prototypical 7TMR, β-arrestins bind phosphatidylinositol 4-phosphate 5-kinase Iα (PIP5K Iα), a PIP2-producing enzyme. Furthermore, β-arrestin2 is required to form a complex with PIP5K Iα and agonist-occupied β2-AR and β-arrestins synergize with the kinase to produce PIP2 in response to isoproterenol stimulation. Interestingly, β-arrestins themselves bind PIP2 and a β-arrestin mutant deficient in PIP2 binding no longer internalizes 7TMRs, fails to interact with PIP5K Iα, and is not associated with PIP kinase activity assayed in vitro. However, a chimaeric protein in which the core kinase domain of PIP5K Iα has been fused to the same β-arrestin mutant rescues internalization of β2-ARs. Collectively, these data support a model in which β-arrestins direct the localization of PIP5K Iα and PIP2 production to agonist-activated 7TMRs, thereby regulating receptor internalization.
The seven transmembrane receptors are integral membrane proteins defined by seven α-helical membrane-spanning segments and comprise the largest group of mammalian cell-surface receptors with nearly one thousand members (1,2). This superfamily is responsive to diverse stimuli, with individual receptors responding to hormones, neurotransmitters, and odorants among other signals. While the signaling cascades initiated by these receptors are critical for intracellular responses to changes in the extracellular environment, termination of signal transduction and proper regulation of responses to stimuli are equally important for proper cellular function. Failures in the regulation of these pathways can lead to undesirable physiological consequences including tumorigenesis (3), vascular hypertrophy (4), and heart failure (4,5). The agonist-specific process by which 7TMRs are silenced is a highly conserved, two-phase process known as homologous desensitization (6). The agonist-occupied 7TMR is phosphorylated on intracellular serine and threonine residues by a G protein-coupled receptor kinase (GRK) (7), followed by the recruitment of arrestin proteins (rod and cone arrestins in the visual system and the ubiquitously expressed isoforms β-arrestin1 and β-arrestin2) to the GRK-phosphorylated receptors. Binding of arrestins sterically occludes the sites of receptor interaction with heterotrimeric G proteins and limits the 7TMR’s responsiveness to repeated stimulation (8,9). Furthermore, recent studies have shown β-arrestins scaffold multiple isoforms of phosphodiesterases (PDEs) (10) and diacylglycerol kinases (DGKs) (11), enzymes which metabolize the second messenger molecules cAMP and diacylglycerol respectively, further enhancing the efficiency of arrestin-mediated 7TMR signal quenching.
While desensitization of 7TMRs begins silencing G protein signaling within seconds of activation, it has been well established that over the span of several minutes, agonist-occupied receptors are also internalized from the cell surface (12). This sequestration not only confines the receptor in a cellular compartment inaccessible to extracellular ligands and membrane-associated G proteins, but also serves to initiate processes such as receptor dephosphorylation and recycling, or proteolytic degradation (1,13). In addition, 7TMR internalization is also required for various signaling pathways, including the β-arrestin dependent, G protein-independent activation of MAP kinases (14–17). 7TMRs are capable of internalization by various mechanisms. However, the best studied pathway for receptor endocytosis is via clathrin-coated pits, where β-arrestins serve as multivalent adaptor proteins to connect the 7TMR cargo to clathrin (18) and the β2 adaptin subunit of the heterotetrameric AP-2 adaptor complex (19,20). β-arrestins are integral components of this process as demonstrated by the severe impairment of 7TMR internalization in tissues from β-arrestin knockout mice (21) as well as cells transfected with siRNA to deplete β-arrestin expression (22). Moreover, this endocytosis requires the interaction of β-arrestins with membrane phospholipids, particularly PIP2 and PIP3. Previous experiments showed that elimination of the β-arrestin2 phosphoinositide binding pocket by the mutation of three basic residues created a mutant protein (β-arrestin2 RRK/Q) indistinguishable from wildtype β-arrestin2 with respect to interactions with 7TMRs and clathrin, but which did not internalize activated receptors (23).
Formation of a clathrin-coated pit begins with a nucleation event involving AP-2 and clathrin with PIP2, concentrations of which are elevated in the plasma membrane (24,25). As its outer clathrin layer begins to polymerize, the budding vesicle incorporates additional PIP2-binding proteins involved in trafficking and endocytosis, including AP180/CALM (26), epsin (27) and the large GTPase dynamin (28). The importance of PIP2 in these events makes its production by type I PIP5Ks a key factor in clathrin-coated pit-mediated internalization (29). Altering PIP2 homeostasis, either by genetic knock-out of PIP5K isoforms (30) or overexpression of a membrane-targeted phosphatidylinositol 5-phosphatase (24) causes gross defects in clathrin-coated vesicle endocytosis. Enzymatic activity of the PIP5Ks is positively regulated by many factors, including the small GTPase ARF6 (24,31) as well as phosphatidic acid (32) which has been shown to up regulate PIP5K activity 20-fold in vitro (33). Interestingly, PIP5K Iα has been shown to interact with DGKζ in an agonist-dependent manner, suggesting a mechanism for enhanced PIP2 production via co-localization of PIP5K Iα with a phosphatidic acid source (34).
As β-arrestins serve as a hub for components involved in 7TMR internalization, such as clathrin and AP-2, as well as the signaling proteins ARF6 (35) and the DGK family of enzymes, we questioned whether β-arrestins might coordinate PIP5K activity. Specifically, we hypothesized β-arrestins may affect the PIP5K Iα isoform, given its role in agonist-dependent internalization of receptors in non-neuronal cell types (36). Thus, we examined the roles of β-arrestins in the agonist-induced generation of PIP2 and the association of PIP5K Iα with the β2-AR. Additionally we investigated the role of PIP2 in regulating the association of β-arrestin2 and PIP5K Iα, utilizing the properties of the β-arrestin2 RRK/Q mutant. Ultimately, it is the goal of these studies to better understand how the interplay of PIP2, PIP5K Iα and β-arrestins coordinate internalization of agonist-stimulated 7TMRs.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Tissue culture reagents and Lipofectamine 2000 transfection reagent were purchased from Invitrogen. The radiolabeled compounds [3H]CGP-12177, [γ-32P]ATP and 32P-orthophosphate were purchased from Perkin Elmer. Chemically synthesized siRNAs were from Dharmacon and GeneSilencer transfection reagent was bought from Gene Therapy Systems. PI(4)P and purified lipid standards were sold by Avanti Polar Lipids. Thin layer chromatography plates were from Whatman International Ltd. Site-directed mutagenesis was done using a QuikChange II kit bought from Stratagene. All other materials were purchased from Sigma.
