

Abstract
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the two most common neurodegenerative disorders, and are characterized by deposition of specific proteins in the brain. If similar abnormal protein deposits are present in the eye, it would facilitate noninvasive diagnosis and monitoring of disease progression. We therefore evaluated expression of proteins associated with AD and PD pathology in postmortem eyes and brains in a case-control study. Eyes from 11 cases of AD, 6 cases of PD or PD with dementia, and 6 age-matched controls were retrieved from the autopsy archives of The Johns Hopkins Hospital. Immunostains for β-amyloid, phospho-tau and α-synuclein and Congo red stains were performed in the same laboratory in both brains and eyes. No amyloid deposits or abnormal tau accumulations were detected in the lens, retina or other structures in the eyes of AD patients. Eyes also lacked definite Lewy bodies or Lewy neurites in either PD or AD cases. Patchy cytoplasmic α-synuclein positivity was seen in the retina of AD, PD and control cases, but did not correlate with the presence or extent of Lewy body pathology in the brain. Abnormal protein aggregations characteristic of AD and PD are thus not commonly present in the retinas or lens of affected patients when assayed using the same protocols as in the brain. This suggests that β-amyloid, phospho-tau nd α-synuclein either do not deposit in the eye in a manner analogous to brain, or are present at lower levels or in different forms.
Keywords: amyloid, eye, lens, synuclein, tau
INTRODUCTION
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are the two most common neurodegenerative disorders, with AD accounting for approximately two-thirds of cases overall (26). The characteristic microscopic finding in AD is neuronal loss in the cerebral cortex accompanied by the accumulation of neurofibrillary tangles (NFTs) and the formation of neuritic plaques (NPs). NFTs are abnormal aggregations of hyperphosphorylated tau proteins in the neuronal bodies and processes, while non-phosphorylated tau is present in both AD and normal brains (12). NPs are complex lesions of the cortical neuropil containing extracellular β-amyloid (Aβ) protein deposits, dystrophic neurites, activated microglia and fibrillary astrocytes (29). Pathological findings associated with PD include loss of dopaminergic neurons and the presence of Lewy bodies (LBs), which are intraneuronal cytoplasmic proteinaceous inclusions made up largely of α-synuclein (10).
Definitive diagnosis of AD and PD still relies on postmortem examination of the brain, but detection of AD- and PD-associated proteins in living patients would facilitate early initiation of treatment and the tracking of therapeutic response. It has been suggested that pathology associated with AD and other neurodegenerative diseases is present in the eye, and that diagnosis of these conditions may be possible through direct optical imaging of abnormal proteins in the lens and retina (6, 13, 16). Such noninvasive tests of ocular tissue would greatly facilitate the diagnosis and treatment of these common diseases.
The concept that AD might affect the retina was first proposed several decades ago. In 1986, Hinton and colleagues compared 4 retinas and 10 optic nerves obtained at autopsy from affected individuals and controls, and concluded that reductions in ganglion cell number and nerve fiber layer thickness, as well as axonal degeneration of the optic nerve, were associated with the neurodegenerative process (9). More recent optical coherence tomography and multifocal electroretinogram studies have confirmed significant nerve fiber layer thinning and functional defects in the retinas of AD patients, although these are often of modest magnitude and present only focally (11, 18, 24). Animal models in which AD-associated proteins are overexpressed provide additional support for the concept that the disease can affect the eye (1, 2, 5, 25, 27, 30), although in such models it is often unclear if the levels or sites of abnormal protein expression faithfully recapitulate those seen in human disease.
Studies of abnormal protein deposition in the retinas of human patients with AD have yielded disparate results. In the initial report of retinal changes in four AD cases by Hinton and colleagues, no NFTs or amyloid deposition was found (9). A subsequent light-microscopic and ultrastructural study by the same group of retinas and optic nerves from 16 AD patients showed degeneration in the retinal ganglion cells but no NFTs, NPs or amyloid angiopathy (3). More recently, immunohistochemical techniques have been applied to answer these questions.
