

Summary
The age-dependent decline in the self-renewal capacity of stem cells plays a critical role in aging, but the precise mechanisms underlying this decline are not well understood. By limiting proliferative capacity, senescence is thought to play an important role in age-dependent decline of stem cell self-renewal, although direct evidence supporting this hypothesis is largely lacking. We have previously identified the E3 ubiquitin ligase Smurf2 as a critical regulator of senescence. In this study, we found that mice deficient in Smurf2 had an expanded hematopoietic stem cell (HSC) compartment in bone marrow under normal homeostatic conditions, and this expansion was associated with enhanced proliferation and reduced quiescence of HSCs. Surprisingly, increased cycling and reduced quiescence of HSCs in Smurf2-deficient mice did not lead to premature exhaustion of stem cells. Instead, HSCs in aged Smurf2-deficient mice had a significantly better repopulating capacity than aged wild-type HSCs, suggesting that decline in HSC function with age is Smurf2-dependent. Furthermore, Smurf2-deficient HSCs exhibited elevated long-term self-renewal capacity and diminished exhaustion in serial transplantation. As we found that the expression of Smurf2 was increased with age and in response to regenerative stress during serial transplantation, our findings suggest that Smurf2 plays an important role in regulating HSC self-renewal and aging.
Keywords: Smurf2, Hematopoietic stem cells, Self-renewal, Aging, Senescence
Introduction
Aging is a complex process of gradual deterioration of physiological functions that involves virtually all cells and tissues. Tissue-specific stem cells, which are capable of self-renewal to preserve stem cell pools as well as differentiation into a variety of effector cells, play a central role in the normal homeostatic maintenance and regenerative repair of tissues throughout the lifetime of an organism. With advancing age, the self-renewal capacity of stem cells invariably declines, eventually leading to the accumulation of unrepaired, damaged tissues in old organisms (Rando 2006; Sharpless & DePinho 2007; Rossi et al. 2008). Aging of the hematopoietic system is driven by many intrinsic and extrinsic factors that adversely affect the production and functions of blood cells (Van Zant & Liang 2003; Chambers & Goodell 2007; Rossi et al. 2008). Increasing evidence suggests that attenuated hematopoietic stem cell (HSC) function plays an important role in hematopoietic aging, including anemia, decreased immune function and increased propensity for myeloid malignancies. The ability of HSCs to repopulate and reconstitute lethally-irradiated recipients is limited in serial transplantation, reflecting a finite potential of HSC self-renewal under regenerative stress (Ogden & Mickliem 1976; Harrison & Astle 1982). In aged mice, HSCs undergo distinct changes: they exhibit decreased repopulating ability, even though the number of phenotypic HSCs based on staining of cell surface markers increases with age in some laboratory mouse strains. Further, aged HSCs show a differentiation preference towards myeloid lineages at the expense of lymphoid differentiation (Morrison et al. 1996; Sudo et al. 2000; Liang et al. 2005; Rossi et al. 2005; Chambers et al. 2007). In humans, aged HSCs also show increased frequency and a bias towards myeloid potential (Pang et al. 2011).
The molecular mechanisms responsible for age-dependent decline in HSC self-renewal are complex and many factors have been implicated (Van Zant & Liang 2003; Chambers & Goodell 2007; Rossi et al. 2008). Senescence, a state of irreversible growth arrest that most cells enter at the end of their replicative lifespan, has been proposed to contribute to aging by reducing the renewal capacity of stem cells (Sharpless & DePinho 2004; Campisi 2005). Although direct evidence supporting the senescence theory of aging is lacking, several senescence regulators including p16INK4a (p16) and p53 have been found to play important roles in aging. The expression of p16 increases with age in many human and rodent tissues (Krishnamurthy et al. 2004; Ressler et al. 2006). Age-associated increase of p16 in mice coincides with a decline in the renewal capacity of stem cells in bone marrow, brain, and pancreas (Janzen et al. 2006; Krishnamurthy et al. 2006; Molofsky et al. 2006), although p16 up-regulation in aged HSCs has been challenged (Attema et al. 2009). HSCs in old mice lacking p16 have increased regenerative potential, suggesting that p16 plays a critical role in limiting HSC self-renewal (Janzen et al. 2006). In p53-null mice, the number of HSCs increases and they perform better in competitive repopulation, suggesting an enhanced self-renewal capacity (TeKippe et al. 2003; Liu et al. 2009). Moreover, mice with one wild-type allele and one mutant allele of p53 that lacks the N-terminal transactivation domain maintain cancer protection, but age prematurely including impairment of HSCs (Tyner et al. 2002; Maier et al. 2004; Dumble et al. 2007). These studies support an emerging link between senescence regulation and aging, and show the potential importance of senescence regulation in the aging of HSCs.
We have recently shown that the E3 ubiquitin ligase Smurf2 plays a critical role in regulating the senescence response in human and mouse cells (Zhang & Cohen 2004; Kong et al. 2011; Ramkumar et al. 2012). Adventitious expression of Smurf2 is sufficient to induce senescence in early passage cells (Zhang & Cohen 2004; Ramkumar et al. 2012), while deficiency in Smurf2 expression impairs the senescence response in culture and in vivo (Kong et al. 2011; Ramkumar et al. 2012). In this study, we found that Smurf2 deficiency led to increased proliferation and an expanded HSC compartment in bone marrow. Surprisingly, increased proliferation did not lead to early HSC exhaustion. Instead, Smurf2-deficient HSCs showed better repopulating ability and multi-lineage potential than wild-type cells with advancing age or under regenerative stress, suggesting a functional role of Smurf2 in the regulation of HSC self-renewal and aging.