DNA Plasmids
FLAG-β-arrestin2 RRK/Q was created by introducing previously described point mutations (23) in a FLAG-tagged rat β-arrestin2 plasmid using a QuikChange II Site-Directed Mutagenesis Kit.
The FLAG-β-arrestin2 RRK/Q-5-kinase domain fusion protein (RRK5K) was engineered by PCR with rat FLAG-β-arrestin2 RRK/Q as the template and primers replacing the stop codon with a HinDIII restriction cut site. The PCR product was subsequently cut with XbaI and HinDIII and ligated into pcDNA 3.1 zeo− vector. The core kinase domain of PIP5K Iα (residues 59–438) was created by PCR with primers (5′-3′): AAAAAGCTTGGTGGCGATCCCGCGGTCCC TTCC and AAAAAGCTTCTAAACAAACCTGTAAGACTG, cut with HinDIII and inserted into the plasmid described above. DNA sequencing at the Duke University DNA Analysis Facility confirmed the finished plasmid encoding FLAG-β-arrestin2 RRK/Q followed by two glycine residues and the PIP5K Iα kinase domain with a terminal stop codon. Plasmid encoding His6-Myc-tagged PIP5K Iα was graciously provided by Dr. Matt Topham at the Huntsman Cancer Institute, University of Utah. All other plasmids used were created previously in the Lefkowitz lab.
Cell culture and Immunoprecipitation
HEK293 cell culture conditions, transient transfection and SDS-PAGE were as described previously (22). Breifly, cells were treated with isoproterenol or propranolol as indicated. Cells were washed once with PBS at 4°C and were harvested by gentle scraping, pelleted and resuspended in glycerol lysis buffer including protease inhibitors. Lysates were normalized for equal protein concentrations and immunoprecipitated with polyclonal rabbit β-arrestin antibody (A1CT) that has been previously described (37). Immunoprecipitation reactions were incubated at 4°C for 3 hours, washed 3 times with glycerol lysis buffer and then resuspended in SDS running buffer. Samples were subjected to SDS-PAGE analysis and Western blotting with polyclonal goat PIP5K Iα antibody (N-20, Santa Cruz Biotechnology).
In vitro binding experiments
HEK293 cells in 10-cm culture dishes transfected with His6-Myc-PIP5K Iα or empty vector were lysed in 1.0 ml glycerol lysis buffer (5.0mM HEPES, 250mM NaCl, 0.5% NP-40, 10% glycerol) containing 0.1% maltoside and protease inhibitor tablets (Roche Applied Science). Cells were incubated with lysis buffer for 1hour at 4°C for maximum solubilization of PIP5K Iα. Cell debris was cleared by centrifugation (20,000 × g for 20 min) and then tumbled with 10 μl of anti-Myc beads (Covance) for 1 hour at 4°C. Myc beads containing the bound PIP5K were then washed 3 × 1.0 ml of glycerol lysis buffer. The beads were subsequently suspended in 1.0 ml of buffer and then purified β-arrestin2 (previously described in (38)) was added to these beads at a final concentration of 1.0 μM. In conditions containing IP6, an equimolar concentration was used (1.0 μM) with respect to β-arrestin2. This mixture was tumbled at 4°C for 2 hours and the beads were then washed 5 × 1.0 ml of glycerol lysis buffer. Beads were suspended in sample buffer and analyzed by Western blotting. Detection of β-arrestin2 bound to PIP5K-beads was performed by A1CT antibody (37).
Whole-cell Lipid Radiolabeling and Separation
Radiolabeling and thin layer chromatography experiments were done as previously described (11). The solvent system used for phospholipid separation by TLC was a mixture of 46:17:15:14:8 (CHCl3: Acetone: MeOH: Glacial Acetic Acid: H2O). Phosphorimager screens were quantified on a Typhoon phosphorimager (Amersham) with volumes normalized for total protein content. Individual lipid species were identified by comparison to iodine stained lipid standards.
In Vitro PI(4)P Kinase Assay
Kinase assays were done as described previously (39). FLAG-β-arrestin (or FLAG-β-arrestin mutant) immunoprecipitates were washed and incubated 20 min at 37°C in 50 μL of kinase buffer (25 mM HEPES, 25 mM KCl, 2.5 mM magnesium acetate, 150 mM potassium glutamate, 10 μM CaCl2, 10 μg PI(4)P, 200 μM ATP and 10 μCi [γ-32P]ATP. Reactions were quenched by the addition of 600 μL CHCl3: MeOH (2:1) and prepared for TLC as described (11).
β2-AR Sequestration Assay
Whole-cell saturation binding experiments were done essentially as described (40). Briefly, HEK293 cells stably expressing FLAG-β2-AR were either unstimulated or treated 30 minutes with 10 μM isoproterenol in triplicate, washed twice with ice-cold PBS, and resuspended in 0.2% EDTA solution. These samples were normalized for total protein and equivalent microgram amounts were incubated with 10 nM [3H]CGP-12177 at 4°C for four hours. After the incubation, the samples were washed and collected using a cell harvester with Whatman GF/C glass microfiber filters and subsequently counted by liquid scintillation spectrometry.
Statistical Analysis
All statistics presented were from data analyzed using GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
RESULTS
To test whether β-arrestins can interact with PIP5K Iα, HEK293 cells with endogenous complements of β-arrestin2, PIP5K Iα and β2-AR were stimulated in a time course with 10 μM isoproterenol. With all proteins expressed at physiological levels, immunoprecipitation of β-arrestin2 with the anti-β-arrestin antibody A1CT (37) co-immunoprecipitated PIP5K Iα with isoproterenol stimulation showing a maximum effect (3.12 +/− 0.26 fold increase in association) at 5 minutes (Figure 1A–B). A ten minute preincubation of the cells with 100mM propranolol, a β2-AR antagonist, effectively blunted the isoproterenol response, verifying that the augmented association of β-arrestin and PIP5K Iα is specifically due to 7TMR stimulation. In support of this finding, FLAG epitope-tagged β-arrestin1 and 2 were overexpressed in HEK293 cells endogenously expressing PIP5K Iα. β-arrestin immunoprecipitation showed a preferential binding of PIP5K Iα to FLAG-β-arrestin2 over FLAG-β-arrestin1, with both isoforms of β-arrestin interacting with PIP5K Iα at levels significantly above vector controls (Figure 1C–D). Furthermore, stimulation of endogenous β2-ARs with 10 μM isoproterenol increased the amount of PIP5K Iα co-immunoprecipitated with FLAG-β-arrestin2 by an average of 2.46 +/− 0.11 fold relative to non-stimulated vector control samples. Comparable results were observed for HEK293 cells treated with 50 μM carbachol acting through endogenous M1 muscarinic receptors (Supplemental Figure 1A–B). FLAG-β-arrestin2 was also observed co-immunoprecipitating with Myc epitope-tagged PIP5K Iα in the same HEK293 experimental system and the protein-protein interaction was enhanced by multiple 7TMR-stimulating compounds: thrombin, carbachol, and isoproterenol (Supplemental Figure 2). Taken together, these findings show that endogenous and overexpressed β-arrestins can interact with PIP5K Iα in living cells and their association can be potentiated through 7TMR stimulation.