Loffler and colleagues examined non-phosphorylated tau, amyloid precursor protein and Aβ in 24 eyes of varying ages, and found increased anti-amyloid precursor protein (APP) (but not Aβ) immunoreactivity in retinas of older individuals (17). However, none of the 24 cases were known to have AD. Leger and colleagues examined 19 enucleated eyes of patients 49–87 years of age, two of whom had a clinical dementia, and found increasing tau-positive ganglion cells with aging but no phospho-tau or Aβ in the retinas (16). In another study, cryosections from postmortem globes of eight patients with a definite AD diagnosis and five with possible or probable AD, as well as controls, were examined by Koronyo-Hamaoui and colleagues using thioflavin-S and curcumin stains, as well as Aβ immunohistochemistry. These investigators found evidence for Aβ deposition in the retina of patients with dementia but not in controls (14). Finally, Schon and colleagues examined postmortem retinas of six patients who had suffered from AD and from from progressive supranuclear palsy (28). While they could detect hyperphosphorylated tau, they did not observe any fibrillar tau or Aβ aggregates.
The lens has also been proposed as an intraocular site in which AD-related amyloid can accumulate. In 2003, Goldstein and colleagues reported using Western blot and mass spectrometry analysis of postmortem eyes from individuals with and without AD that Aβ accumulated to a greater degree in those with the neurodegenerative condition. They also localized this protein to the supranuclear region using Congo red staining and Aβ immunohistochemistry, and suggested that its accumulation in the cytosol of the supranuclear cortical lens fiber cells leads to formation of supranuclear cataracts in AD patients (7). Subsequently, the same group has identified Aβ deposits in the lenses of Down syndrome patients, who generally manifest early-onset AD (23).
Thus, while some groups have identified AD-associated proteins in the retina or lens of affected patients, others have not. In many of the studies described above, only a few AD cases were examined, or the status of commonly used markers such as phosphorylated tau were not assessed. Techniques such as cryosection or curcumin staining not commonly used in routine AD diagnosis were also often utilized. Indeed, in most of the studies only eye tissues were examined, without confirmation using the same techniques that protein deposits were present in both the brain and the eye of each patient. This makes it difficult to directly compare the levels and appearance of AD-related proteins in the brain to those in the retina and lens in individual cases. Finally, little has been reported on deposition of abnormal proteins in the eyes of PD patients (19). We therefore evaluated the expression of proteins associated with AD and PD pathology in paired postmortem eyes and brains, as well as in controls, using the same standard diagnostic techniques in all tissues.
MATERIALS AND METHODS
Eyes enucleated at autopsy from 11 cases of AD (including one case of Down syndrome), 6 cases of PD or PD with dementia, 1 case of incidental NFTs (neurofibrillary degeneration) and 6 age-matched controls were retrieved from the brain resource center and autopsy archives of The Johns Hopkins Hospital. Neuropathological changes in all the cases had been previously confirmed at our center. All brains were reviewed by one or more board certified neuropathologists (JT, CGE), while eyes were reviewed by an ophthalmic pathologist (CGE). The AD cases included nine cases of definite AD with a CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) plaque score C and Braak tangle score IV-VI/VI, and two cases of probable AD with a CERAD score B and Braak score II-IV/VI (male : female = 3:7; median age: 82 years; range 61–92 years) (4, 22). The PD cases included four cases of PD and two cases of PD with dementia (male : female = 5:1; median age: 76 years; range 68–97 years) (20). PD cases with three different LB distribution patterns (brain stem, limbic and neocortical) were examined to investigate the relationship between the α-synuclein expression in the retina and the severity of the LB pathology in the brain. There were six aged-matched controls (male : female = 2:4; median age: 74 years; range 66–86 years) and one case with incidental NFTs (neurofibrillary degeneration) in the limbic region (male; 55 years old). The case with incidental tangles did not have any history of cognitive impairment or motor deficits. An additional four eyes from patients diagnosed with Down syndrome (ages 2 weeks, 6 years, 10 years and 36 years) were retrieved from the Wilmer Ophthalmic Pathology laboratory.