Results
Increased expression of Smurf2 in mouse bone marrow (BM) during aging
We have shown previously that Smurf2 is an important regulator of senescence (Zhang & Cohen 2004; Kong et al. 2011; Ramkumar et al. 2012). To investigate whether Smurf2 plays a role in aging, we first examined the expression of Smurf2 in mouse BM and the LSK (Lin−Sca-1+c-kit++; Lin−: negative for lineage markers B220, CD3, CD11b, CD19, Gr-1 and Ter-119) population that is enriched for HSCs (Ikuta & Weissman 1992; Okada et al. 1992; Osawa et al. 1996). We found that Smurf2 expression was increased in total BM and LSK cells of aged (24-month) C57BL/6 mice compared to young (2-month) mice (Fig. 1).
Fig. 1.
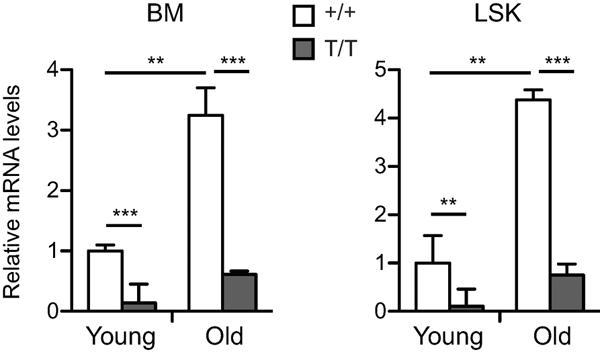
Increased Smurf2 expression in aged mice. Quantitative RT-PCR analysis of Smurf2 expression in bone marrow (BM) and sorted LSK (Lin−Sca1+c-kit++) cells of young (2-month) and old (24-month) wild-type (+/+) and Smurf2T/T (T/T) mice. Relative expression in young wild-type cells was set to be 1 after normalization with β-actin. Error bars are SD of 3 independent experiments. Student's t-test is used for statistical analysis. **: P<0.01, ***: P<0.001.
We have generated a Smurf2-deficient mouse model (Smurf2T/T, T for the trapped allele), in which a gene-trapping cassette is inserted into intron 1 of Smurf2 to disrupt its normal splicing (Ramkumar et al. 2012). The expression of Smurf2 was significantly reduced in total BM and LSK cells of Smurf2-deficient mice compared to wild-type (WT) mice (Fig. 1). Because of the hypomorphic nature of the trapped Smurf2 allele, there were residual normal splicing and Smurf2 expression in BM, LSK cells (Fig. 1), common lymphoid progenitors, multi-potent progenitors and HSCs (Fig. S1A) of Smurf2-deficient mice, similar to what we have found previously in other tissues (Ramkumar et al. 2012). Reduced Smurf2 expression in this mouse model allowed us to investigate whether Smurf2 has a functional role in HSC aging.
Expansion of long-term HSCs (LT-HSCs) in Smurf2 deficient mice
To investigate the role of Smurf2 in HSC aging, we used flow cytometry to enumerate BM populations enriched for LT-HSCs (Lin−Sca1+c-kit++CD150+Flt3−), short-term HSCs (ST-HSCs: Lin−Sca1+c-kit++CD150−Flt3−) and multi-potent progenitors (MPPs: Lin−Sca1+c-kit++CD150−Flt3+) (Adolfsson et al. 2001; Wilson et al. 2007; Papathanasiou et al. 2009) (Fig. 2A). We noticed a statistically significant increase (1.28-fold, P=0.026) in the total live BM cells collected from long bones of hind and forelegs of 2-month old Smurf2-deficient mice compared to age-matched WT mice (Fig. 2B), whereas gross body weights were not significantly different between WT and Smurf2-deficient mice (Fig. S1B). Although no significant difference in the frequencies of LT-HSCs, ST-HSCs, MPPs or LSK population was found between young WT and Smurf2-deficient mice (Fig. 2C), the total number of LT-HSCs in young Smurf2-deficient mice was significantly increased (1.64-fold, P=0.038) compared to WT mice (Fig. 2D). ST-HSCs, MPPs or LSK cells were also increased (1.30 to 1.45-fold) in young Smurf2-deficient mice, although the increases were not statistically significant (Fig. S2A).
Fig. 2.

Increased bone marrow (BM) cellularity and expanded long-term hematopoietic stem cell (LT-HSC) population in Smurf2-deficient mice. (A) Representative flow cytometry analysis of HSCs in BM of 2-month old wild-type (+/+) and Smurf2T/T (T/T) mice. Live lineage negative (Lin−) cells are gated and displayed for staining of Sca1 and c-Kit (left panels). LSK (Lin−Sca1+c-kit++) population is gated and displayed for staining of CD150 and Flt3 (right panels). LT-HSCs are defined as Lin−Sca1+c-kit++CD150+Flt3−, while Lin−Sca1+ckit++CD150−Flt3− and Lin−Sca1+c-kit++CD150−Flt3+ are used to enumerate short-term HSCs (ST-HSCs) and multi-potent progenitors (MPPs), respectively. (B) Average live BM cell numbers in young (2-month, N=16) and old (24 to 30-month, N=7) mice. (C) Frequencies of LT-HSC, ST-HSCs, MPP and LSK cells as percent of live BM cells in young (2-month old, N=11) mice. (D) Average LT-HSC numbers in young (2-month, N=11) and old (24 to 30-month, N=6) mice. (E) Frequencies of LT-HSC, ST-HSCs, MPP and LSK cells as percent of live BM cells in old (24 to 30-month, N=6) mice. Error bars are SD and Student's t-test is used for statistical analysis.