Figure 1.
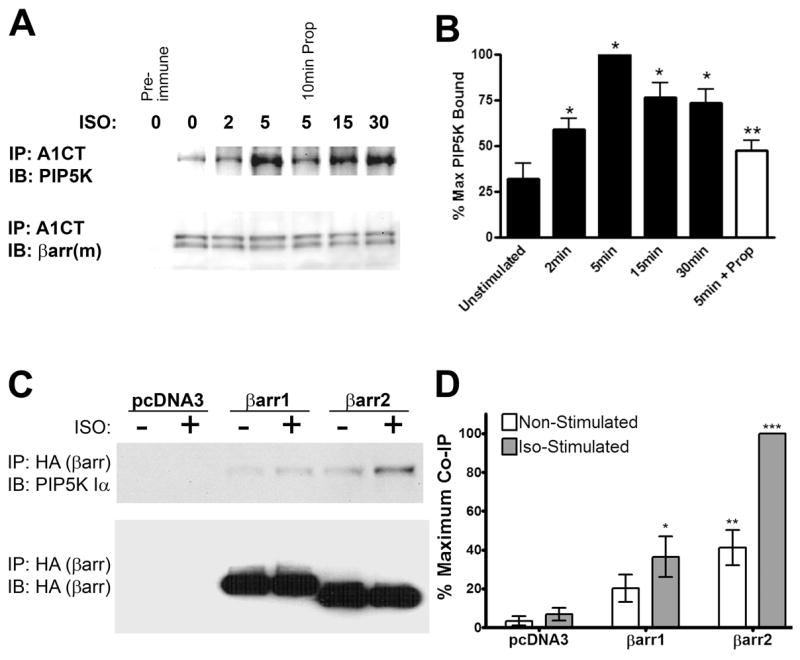
PIP5K Iα Co-immunoprecipitates with β-arrestins in HEK293 Cells. A. Western blots of endogenous β-arrestin immunoprecipitated with A1CT antibody and co-immunoprecipitated endogenous PIP5K Iα from HEK293 cells stimulated in a time course with 10 μM isoproterenol. Where indicated, cells were pre-incubated for 10 minutes with 100 μM propranalol. Immunoblots shown are representative of three independent experiments. B. Quantification of the isoproterenol time course experiments. Data were normalized as a percentage of the maximum observed co-immunoprecipitation of PIP5K Iα for each experiment. Statistical significance for time course samples was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (*, p < 0.05 vs. non-stimulated). Statistical significance for the propranalol treatment was determined by a paired t test comparing 5 minute isoproterenol-stimulated samples with or without propranalol preincubation (***, p < 0.001). C. Western blots of FLAG immunoprecipitates and lysates from HEK293 cells endogenously expressing PIP5K Iα transfected with FLAG β-arrestin1 or FLAG β-arrestin2 as shown. Where indicated, endogenous β2-ARs were stimulated with 10 μM isoproterenol (ISO) for five minutes. Immunoblots shown are representative of four independent experiments. B. Quantification of PIP5K Iα co-immunoprecipitation with FLAG-β-arrestins. All data were normalized as a percentage of the maximum observed co-immunoprecipitation of PIP5K Iα for each experiment. Statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (*, p < 0.05, **, p < 0.01, ***, p < 0.001 vs. non-stimulated control).
While the data indicate β-arrestin and PIP5K Iα can be found together in protein complexes isolated from cells, they do not address whether this interaction is direct or indirect. Notably in this regard, PIP5K Iα is capable of interacting with DGKζ (34) and AP-2 (39) both of which are known β-arrestin binding partners. Therefore, we screened a panel of β-arrestin2 deletion mutants to map regions required to bind PIP5K Iα and compared them with those previously published for other β-arrestin interactors. Interestingly, endogenous PIP5K Iα was observed co-immunoprecipitating with elements of both the N- and C- terminal halves of β-arrestin2 in addition to the full-length protein (Figure 2A). This stands in contrast to the DGKs and AP-2, binding partners common to both β-arrestin and PIP5K Iα, which map to discrete loci in the primary sequence (Figure 2B). To directly test the hypothesis that β-arrestin2 and PIP5K Iα can interact directly, we affinity purified Myc-tagged PIP5K Iα from HEK293 cells and incubated these PIP5K immunoprecipitates with purified recombinant β-arrestin2 in vitro (Figure 2C). A fivefold increase in β-arrestin2 binding was observed when incubated with Myc-tagged PIP5K Iα as opposed to control immunoprecipitates from cells transfected with empty vector. Furthermore, the interaction was effectively blocked by the inclusion of inositol hexaphosphate (IP6) in the binding buffer at an equimolar concentration with β-arrestin2. This latter finding is similar to previously reported effects of IP6 on the interactions of β-arrestins with 7TMRs and clathrin (23).
Figure 2.