The eyes and brains removed at autopsy were fixed in buffered formalin, processed in the Johns Hopkins Hospital clinical laboratories and embedded in paraffin. In each case, the superior and inferior caps were removed to generate a pupil-optic (PO) nerve block in which the anterior chamber and lens, peripheral nasal retina, posterior retina including the macular region and optic nerve head, and peripheral temporal retina were represented. Several widely spaced, 4-μm-thick hematoxylin and eosin (H&E) sections of eyes were examined. Immunohistochemistry was performed on eye sections and selected brain sections for Aβ (Dako, Carpinteria, CA, USA; mouse monoclonal antibody, clone 6F/3D; 1:100 dilution) and phospho-tau (Research Diagnostic Inc., Flanders, NJ, USA; mouse monoclonal antibody, clone AT8; 1:50 dilution) on all the AD and control cases. Congo red stains were performed on all the AD and control eye sections and examined in brightfield and polarized light to detect the presence of amyloid. Immunohistochemistry was performed on eye sections and selected brain sections for α-synuclein (Transduction Laboratory, San Jose, CA, USA; mouse monoclonal antibody, clone 42; 1:1000 dilution) on all cases. The tissues were pretreated with formic acid for 20 minutes prior to the immunostains for Aβ and α-synuclein. All of these tests were performed using automated stainers in our routine diagnostic laboratory, with appropriate positive and negative controls included on each run.
RESULTS
No amyloid deposits or abnormal tau accumulations were detected in the eyes of AD patients
Patient demographics, clinicopathological diagnosis and the results of special stains and immunohistochemical analysis from eyes in the 24 cases are summarized in Table 1. Immunohistochemical changes consistent with the neuropathological diagnoses were present in the brains of all affected study patients, including Congo red, Aβ and phospho-tau staining in individuals with AD (Figure 1A–D), and α-synuclein staining in those with PD (Figure 1E). Degenerative changes consistent with superficial cortical cataracts were observed on H&E stained sections in 5 of 11 AD and 3 of 6 control cases (Figure 2A,B). We stained eye sections using the same protocols as the brains in Figure 1. On Aβ immunostains, the superficial cortical region of the lenses from five AD and three control cases demonstrated a faint granular staining pattern (Figure 2C,D). Congo red stains showed variable orange-red staining of lens fibers in both AD and control groups, but all cases were completely negative for birefringent congophilic material. Given the negative Congo red stains, we believe that the very weak Aβ staining seen in a subset of AD and control cases was most likely nonspecific and unrelated to AD pathology.
Table 1.
Summary of demographics and staining results for abnormal protein deposits in the eyes of neurodegenerative disease patients and controls.