In aged (24 to 30-month) mice, we also found an increase in BM cellularity in Smurf2-deficent mice (1.59-fold, P=0.0007) compared to WT mice (Fig. 2B). Similar to what we found in young mice, the frequencies of LT-HSCs, ST-HSCs, MPPs and LSK population were not significantly different between aged WT and Smurf2-deficient mice (Fig. 2E). Although the average numbers of LT-HSCs, STHSCs, MPPs and LSK populations increased (1.73 to 2.21-fold) in aged Smurf2-deficient mice, there were great variations among individual aged mice and the increases were not significantly different from those in WT mice (Fig. 2D and Fig. S2B).
Complete blood count analyses of peripheral blood showed no significant difference between age-matched WT and Smurf2-deficient mice at either young (2-month) or old (18 to 20-month) age in various lineages, including white blood cells, red blood cells, lymphocytes, granulocytes, monocytes and platelets (Fig. S3). These results suggest that differentiation and commitment of HSCs to various lineages are not affected by Smurf2 deficiency.
Enhanced cell proliferation of LT-HSCs in Smurf2-deficient mice
The observed increases in BM cells and LT-HSCs in Smurf2-deficient mice suggest an enhanced cell proliferation of these cells. Supporting this notion, we found that the fraction of BM cells in the S phase of cell cycle was increased in 2-month old Smurf2-deficient mice compared to WT mice (Fig. S4A; 11.9±0.7% in Smurf2-deficient vs. 8.1±0.4% in WT, P=0.0012). To further investigate whether cell proliferation is enhanced in Smurf2-deficient LT-HSCs and progenitor populations, we gave young mice 4 injections of bromodeoxyuridine (BrdU) in a span of 24 hours to label LT-HSCs (Passegue et al. 2005). BrdU was incorporated in a significant portion of LT-HSCs (Lin−Sca1+c-kit++CD150+) using this labeling method (Fig. 3A). We found that BrdU incorporation was significantly increased in Smurf2-deficient LT-HSCs (1.8-fold, P=0.019) compared to WT LT-HSCs (Fig. 3B). Increased BrdU incorporation was also observed in Smurf2-deficient LSK and ST-HSC/MPP (Lin−Sca1+c-kit++CD150−) populations compared to WT cells (Fig. S4B).
Fig. 3.
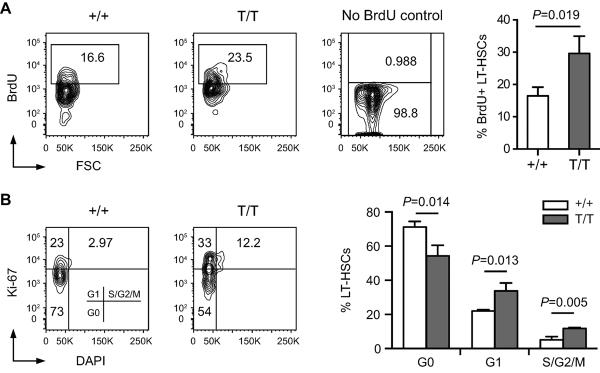
Increased proliferation and decreased quiescence of Smurf2-deficient hematopoietic stem cells (HSCs). (A) Representative BrdU incorporation analysis of HSCs in wild-type (+/+) and Smurf2T/T (T/T) mice. Long-term HSCs (Lin−Sca1+c-kit++CD150+) are gated and displayed for BrdU staining. BM from mice without BrdU injection was stained for BrdU as a control. Average of 3 independent experiments is shown at right. (B) Representative cell cycle analysis of HSCs in wild-type (+/+) and Smurf2T/T (T/T) mice. Long-term HSCs (Lin−Sca1+c-kit++CD150+) are gated and displayed for staining of Ki-67 and DAPI. Average of 3 independent experiments is shown at right. Error bars are SD and Student's t–test is used in statistical analysis.
As most LT-HSCs in adult mice remain quiescent (Cheshier et al. 1999), we combined Ki-67 staining with cell cycle analysis to determine the frequencies of LT-HSCs that are quiescent or cycling (Fig. 3B). Smurf2-deficient LT-HSCs (Lin−Sca1+c-kit++CD150+) exhibited decreased frequency in the G0 phase (54.2% in Smurf2-deficient vs. 71.2% in WT; P=0.014), and increased frequencies in the G1 (33.8% in Smurf2-deficient vs. 22.1% in WT; P=0.013) and S-G2/M phases (11.8% in Smurf2-deficient vs. 5.1% in WT; P=0.005) (Fig. 3B). Similarly, Smurf2-deficient LSK and ST-HSC/MPP (Lin−Sca1+c-kit++CD150−) populations also showed significantly increased cycling and decreased quiescence (Fig. S4C). We did not observe LT-HSCs in sub-G1 phase, suggesting that there is no apoptosis in these cells. Collectively, these data indicate that cell proliferation is enhanced in Smurf2-deficient LT-HSCs.