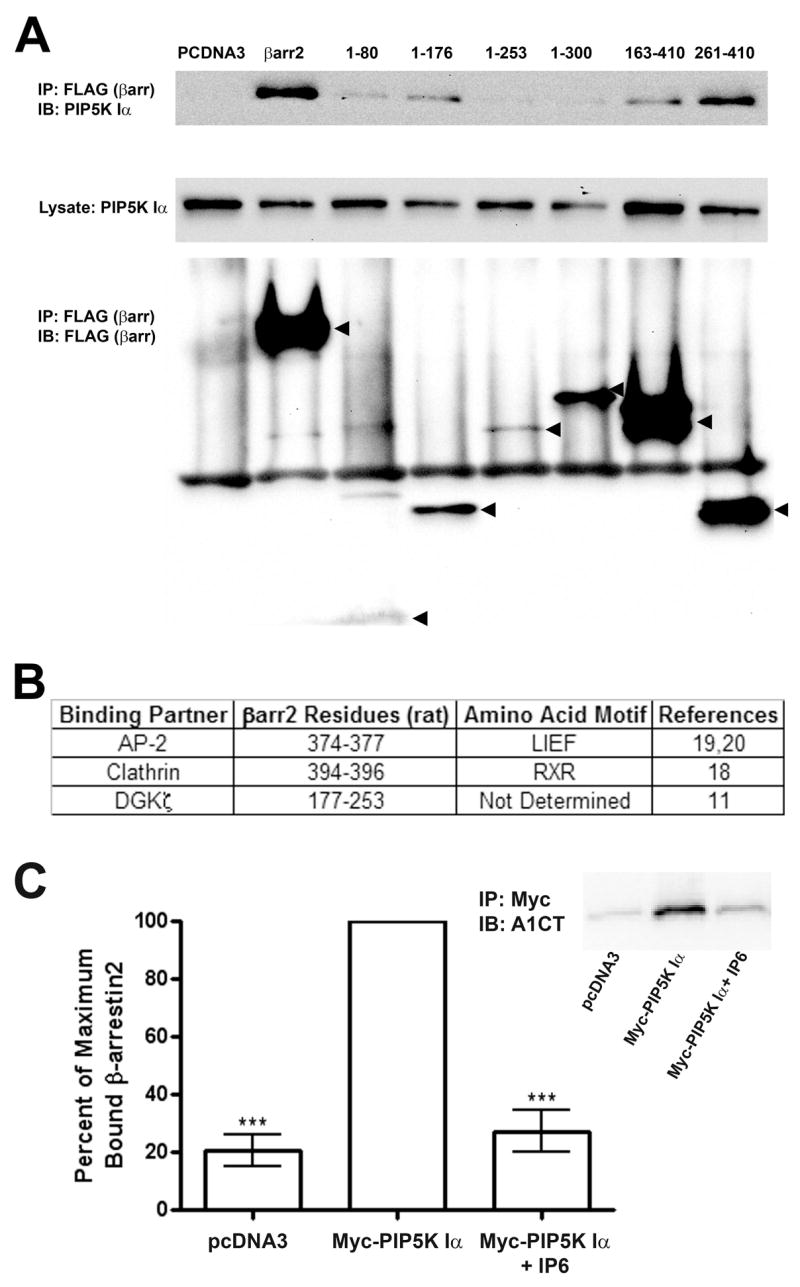
β-arrestin2 Directly Binds PIP5K Iα and is Independent of Association with DGKζ AP-2, or Clathrin. A. Western blots of endogenous PIP5K Iα (Upper and Middle panels) co-immunoprecipitated in FLAG immunoprecipitates from HEK293 cells were transfected with empty vector (pcDNA3), full-length FLAG-β-arrestin 2, or FLAG-tagged β-arrestin2 truncation mutants (Lower panel). Images shown are representative of three independent experiments. B. Table illustrating β-arrestin2 amino acid residues required for interacting with previously mapped binding partners. C. Quantification of purified recombinant β-arrestin2 co-immunoprecipitating with His6-Myc-PIP5K Iα affinity purified from HEK293 cells. Data from three independent experiments were normalized as a percentage of the maximum amount of β-arrestin2 detected by A1CT antibody for each experiment. Statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons ( **, p < 0.01, vs. PIP5K Iα immunoprecipitates without IP6). Inset: Representative A1CT immunoblot of purified β-arrestin2 co-immunoprecipited under the experimental conditions.
As β-arrestins are known to recruit enzymes such as Src (41) and DGKζ (11) into multi-protein signalasomes with activated receptors, we reasoned β-arrestin2 may similarly mediate the interaction of PIP5K Iα with 7TMRs. Endogenous PIP5K Iα co-immunoprecipitated with stably overexpressed β2-ARs in HEK293 cells in an agonist-dependent manner as demonstrated by dithiobis(succinimidyl)propionate (DSP) cross-linking experiments and this interaction is augmented 2.71 +/− 0.51 fold by stimulation with isoproterenol (Figure 3). Depletion of β-arrestin2 by RNAi had no significant affect on the amount of PIP5K Iα associated with the receptor under basal conditions. However, isoproterenol treatment of the cells transfected with β-arrestin2 RNAi failed to show agonist-stimulated recruitment of PIP5K Iα to the β2-AR (1.12 +/− 0.17 fold of CTL basal), indicating a requirement for β-arrestin2 in facilitating this enzyme-receptor interaction. This phenomenon was also observed in cells transfected with a second siRNA targeting β-arrestin2, reinforcing the specificity of the RNAi results (Supplemental Figure 3).
Figure 3.

Interaction of PIP5K Iα with the β2-AR is Agonist- and β-arrestin-dependent. A. Western blots of FLAG immunoprecipitates with DSP cross-linking from HEK293 cells stably expressing FLAG-β2-AR transfected with either control (CTL) or β-arrestin2 specific siRNA (βarr2). Immunoblots were probed for endogenous PIP5K Iα co-immunoprecipitating with the receptor and subsequently reprobed with A1CT and β2-AR antibodies. Cells were stimulated 5 minutes with 10 μM isoproterenol as indicated. Images shown are representative of three independent experiments. B. Densitometry quantification of receptor-associated PIP5K Iα with data normalized to non-stimulated control samples. Statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (**, p < 0.01 vs. non-stimulated control).
With the finding that β-arrestin2 is required to recruit PIP5K Iα to β2-ARs in response to agonist, we hypothesized β-arrestins may affect isoproterenol-stimulated PIP5K activity and PIP2 production in cells. Whole-cell 32P-orthophosphate labeling of HEK293 cells followed by lipid extraction and TLC separation showed isoproterenol stimulation of endogenous β2-ARs yielded no net change in radiolabeled PIP2 levels when β-arrestin2 and PIP5K Iα are expressed at endogenous levels (Figure 4). Overexpression of either β-arrestin2 or PIP5K Iα alone also showed no significant effect on the radiolabeled pool of PIP2 in either basal or stimulated samples. However, increasing expression of both proteins revealed a significant isoproterenol-stimulated elevation of 32P-PIP2 (2.1 +/− 0.3 fold versus basal control cells). Similar results were seen with carbachol stimulation of endogenous M1 muscarinic receptors in the same experimental system (Supplemental Figure 4). Thus, β-arrestin2 and PIP5K Iα show a synergistic effect on agonist-induced PIP2 production in cells, consistent with the β-arrestin-mediated scaffolding of PIP5K Iα to activated receptors.