Enhanced repopulation ability of HSCs in aged Smurf2-deficient mice
As quiescence has been postulated to prevent premature stem cell exhaustion during aging (Orford & Scadden 2008), our observation of decreased frequency of quiescent LT-HSCs in Smurf2-deficient mice suggests that Smurf2-deficient HSCs may experience premature exhaustion. To determine the self-renewal ability and multi-lineage potential of HSCs, we used competitive repopulation (Harrison 1980), in which donor BM cells of either WT or Smurf2-deficient mice (CD45.2+) were mixed at a 1:1 ratio with competitor BM cells of 2-month old CD45.1+/CD45.2+ WT mice and injected into lethally-irradiated congenic recipient mice (CD45.1+). Contribution of donor, competitor and recipient in peripheral blood and BM of reconstituted recipients was determined by flow cytometry at different time post transplantation. As we have found previously that ~30% of Smurf2-deficient mice develop tumor spontaneously after 12-month of age, and ~70% of tumors are B-cell lymphomas in spleen (Ramkumar et al. 2012), only mice that were tumor free were used in this study. We selected aged mice that were physically fit without any sign of stress or illness and did not have visible tumors upon necropsy. Furthermore, we used flow cytometry to characterize spleen from these aged mice to make sure that there was no lymphoma as described (Ramkumar et al. 2013). Smurf2-deficient HSCs of young (2-month) mice were more efficient in repopulating B and myeloid cells than young WT HSCs and as efficient as WT HSCs in repopulating T cells in the peripheral blood of the reconstituted recipient mice (Fig. 4A). Interestingly, Smurf2-deficient HSCs of aged (24-month) animals showed significantly better repopulation ability than the age-matched WT HSCs (Fig. 4A), despite the decreased quiescence and increased cycling of Smurf2-deficient LT-HSCs. In fact, old Smurf2-deficient HSCs had similar repopulating ability as the young HSCs (Fig. 4A). Furthermore, analysis of BM and LSK population 6 months after transplantation showed increased contribution from aged Smurf2-deficient donor compared to WT donor (Fig. 4B). Collectively, these results indicate that HSCs in aged Smurf2-deficient mice have better repopulation ability and suggest that Smurf2 deficiency protects HSCs from early exhaustion during aging.
Fig. 4.
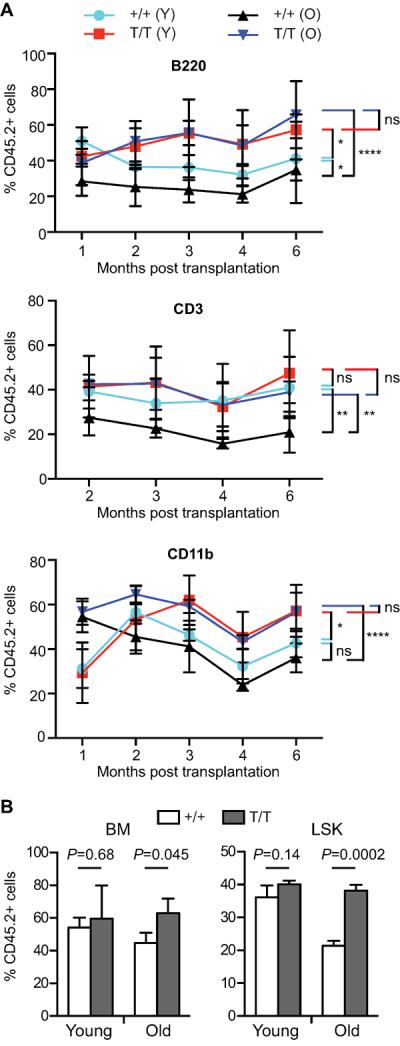
Hematopoietic stem cells (HSCs) in aged Smurf2-deficient mice exhibit enhanced repopulating ability. Donor bone marrow (BM) from wild-type (+/+) or Smurf2T/T (T/T) mice (CD45.2+) mixed at 1:1 ratio with competitor BM of 2-month old wild-type (CD45.1+/CD45.2+) mice are injected into lethally-irradiated recipient mice (CD45.1+). Contribution of donors is determined by CD45 staining. Both young (2-month) and old (24-month) donors were used. (A) Donor (CD45.2+) contributions to B (B220+), T (CD3+) and myeloid (CD11b+) lineages in peripheral blood of recipient mice (N=4) at indicated time post BM transplantation. Two-way ANOVA is used for statistical analysis. *: P<0.05, **: P<0.01, ****: P<0.0001, ns: not significant. (B) Donor contribution to BM and LSK (Lin−Sca1+c-kit++) cells in recipient mice (N=3) at 6 months post BM transplantation. Error bars are SD. Student's t–test is used in statistical analysis.
Smurf2-deficient LT-HSCs exhibit better long-term self-renewal capacity under regenerative stress
To investigate the long-term self-renewal and multi-lineage potential of HSCs, we used BM serial transplantation, in which LT-HSCs in donor BM are able to reconstitute lethally-irradiated recipients in successive but limited transplantations, reflecting the finite potential of HSC self-renewal (Ogden & Mickliem 1976; Harrison & Astle 1982). As the frequency of LT-HSCs in Smurf2-deficient mice was not significantly different from WT mice, we used the same number of BM cells from 2-month old WT and Smurf2-deficient mice in transplantation. We used either female or male donors in two independent experiments. During the 1st, 2nd or 3rd transplantation, 100% of recipient mice receiving donor BM from WT or Smurf2-deficient mice survived. Recipient mice from the 3rd transplantation were followed for a year and none of them died during observation. Staining of BM cells in the recipient mice during these successive transplantations revealed that >75% of the LT-HSC population had been reconstituted by the donor (Fig. S5A). 3 months after transplantation, complete blood count analysis of peripheral blood of recipient mice showed that various blood cell lineages were successfully reconstituted by both the WT and Smurf2-deficient donors (Fig. S5B).
During the 4th transplantation, all recipients receiving BM from WT mice died within 3 weeks post transplantation. In contrast, recipients receiving BM from Smurf2-deficient mice showed significantly better survival (Fig. 5A; 63–67% survival, P<0.001), suggesting that Smurf2-deficient HSCs have an enhanced long-term self-renewal capacity. None of these recipient mice died during 1-year of observation. We carried out a 5th transplantation with Smurf2-deficient donors, and found that 60% of the recipient mice survived for one year post transplantation without signs of illness or stress (Fig. S6).
Fig. 5.
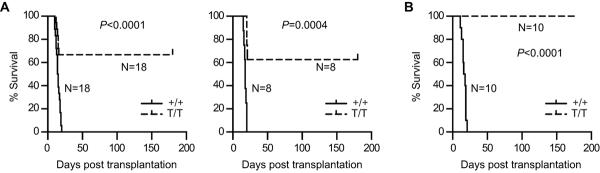
Increased long-term self-renewal capacity and multi-lineage potential in Smurf2-deficient hematopoietic stem cells (HSCs). Bone marrow (BM) cells of wild-type (+/+) and Smurf2T/T (T/T) mice are used as donors in serial BM transplantation. (A) Kaplan-Meier survival curves of recipient mice receiving BM from 2-month old donors in the 4th transplantation cycle. Two independent serial transplantation experiments using male (left panel) or female (right panel) donors are shown. (B) Kaplan-Meier survival curves of recipient mice receiving BM from 24-month old donors in the 4th transplantation cycle. The log-rank test is used for statistical analysis.
Similarly we carried out BM serial transplantation using aged mice as donors. All recipients receiving donor BM from either aged WT or Smurf2-deficient mice survived during the first 3 cycles of transplantation. Complete blood count analysis of recipient peripheral blood 3 months after transplantation showed that various blood cell lineages were successfully reconstituted by both the WT and Smurf2-deficient donors (Fig. S5C). During the 4th transplantation, all recipients receiving BM from WT mice died within 3 weeks post transplantation, while 100% recipients receiving BM from Smurf2-deficient mice survived more than 6 months post transplantation (Fig. 5B; P<0.0001). Collectively, these results indicate that Smurf2-deficient LT-HSCs have a better capacity in the long-term self-renewal and multi-lineage potential than the WT LT-HSCs. Furthermore, this stem cell-intrinsic functional enhancement is maintained in aged Smurf2-deficient mice.
Smurf2 regulates p16 expression during aging and under regenerative stress
Enhanced long-term self-renewal of Smurf2-deficient HSCs despite increased cycling and decreased quiescence in these cells prompted us to examine the expression of cyclin-dependent kinase inhibitors, as several of them have been found to regulate HSC self-renewal. HSCs lacking p18INK4c (p18) have increased self-renewal ability accompanied by increased cycling (Yuan et al. 2004), and p16 has been shown to limit HSC self-renewal during aging (Janzen et al. 2006). We found that p18 expression was not changed, while the expression of p16 and p19Arf (p19), which are located in the same INK4a locus, was increased in BM of aged (24-month) WT C57BL/6 mice compared to young (2-month) mice (Fig. 6A), consistent with a previous report (Krishnamurthy et al. 2004). Interestingly, the expression of p16 and p18 was increased in LSK cells with age, whereas p19 expression was undetectable in LSK cells (Fig. 6A). As our recent studies show that Smurf2 regulates the expression of p16 during senescence (Kong et al. 2011), we found that p16 expression was significantly suppressed in BM and LSK cells in Smurf2-deficient mice (Fig. 6A). In contrast, the expression of p19 showed a slight decrease and p18 was largely unchanged in Smurf2-deficient cells (Fig. 6A).
Fig. 6.

Age or regenerative stress-induced p16 expression is attenuated in Smurf2-deficient mice. (A) Quantitative RT-PCR analysis of gene expression in bone marrow (BM) and sorted LSK (Lin−Sca1+c-kit++) cells of young (2-month) and old (24-month) wild-type (+/+) and Smurf2T/T (T/T) mice. Relative expression in young wild-type cells was set to be 1 after normalization with β-actin. Only P<0.05 are indicated in pairwise comparison. (B) Quantitative RT-PCR analysis of gene expression in BM of donors and successive recipients in serial transplantation. Relative expression in wild-type donors is set to be 1 after normalization with β-actin. Error bars are calculated from standard deviations of three independent experiments. Student's t-test is used for statistical analysis. *: P<0.05, **: P<0.01, ***: P<0.001, ns: not significant.
We then analyzed the expression of these genes as well as Smurf2 in BM of reconstituted recipients during BM serial transplantation. We found that Smurf2 expression was increased during successive transplantation in mice receiving WT donor BM, concomitant with an increase in the expression of p16, p18 or p19 (Fig. 6B). While the increase in p18 and p19 expression was not consistently affected by Smurf2 deficiency, the increase in p16 expression was greatly attenuated in mice receiving the Smurf2-deficient donor BM (Fig. 6B), suggesting a possible link between p16 and Smurf2-mediated regulation of HSC self-renewal and aging.