Figure 4.

Overexpression of β-arrestin and PIP5K Iα Synergistically Enhances Isoproterenol-dependent PIP2 Production. A. Representative phosphorimager image of whole-cell 32P-labeled lipids from HEK293 cells after separation by TLC. Cells were co-transfected with FLAG-β-arrestin 2 and His6-Myc-PIP5K Iα and stimulated 5 minutes with 10 μM isoproterenol as indicated. Spots were identified by migration relative to iodine-stained lipid standards. B. Densitomitry quantification of radiolabeled PIP2 from the above images. Spot volumes were adjusted for equivalent total protein in each sample and normalized to control transfected non-stimulated cells. Values shown represent the mean +/− SE from three independent experiments. Statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (*, p < 0.05 vs. non-stimulated control).
To further investigate the role of β-arrestins in PIP5K Iα activation and targeting, we examined the properties of a rat β-arrestin2 mutant with the amino acids R234, R238 and K252 mutated to glutamine (RRK/Q). This is an orthologue of a previously described bovine β-arrestin2 mutant deficient in phosphoinositide binding, but which retains the ability to interact with clathrin and a 7TMR (rhodopsin) in vitro (23). However, despite binding receptors and clathrin equally as well as wildtype β-arrestin2, RRK/Q was significantly impaired in its ability to direct receptor internalization when transfected into COS cells. This deficiency was attributed to PIP2 and PIP3 providing additional sites of membrane attachment for β-arrestins recruited to 7TMRs. However, when we tested the RRK/Q mutant for interaction with PIP5K Iα, it did not show the agonist-stimulated interaction exhibited by the wildtype protein (Figure 5A–B). Even more dramatically, when FLAG-β-arrestin2 and FLAG-RRK/Q immunoprecipitates were assayed for endogenous co-immunoprecipitated PI(4)P kinase activity in vitro, wildtype β-arrestin2 was associated with robust kinase activity (20.0 +/− 4.0 fold over vector control) whereas β-arrestin2 RRK/Q was indistinguishable from the vector control (2.1 +/− 0.7 fold increase) (Figure 5C).
Figure 5.

Fusion of the Kinase Domain of PIP5K Iα to β-arrestin2 RRK/Q Rescues β2-AR Endocytosis. A. Immunoblots of Myc immunoprecipitates from HEK293 cells overexpressing His6-Myc-PIP5K Iα and either pcDNA3 vector, rat β-arrestin2-GFP (βarr2-GFP), or the β-arrestin2 mutant RRK/Q-GFP. Where indicated, cells were stimulated with 10 μM isoproterenol for five minutes. Images shown are representative of four independent experiments. B. Densitometry data from the PIP5K Iα IPs normalized to non-stimulated β-arrestin2-GFP samples. Statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (*, p < 0.05 vs. non-stimulated control). C. β-arrestin2 and RRK/Q immunoprecipitates were assayed in vitro for associated PI(4)P kinase activity. Data shown are the average +/− SE of three independent experiments with the inset shown as a representative image. Statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (**, p < 0.01 vs. pcDNA3 control). D. Internalization of β2-AR in response to isoproterenol-stimulation measured by whole-cell saturation binding of [H3]CGP-12177. HEK293 cells stably expressing the β2-AR were transfected with siRNA targeting the endogenous human β-arrestin2 and co-transfected with rat β-arrestin2-GFP, RRK/Q-GFP, or RRK5K (upper panel). Internalization for each sample was quantified as the percentage loss of [H3]CGP-12177 binding after 30 minute stimulation with 10 μM isoproterenol. Data shown are the average +/− SE of three independent experiments and statistical significance was determined using a one-way ANOVA with a Bonferroni post-hoc test to correct for multiple comparisons (*, p < 0.05 vs. pcDNA3; **, p < 0.01 vs. RRK/Q).
These data suggested that rather than elimination of a secondary membrane attachment point, a defect in receptor localized of PIP2 production and subsequent loss of PIP2-mediated processes such as clathrin-coated pit formation may be responsible for the mutant’s inability to internalize 7TMRs. Seeking to test this hypothesis, we constructed a chimaeric protein with FLAG-RRK/Q fused to the core catalytic domain of PIP5K Iα. This construct named RRK5K was expressed in HEK293 cells and unlike the RRK/Q mutant, FLAG immunoprecipitates of RRK5K promoted significant 32P phosphorylation of PI(4)P in vitro, indicating the fusion protein possessed PIP5K catalytic activity as intended (Supplemental Figure 5).
We next sought to determine if the incorporation of PIP5K activity in the RRK5K mutant was sufficient to rescue the loss of 7TMR internalization reported for the β-arrestin2 RRK/Q. HEK293 cells stably expressing the β2-AR were treated with β-arrestin2 siRNA to minimize the effect of endogenous β-arrestins on receptor sequestration. Concurrently, these cells were transfected with plasmids encoding either β-arrestin2-GFP, RRK/Q-GFP, or FLAG-RRK5K. All DNA constructs used originated from rat β-arrestin2 and consequently were unaffected by siRNA directed against human β-arrestin2 (Figure 5D, upper panel). These samples, either non-stimulated or isoproterenol-treated, were incubated with [3H]CGP 12177, a hydrophilic β2-AR ligand, and assayed under whole-cell saturation binding conditions. Receptor internalization was defined in these experiments as an isoproterenol-stimulated loss of cell-surface bound [3H]CGP relative to the non-stimulated counts for each transfection condition. Cells transfected with empty vector showed low levels of β2-AR internalization (7.1 +/− 3.6 %) consistent with previous β-arrestin2 RNAi experiments (22) while expression of β-arrestin2-GFP enhanced sequestration to 27.8 +/− 2.7%. As described previously (23), receptor endocytosis in RRK/Q transfected cells was negligible with 3.3 +/− 4.8% internalization. However, cells expressing the RRK5K mutant exhibited robust β2-AR sequestration (33.0 +/− 6.7%) suggesting that the chimaeric fusion of PIP5K activity with the RRK/Q mutant is sufficient to circumvent the defective interaction with PIP5K Iα and rescue β2-AR endocytosis. In a larger context, these findings also illustrate the importance of the interaction of β-arrestin with phosphoinositides and how this may affect protein-protein interactions and other β-arrestin-mediated processes.