Discussion
An important characteristic of HSCs in adult animals is their relative quiescence (Cheshier et al. 1999; Mahmud et al. 2001). It is thought that quiescence prevents premature stem cell exhaustion during aging (Orford & Scadden 2008). Many quiescence regulators have been found to control stem cell aging. For example, cyclin-dependent kinase inhibitor 1A (CDKN1A or p21) is required to maintain quiescence of HSCs. In the absence of p21, increased cell cycling leads to premature exhaustion and impaired self-renewal of HSCs (Cheng et al. 2000). Conversely, deletion of transcriptional factor ELF4, a negative regulator of quiescence, leads to increased quiescence and reduced exhaustion of HSCs (Lacorazza et al. 2006). In this study, we found that Smurf2 deficiency led to enhanced cell proliferation in BM. HSCs in Smurf2-deficient mice exhibited increased cell cycling and decreased quiescence. Surprisingly, increased proliferation of HSCs in Smurf2-deficient mice did not lead to premature stem cell exhaustion. Instead, Smurf2-deficient HSCs showed increased long-term self-renewal and multi-lineage repopulating capacity under regenerative stress in serial transplantation compared to WT HSCs. Furthermore, HSCs in aged Smurf2-deficient mice displayed better repopulating capability than HSCs in aged WT mice. Taken together, our results indicate that Smurf2 deficiency mitigates age-dependent decline in HSC self-renewal and function despite increased HSC cycling.
We found that Smurf2 expression was increased in BM and LSK population enriched for HSCs with advancing age or in response to regenerative stress in serial transplantation, concomitant with increases in the expression of p16, p19 and p18. HSCs lacking p18 have increased long-term self-renewal capacity, even though these cells show increased proliferation (Yuan et al. 2004). Our finding that p18 expression was unchanged by Smurf2 deficiency suggests that p18 is unlikely responsible for Smurf2-regulated decline in HSC self-renewal with age. It is documented that p16 expression increases with age in many human and rodent tissues, including BM and HSCs (Krishnamurthy et al. 2004; Janzen et al. 2006; Ressler et al. 2006), although age-dependent increase in p16 in HSCs has been challenged (Attema et al. 2009). In old mice, p16−/− HSCs perform much better than their WT counterparts in serial transplantation, suggesting that age-dependent increase of p16 expression limits HSC self-renewal (Janzen et al. 2006). We have recently found that Smurf2 regulates p16 expression during senescence (Kong et al. 2011). In Smurf2-deficient mice, age or regenerative stress-induced p16 expression was significantly attenuated, suggesting a possible link between HSC self-renewal and p16 expression regulated by Smurf2. Supporting this notion, we found that Smurf2-deficient HSCs of aged mice substantially outperformed aged WT HSCs in competitive repopulation and serial transplantation. In fact, old Smurf2-deficient HSCs performed as well as young HSCs, a phenotype similarly observed in old p16−/− HSCs (Janzen et al. 2006). However, Smurf2-deficient HSCs in young mice behaved differently from young p16−/− HSCs, which are more readily depleted in serial transplantation than WT HSCs (Janzen et al. 2006). In contrast, HSCs in young Smurf2-deficient mice also outperformed WT HSCs in serial transplantation and competitive repopulation. It is possible that Smurf2 limits HSC self-renewal in a p16-dependent manner only in aged mice, or Smurf2 deficiency protects age-dependent decline in HSC self-renewal independently of p16. Additional studies are needed to address the precise role of p16 in Smurf2-mediated regulation of HSC self-renewal and aging.
In complex organisms such as mammals, many somatic tissues such as bone marrow are capable of renewal, repair, and even regeneration. Renewable tissues allow the replacement of damaged cells, offering a clear advantage over post-mitotic tissues. It is important for stem cells to maintain life-time self-renewal and proliferation. In this study, we found that Smurf2 expression was increased with advancing age in BM and LSK cells. More importantly, HSCs in old Smurf2-deficient mice maintain their self-renewal capacity at the level similar to young HSCs, suggesting that functional decline in HSC self-renewal during aging is Smurf2-dependent. We have previously shown that Smurf2 is an important regulator of senescence (Zhang & Cohen 2004; Kong et al. 2011; Ramkumar et al. 2012). Increased expression of Smurf2 is sufficient to induce senescence (Zhang & Cohen 2004; Ramkumar et al. 2012), while loss of Smurf2 expression impairs the senescence response in culture and in vivo (Kong et al. 2011; Ramkumar et al. 2012). Senescence is proposed to contribute to aging by depleting the renewal capacity of tissues and/or by interfering with tissue homeostasis and functions (Sharpless & DePinho 2004; Campisi 2005). Although cells with characteristics of senescence have been detected to accumulate with age in vivo, their functional contribution to aging is still not completely clear (Sharpless & DePinho 2004; Campisi 2005). Our study provides evidence for Smurf2 in regulating HSC self-renewal and aging, further strengthening the emerging link between senescence and stem cell aging.
Experimental Procedures
Smurf2-deficient mice
Smurf2-deficient mice as described previously (Ramkumar et al. 2012) had been backcrossed to C57BL/6 (CD45.2+) for more than 10 generations. All mouse studies were carried out according to guidelines approved by the Institutional Animal Care and Use Committee of University of Massachusetts Medical School.
Complete blood count
Peripheral blood was bled from mouse tail veins into EDTA-coated tubes and complete blood count analysis was performed using the Heska CBC-Diff veterinary hematology system (Heska, Loveland, CO).
Flow cytometry
BM cells were harvested from long leg bones and resuspended in staining medium consisting of biotin-, flavin and phenol red-deficient RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 2% FBS (Hyclone, Logan, UT), 10 mM HEPES (pH 7.2), 1 mM EDTA and 0.02% sodium azide. After filtering through 70 μm nylon mesh, BM cells were incubated with anti-CD16/32 antibody (BioXCell, West Lebanon, NH) for 10 min on ice to block Fc receptors, and then incubated with primary antibodies for 20 min. The lineage cocktail contained biotin-conjugated antibodies to B220 (RA3-6B2), CD3e (145-2C11), CD11b (M1/70), CD19 (1D3), Ly-6G (RB6-8C5) and Ter-119 (TER-119). Additional antibodies for HSC analysis were Ly-6A/E(Sca1)-FITC or -PerCp-Cy5.5 (D7), CD117(c-Kit)-PE-Cy7 or -APC (2B8), CD135(Flt3)-PE (A2F10) and CD150-APC (mShad150). All antibodies were purchased from eBioscience (San Diego, CA). Cells stained with biotin-labeled antibodies were incubated with streptavidin-eFluor 450 (eBioscience) for 15 min on ice and washed three times with staining medium. After the final wash, cells were resuspended in staining medium with 1 μg/ml propidium iodide to exclude dead cells. Flow cytometry analysis was performed on a 5-laser, 18-detector LSR II FACS (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software (Treestar, Ashland, OR).
Cell cycle and cell proliferation analyses
Mice were injected with 1 mg BrdU intraperitoneally every 6 hours for 24 hours as described previously (Passegue et al. 2005). BM cells were stained for HSCs as described above, fixed and permeabilized using the BrdU flow kit (BD Biosciences). These cells were then stained with anti-BrdU-FITC or anti-Ki-67-PerCp-Cy5.5 (Sola15) antibody for 20 min. Cells stained for Ki-67 were further incubated with DAPI for 30 min at room temperature as described (Yang et al. 2008). Stained cells were analyzed by flow cytometry as described above.
Serial transplantation and competitive repopulation
WT or Smurf2-deficient mice (CD45.2+) were used as donors, and congenic CD45.1+ mice (The Jackson Laboratory, Bar Harbor, ME) were used as recipients. 8 to 10-weeks old Recipient mice were lethally irradiated (10 Gy) using a Cs137 irradiator 24 hours before transplantation, and were treated with antibiotics (0.5 mg/ml neomycin and 100U/ml polymyxin-B) in drinking water 24 hours prior to exposure to radiation until 1 month after transplantation. For serial transplantation, 5×106 donor BM cells were injected retro-orbitally into 8 to 10-week old lethally irradiated recipient mice. The transplantation cycle was repeated every 2 months.
For competitive repopulation, 2-month old WT CD45.1+/CD45.2+ mice were used as competitors. 1×106 donor BM cells were mixed with 1×106 competitor BM cells, and injected into 8 to 10-weeks old lethally irradiated recipient mice. The relative contributions from the donors, competitors or recipients in peripheral blood and BM of reconstituted recipients were analyzed in flow cytometry using antibodies to CD45.2-FITC (104) and CD45.1-PE-Cy7 (A20). Multi-lineage reconstitution in peripheral blood was analyzed using antibodies to B220-APC (RA3-6B2), CD3e-biotin (145-2C11) and CD11b-APC-Cy7 (M1/70, BD Biosciences). Antibodies were purchased from eBioscience unless specified.
Quantitative RT-PCR
Total RNA was isolated from freshly collected BM or FACS sorted LSK cells using Trizol (Invitrogen), and reverse-transcribed using Superscript II reverse transcriptase (Invitrogen). Real-time PCR was carried out on MyiQ iCycler using SYBR Green PCR kit (Bio-Rad, Hercules, CA). The following primers were used: Smurf2 (5'-ATGAAGTCATTCCCCAGCAC-3'; 5'-AACCGTGCTCGTCTCTCTTC-3'), p16 (5'-CGAACTCTTTCGGTCGTACCC-3'; 5'-CGAATCTGCACCGTAGTTGAG-3'), p18 (5'-GGGGACCTAGAGCAACTTACT-3'; 5'-AAATTGGGATTAGCACCTCTGAG-3'), p19 (5'-GCTCTGGCTTTCGTGAACATG-3'; 5'-TCGAATCTGCACCGTAGTTGAG-3') and β-actin (5'-GCTCTTTTCCAGCCTTCCTT-3', 5'-GTGCTAGGAGCCAGAGCAGT-3').
Statistical analysis
Data were presented as mean ± SD. Two-tailed and unpaired Student's t-test was used for pairwise comparisons. Two-way ANOVA was used for multiple comparisons. Kaplan-Meier survival curves were plotted using GraphPad Prism 5.0, and statistical significance was analyzed using the log-rank test. P<0.05 was considered as statistically significant.
Supplementary Material
Acknowledgements
We thank Dr. Jie Song and Henry Huang for providing reagents, Erin Cloherty for excellent technical assistance, and members of Zhang lab for helpful discussions. This work is supported by grants from the National Cancer Institute (R01CA131210) and The Ellison Medical Foundation (AG-NS-0347-06) to H.Z.
Footnotes
Author Contributions C.R., R.M.G. and H.Z. designed the experiments, C.R., Y.K., S.E.T. and R.M.G. performed the experiments, C.R., R.M.G. and H.Z. analyzed the data. C.R. and H.Z. wrote the manuscript.
The authors declare no conflict of interest.