DISCUSSION
Both β-arrestins and PIP2 produced by the PI(4)P 5-kinase family of enzymes are required for the efficient endocytosis of agonist-stimulated 7TMRs via clathrin-coated pits. In this report we have shown that β-arrestins (preferentially β-arrestin2) bind PIP5K Iα and mediate the association of PIP5K Iα with β2-ARs, with both processes potentiated by 7TMR stimulation. Also, as evidenced by the deficiencies of the RRK/Q mutant, formation of the β-arrestin-PIP5K Iα complex is dependent on the ability of β-arrestin to interact with PIP2/3 and disruption of this association effectively eliminates 7TMR internalization. Collectively, these data suggest a positive feedback mechanism where β-arrestin binding to phosphoinositides enhances 7TMR internalization by putting β-arrestin in a conformation to promote the interaction with PI5K Iα consequently elevating local PIP2 concentrations (Figure 6). It is also likely that other β-arrestin-associated signaling such as ARF6 and phosphatidic acid produced by DGKs further up-regulate PIP5K Iα enzymatic activity. Recent data by Kawasaki et al. showed that an interaction between AP-2 and catalytically active DGKδ are required for efficient clathrin-mediated endocytosis in HeLa and COS7 cells (42). These findings also support a model for receptor internalization where clathrin-coated pit formation is triggered by DGK-mediated upregulation of PIP5K activity in a process scaffolded by β-arrestins and the endocytic machinery. Thus, it appears the β-arrestins have adopted multiple roles in mediating internalization of stimulated receptors; connecting 7TMRs to the endocytic machinery and coordinating PIP5K Iα localization and activity.
Figure 6.
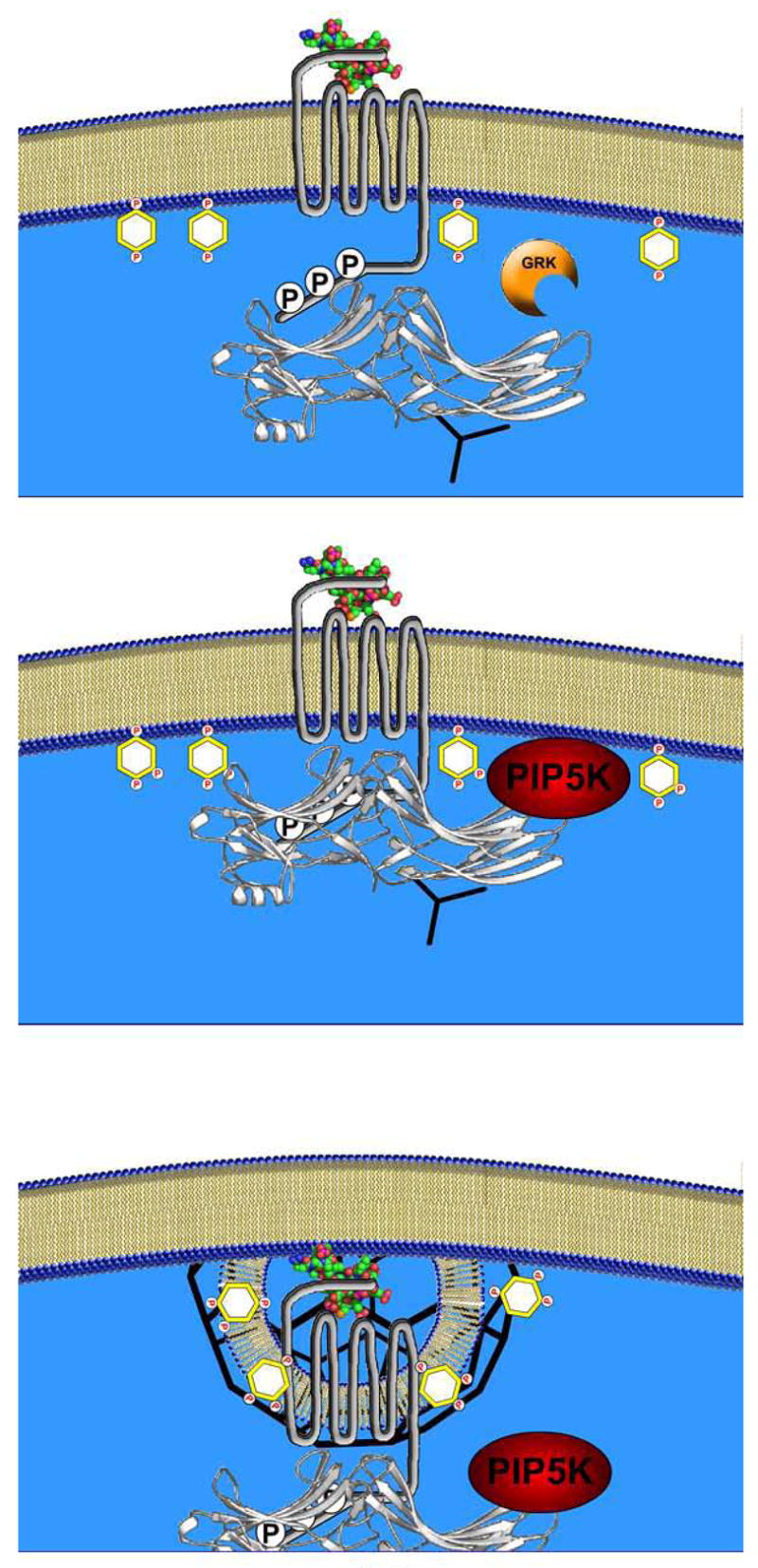
A Model of β-arrestin-PIP5K Iα Coordinated Clathrin-Coated Pit Formation and Receptor Endocytosis. Upper panel. Illustration of β-arrestin (white ribbon structure) recruitment of clathrin (black triskelion) to a GRK-phosphorylated, agonist-occupied 7TMR. Middle panel. β-arrestin binding to the receptor and interactions with PIP2 and PIP3 in the plasma membrane can promote an association with PIP5K Iα. Lower panel. β-arrestin-bound PIP5K Iα localizes to the activated 7TMR where it may enrich the local pool of PIP2. This elevated PIP2 concentration can promote clathrin-AP2 nucleation and 7TMR sequestration as well as other PIP2-dependent processes including the possibility of initiating a feed-forward mechanism of β-arrestin-PIP5K Iα interaction.
As mentioned above, in the whole-cell radiolabeled lipid extracts (Figure 4), there was no change in bulk PIP2 levels observed with isoproterenol stimulation when β-arrestins and PIP5K Iα were expressed endogenously. However, this result is not unexpected when the presence of endogenous phosphoinositide 5-phosphatases are taken into consideration. These enzymes, for which there currently are no pharmalogical inhibitors, can counteract agonist-stimulated PIP5K activity to maintain homeostasis. The observation that overexpression of both β-arrestin2 and PIP5K Iα are required for agonist-stimulated PIP2 synthesis, while overexpression of either protein alone had no effect would argue that efficient targeting and allosteric activation of PIP5K are equally as important as increasing kinase expression to overcome repression by the endogenous 5-phosphatases. Furthermore, we propose 7TMR-stimulated PIP2 changes do occur under physiological conditions, but on a local scale at macromolecular receptor signalosomes rather than the global lipid pool.
The discovery of PIP5K Iα as a novel β-arrestin binding partner also generates interesting hypotheses for future study. In addition to regulating the interaction of clathrin and AP-2, PIP2 can promote actin polymerization (43,44) as well as stimulate WASP, ARP2/3 and cofilin and activate many other key proteins regulating cytoskeletal rearrangement (reviewed in (45)). PIP2 also serves as a substrate for PI 3-kinase to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3), which promotes anti-apoptotic signaling and is required for chemotaxis (46). Interestingly, β-arrestin has been implicated as a signaling node in both of these processes. β-arrestin-dependent chemotaxis has been demonstrated for many chemokine 7TMRs including CXCR4 (47), PAR-2 (48), and the AT1aR (49). In the latter case, the β-arrestin chemotactic pathway was shown to be independent of G protein activity. Finally, β-arrestin-dependent events are required for signaling pathways both upstream and downstream of PI 3-kinase. β-arrestin1 is absolutely required for IGF-1-stimulated PI3K activity in MEF cells (50), while β-arrestin2 influences dopaminergic behaviors by promoting inactivation of Akt, the best studied PIP3 effector, by scaffolding Akt with its negative regulator, protein phosphatase 2a (51). Future investigation will determine if any or all of these signaling pathways require β-arrestin-scaffolded PIP5K activity and receptor-localized PIP2 production.
As the lists of β-arrestin binding proteins and signaling pathways continue to expand at a very rapid pace, there is a growing appreciation for how β-arrestin conformational changes govern interactions with binding partners. In this regard, β-arrestin post-translational modification or allosteric binding of a cofactor can alter the conformation of β-arrestin into one or more “active” states, with increased affinity for a particular protein-protein interaction (38). Precedents for this method of regulation have been previously shown for β-arrestin phosphorylation and ubiquitination which can modulate β-arrestin interactions with clathrin and the β2-AR respectively (52,53). The finding described here that a β-arrestin interaction with phosphoinositides is required for PIP5K Iα binding constitutes a novel β-arrestin regulatory mechanism. If in fact phosphoinositide binding alters β-arrestin conformation, additional binding partners may show different affinities for this state as opposed to the “inactive” β-arrestin. Moreover, in our in vitro binding experiments, IP6 was a potent inhibitor of the β-arrestin2-PIP5K Iα interaction. These findings are consistent with previous work by Gaidarov, et al. (23) showing bidirectional regulation of β-arrestin binding to clathrin and 7TMRs by IP6 (inhibitory) and PIP3 (stimulatory) respectively. Based on the data presented here, we propose that PIP5K Iα is another binding partner regulated by β-arrestin binding to either IP6 or PIP2/3 and that reciprocal regulation by inositol polyphosphates and phosphoinositides may be a general mechanism for modulation of additional β-arrestin interactions. Further experiments with the β-arrestin2 RRK/Q mutant in the future will likely help define the physiological consequences of IP6, PIP2 and PIP3 binding on β-arrestin functions.
Supplementary Material
Acknowledgments
Thank you to Dr. Matt Topham for comments on the manuscript. We would also like to thank Donna Addison and Elizabeth Hall for excellent secretarial assistance. This work was supported in part by National Institutes of Health grants HL16037 and HL70631. R.J.L. is an Investigator with the Howard Hughes Medical Institute.
The abbreviations used are
- 7TMR
seven transmembrane receptor
- AP-2
adaptor protein complex 2
- β2-AR
β2-adrenergic receptor
- βarr2
β-arrestin2
- DGK
diacylglycerol kinase
- GRK
G protein-coupled receptor kinase
- Iso
isoproterenol
- PDE
phosphodiesterase
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PIP5K Iα
phosphatidylinositol 4-phosphate 5-kinase type Iα
- RRK/Q
β-arrestin2 R237Q R239Q K251Q
- RRK5K
β-arrestin2 RRK/Q-5-kinase domain fusion protein
References
- 1.Lefkowitz RJ. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 2.Lefkowitz RJ, Shenoy SK. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 3.Marinissen MJ, Gutkind JS. Trends in pharmacological sciences. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 4.Rockman HA, Koch WJ, Lefkowitz RJ. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 5.Petrofski JA, Koch WJ. Journal of molecular and cellular cardiology. 2003;35:1167–1174. doi: 10.1016/s0022-2828(03)00243-8. [DOI] [PubMed] [Google Scholar]
- 6.Freedman NJ, Lefkowitz RJ. Recent progress in hormone research. 1996;51:319–351. discussion 352–313. [PubMed] [Google Scholar]
- 7.Pitcher JA, Freedman NJ, Lefkowitz RJ. Annual review of biochemistry. 1998;67:653–692. doi: 10.1146/annurev.biochem.67.1.653. [DOI] [PubMed] [Google Scholar]
- 8.Pitcher J, Lohse MJ, Codina J, Caron MG, Lefkowitz RJ. Biochemistry. 1992;31:3193–3197. doi: 10.1021/bi00127a021. [DOI] [PubMed] [Google Scholar]
- 9.Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:8879–8882. doi: 10.1073/pnas.84.24.8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ. Science. 2002;298:834–836 8. doi: 10.1126/science.1074683. [DOI] [PubMed] [Google Scholar]