References
- Adolfsson J, Borge OJ, Bryder D, Theilgaard-Monch K, Astrand-Grundstrom I, Sitnicka E, Sasaki Y, Jacobsen SE. Upregulation of Flt3 expression within the bone marrow Lin(−)Sca1(+)c-kit(+) stem cell compartment is accompanied by loss of self-renewal capacity. Immunity. 2001;15:659–669. doi: 10.1016/s1074-7613(01)00220-5. [DOI] [PubMed] [Google Scholar]
- Attema JL, Pronk CJ, Norddahl GL, Nygren JM, Bryder D. Hematopoietic stem cell ageing is uncoupled from p16 INK4A-mediated senescence. Oncogene. 2009;28:2238–2243. doi: 10.1038/onc.2009.94. [DOI] [PubMed] [Google Scholar]
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Goodell MA. Hematopoietic stem cell aging: wrinkles in stem cell potential. Stem cell reviews. 2007;3:201–211. doi: 10.1007/s12015-007-0027-1. [DOI] [PubMed] [Google Scholar]
- Chambers SM, Shaw CA, Gatza C, Fisk CJ, Donehower LA, Goodell MA. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007;5:e201. doi: 10.1371/journal.pbio.0050201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Cheshier SH, Morrison SJ, Liao X, Weissman IL. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci USA. 1999;96:3120–3125. doi: 10.1073/pnas.96.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumble M, Moore L, Chambers SM, Geiger H, Van Zant G, Goodell MA, Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE. Competitive repopulation: a new assay for long-term stem cell functional capacity. Blood. 1980;55:77–81. [PubMed] [Google Scholar]
- Harrison DE, Astle CM. Loss of stem cell repopulating ability upon transplantation. Effects of donor age, cell number, and transplantation procedure. J Exp Med. 1982;156:1767–1779. doi: 10.1084/jem.156.6.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta K, Weissman IL. Evidence that hematopoietic stem cells express mouse c-kit but do not depend on steel factor for their generation. Proc Natl Acad Sci USA. 1992;89:1502–1506. doi: 10.1073/pnas.89.4.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- Kong Y, Cui H, Zhang H. Smurf2-mediated ubiquitination and degradation of Id1 regulates p16 expression during senescence. Aging Cell. 2011;10:1038–1046. doi: 10.1111/j.1474-9726.2011.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacorazza HD, Yamada T, Liu Y, Miyata Y, Sivina M, Nunes J, Nimer SD. The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell. 2006;9:175–187. doi: 10.1016/j.ccr.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Liang Y, Van Zant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106:1479–1487. doi: 10.1182/blood-2004-11-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmud N, Devine SM, Weller KP, Parmar S, Sturgeon C, Nelson MC, Hewett T, Hoffman R. The relative quiescence of hematopoietic stem cells in nonhuman primates. Blood. 2001;97:3061–3068. doi: 10.1182/blood.v97.10.3061. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- Ogden DA, Mickliem HS. The fate of serially transplanted bone marrow cell populations from young and old donors. Transplantation. 1976;22:287–293. doi: 10.1097/00007890-197609000-00010. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, Suda T. In vivo and in vitro stem cell function of c-kit- and Sca-1-positive murine hematopoietic cells. Blood. 1992;80:3044–3050. [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Osawa M, Nakamura K, Nishi N, Takahasi N, Tokuomoto Y, Inoue H, Nakauchi H. In vivo self-renewal of c-Kit+ Sca-1+ Lin(low/-) hemopoietic stem cells. J Immunol. 1996;156:3207–3214. [PubMed] [Google Scholar]
- Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci USA. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathanasiou P, Attema JL, Karsunky H, Xu J, Smale ST, Weissman IL. Evaluation of the long-term reconstituting subset of hematopoietic stem cells with CD150. Stem Cells. 2009;27:2498–2508. doi: 10.1002/stem.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkumar C, Cui H, Kong Y, Jones SN, Gerstein RM, Zhang H. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat Commun. 2013;4:2598–2610. doi: 10.1038/ncomms3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramkumar C, Kong Y, Cui H, Hao S, Jones SN, Gerstein RM, Zhang H. Smurf2 regulates the senescence response and suppresses tumorigenesis in mice. Cancer Res. 2012;72:2714–2719. doi: 10.1158/0008-5472.CAN-11-3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080–1086. doi: 10.1038/nature04958. [DOI] [PubMed] [Google Scholar]
- Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Durr P, Wlaschek M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006;5:379–389. doi: 10.1111/j.1474-9726.2006.00231.x. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci USA. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Jamieson CH, Weissman IL. Stems cells and the pathways to aging and cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004;113:160–168. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192:1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeKippe M, Harrison DE, Chen J. Expansion of hematopoietic stem cell phenotype and activity in Trp53-null mice. Exp Hematol. 2003;31:521–527. doi: 10.1016/s0301-472x(03)00072-9. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee Park S, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- Van Zant G, Liang Y. The role of stem cells in aging. Exp Hematol. 2003;31:659–672. doi: 10.1016/s0301-472x(03)00088-2. [DOI] [PubMed] [Google Scholar]
- Wilson A, Oser GM, Jaworski M, Blanco-Bose WE, Laurenti E, Adolphe C, Essers MA, Macdonald HR, Trumpp A. Dormant and self-renewing hematopoietic stem cells and their niches. Ann N Y Acad Sci. 2007;1106:64–75. doi: 10.1196/annals.1392.021. [DOI] [PubMed] [Google Scholar]
- Yang Q, Kardava L, St Leger A, Martincic K, Varnum-Finney B, Bernstein ID, Milcarek C, Borghesi L. E47 controls the developmental integrity and cell cycle quiescence of multipotential hematopoietic progenitors. J Immunol. 2008;181:5885–5894. doi: 10.4049/jimmunol.181.9.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cohen SN. Smurf2 up-regulation activates telomere-dependent senescence. Genes Dev. 2004;18:3028–3040. doi: 10.1101/gad.1253004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.