- 11.Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ. Science. 2007;315:663–666. doi: 10.1126/science.1134562. [DOI] [PubMed] [Google Scholar]
- 12.Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Progress in neurobiology. 2002;66:61–79. doi: 10.1016/s0301-0082(01)00023-5. [DOI] [PubMed] [Google Scholar]
- 13.Bonifacino JS, Weissman AM. Annual review of cell and developmental biology. 1998;14:19–57. doi: 10.1146/annurev.cellbio.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. The Journal of biological chemistry. 1999;274:1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- 15.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, Lefkowitz RJ. The Journal of biological chemistry. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 16.Pierce KL, Maudsley S, Daaka Y, Luttrell LM, Lefkowitz RJ. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1489–1494. doi: 10.1073/pnas.97.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shenoy SK, Barak LS, Xiao K, Ahn S, Berthouze M, Shukla AK, Luttrell LM, Lefkowitz RJ. The Journal of biological chemistry. 2007;282:29549–29562. doi: 10.1074/jbc.M700852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 19.Laporte SA, Oakley RH, Holt JA, Barak LS, Caron MG. The Journal of biological chemistry. 2000;275:23120–23126. doi: 10.1074/jbc.M002581200. [DOI] [PubMed] [Google Scholar]
- 20.Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SS, Caron MG, Barak LS. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn S, Nelson CD, Garrison TR, Miller WE, Lefkowitz RJ. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1740–1744. doi: 10.1073/pnas.262789099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaidarov I, Krupnick JG, Falck JR, Benovic JL, Keen JH. EMBO J. 1999;18:871–881. doi: 10.1093/emboj/18.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krauss M, Kinuta M, Wenk MR, De Camilli P, Takei K, Haucke V. The Journal of cell biology. 2003;162:113–124. doi: 10.1083/jcb.200301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haucke V. Biochemical Society transactions. 2005;33:1285–1289. doi: 10.1042/BST0331285. [DOI] [PubMed] [Google Scholar]
- 26.Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, Gibson A, Hopkins CR, Evans PR, McMahon HT. Science. 2001;291:1051–1055. doi: 10.1126/science.291.5506.1051. [DOI] [PubMed] [Google Scholar]
- 27.Itoh T, Koshiba S, Kigawa T, Kikuchi A, Yokoyama S, Takenawa T. Science. 2001;291:1047–1051. doi: 10.1126/science.291.5506.1047. [DOI] [PubMed] [Google Scholar]
- 28.Lin HC, Barylko B, Achiriloaie M, Albanesi JP. The Journal of biological chemistry. 1997;272:25999–26004. doi: 10.1074/jbc.272.41.25999. [DOI] [PubMed] [Google Scholar]
- 29.Loijens JC, Boronenkov IV, Parker GJ, Anderson RA. Advances in enzyme regulation. 1996;36:115–140. doi: 10.1016/0065-2571(95)00005-4. [DOI] [PubMed] [Google Scholar]
- 30.Padron D, Wang YJ, Yamamoto M, Yin H, Roth MG. The Journal of cell biology. 2003;162:693–701. doi: 10.1083/jcb.200302051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aikawa Y, Martin TF. The Journal of cell biology. 2003;162:647–659. doi: 10.1083/jcb.200212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tolias KF, Rameh LE, Ishihara H, Shibasaki Y, Chen J, Prestwich GD, Cantley LC, Carpenter CL. The Journal of biological chemistry. 1998;273:18040–18046. doi: 10.1074/jbc.273.29.18040. [DOI] [PubMed] [Google Scholar]
- 33.Moritz A, De Graan PN, Gispen WH, Wirtz KW. The Journal of biological chemistry. 1992;267:7207–7210. [PubMed] [Google Scholar]
- 34.Luo B, Prescott SM, Topham MK. Cell Signal. 2004;16:891–897. doi: 10.1016/j.cellsig.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Claing A, Chen W, Miller WE, Vitale N, Moss J, Premont RT, Lefkowitz RJ. The Journal of biological chemistry. 2001;276:42509–42513. doi: 10.1074/jbc.M108399200. [DOI] [PubMed] [Google Scholar]
- 36.Barbieri MA, Heath CM, Peters EM, Wells A, Davis JN, Stahl PD. The Journal of biological chemistry. 2001;276:47212–47216. doi: 10.1074/jbc.C100490200. [DOI] [PubMed] [Google Scholar]
- 37.Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, Snyder SH, Caron MG, Lefkowitz RJ. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 38.Xiao K, Shenoy SK, Nobles K, Lefkowitz RJ. J Biol Chem. 2004;279:55744–55753. doi: 10.1074/jbc.M409785200. [DOI] [PubMed] [Google Scholar]
- 39.Krauss M, Kukhtina V, Pechstein A, Haucke V. Proc Natl Acad Sci U S A. 2006;103:11934–11939. doi: 10.1073/pnas.0510306103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker JG, Hall IP, Hill SJ. Br J Pharmacol. 2002;137:400–408. doi: 10.1038/sj.bjp.0704855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 42.Kawasaki T, Kobayashi T, Ueyama T, Shirai Y, Saito N. Biochem J. 2008;409:471–479. doi: 10.1042/BJ20070755. [DOI] [PubMed] [Google Scholar]
- 43.Yin HL, Janmey PA. Annual review of physiology. 2003;65:761–789. doi: 10.1146/annurev.physiol.65.092101.142517. [DOI] [PubMed] [Google Scholar]
- 44.Weernink PA, Meletiadis K, Hommeltenberg S, Hinz M, Ishihara H, Schmidt M, Jakobs KH. The Journal of biological chemistry. 2004;279:7840–7849. doi: 10.1074/jbc.M312737200. [DOI] [PubMed] [Google Scholar]
- 45.Sechi AS, Wehland J. Journal of cell science. 2000;113(Pt 21):3685–3695. doi: 10.1242/jcs.113.21.3685. [DOI] [PubMed] [Google Scholar]
- 46.Toker A, Cantley LC. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 47.Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7478–7483. doi: 10.1073/pnas.112198299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ge L, Ly Y, Hollenberg M, DeFea K. The Journal of biological chemistry. 2003;278:34418–34426. doi: 10.1074/jbc.M300573200. [DOI] [PubMed] [Google Scholar]
- 49.Hunton DL, Barnes WG, Kim J, Ren XR, Violin JD, Reiter E, Milligan G, Patel DD, Lefkowitz RJ. Molecular pharmacology. 2005;67:1229–1236. doi: 10.1124/mol.104.006270. [DOI] [PubMed] [Google Scholar]
- 50.Povsic TJ, Kohout TA, Lefkowitz RJ. The Journal of biological chemistry. 2003;278:51334–51339. doi: 10.1074/jbc.M309968200. [DOI] [PubMed] [Google Scholar]
- 51.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 52.Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ. The Journal of biological chemistry. 1997;272:31051–31057. doi: 10.1074/jbc.272.49.31051. [DOI] [PubMed] [Google Scholar]
- 53.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.