| Case | Age/sex | Clinicopathological diagnosis | Congo red | Aβ | Tau | α-Syn |
|---|---|---|---|---|---|---|
| 1 | 82/F | Definite AD (C, VI) | Neg | Neg | Neg | * |
| 2 | 72/F | Definite AD (C, VI) | Neg | ‡ | Neg | † |
| 3 | 92/F | Definite AD (C, VI) | Neg | Neg | Neg | * |
| 4 | 92/M | Definite AD (C, VI) | Neg | Neg | Neg | * |
| 5 | 82/M | Definite AD (C, VI) | Neg | Neg | Neg | † |
| 6 | 86/M | Probable AD (B, II) | Neg | Neg | Neg | † |
| 7 | 82/F | Definite AD (C, V) | Neg | ‡ | Neg | Neg |
| 8 | 70/F | Definite AD (C, IV) | Neg | ‡ | Neg | Neg |
| 9 | 79/F | Definite AD (C, IV) | Neg | ‡ | Neg | Neg |
| 10 | 61/F | Definite AD (C, V) Down syndrome | Neg | Neg | Neg | * |
| 11 | 83/F | Probable AD (B, IV) | Neg | ‡ | Neg | Neg |
| 12 | 76/F | PD with dementia | — | — | — | † |
| 13 | 85/M | PD | — | — | — | * |
| 14 | 65/M | PD with dementia | — | — | — | Neg |
| 15 | 97/M | PD | — | — | — | * |
| 16 | 68/M | PD | — | — | — | Neg |
| 17 | 75/M | PD | — | — | — | * |
| 18 | 54/M | NFD (0, IV) | Neg | ‡ | Neg | † |
| 19 | 84/M | Control | Neg | Neg | Neg | Neg |
| 20 | 66/M | Control | Neg | ‡ | Neg | Neg |
| 21 | 71/F | Control | Neg | ‡ | Neg | * |
| 22 | 70/F | Control | Neg | ‡ | Neg | * |
| 23 | 86/F | Control | Neg | Neg | Neg | Neg |
| 24 | 77/F | Control | Neg | Neg | Neg | † |
Frequent cells with diffuse cytoplasmic α-synuclein immunoreactivity in the ganglion cell layer and inner nuclear layer and moderate immunoreactivity in the inner plexiform layer.
Rare cells with diffuse cytoplasmic α-synuclein immunoreactivity in the ganglion cell layer and/or inner nuclear layer.
Cortical cataract with weak granular β-amyloid immunoreactivity.
—, not performed. F = female; M = male; AD = Alzheimer’s disease; PD = Parkinson’s disease: NFD = neurofibrillary degeneration.
Figure 1. Brain pathology of Alzheimer’s and Parkinson’s disease.
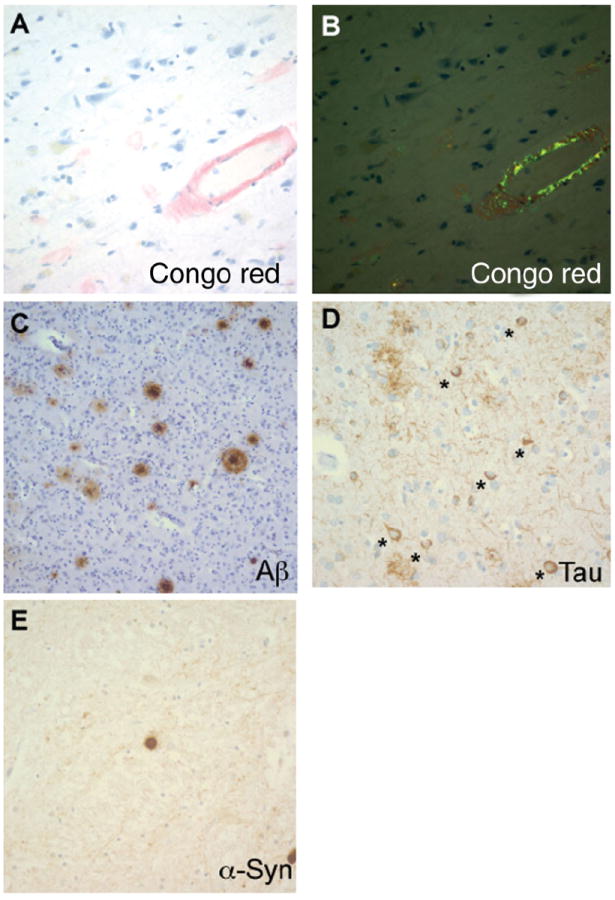
A. Amyloid angiopathy and amyloid plaques on Congo red stain. B. Apple green birefringence of Congo red stains under polarized light. C. Amyloid plaques on the Aβ immunostain. D. Neurofibrillary tangles on the phospho-tau immunostain; * indicates neurons with tangles. E. Lewy body on the α-synuclein immunostain.
Figure 2. Cortical cataracts and the weak Aβ immunoreactivity were present in some AD and control cases.
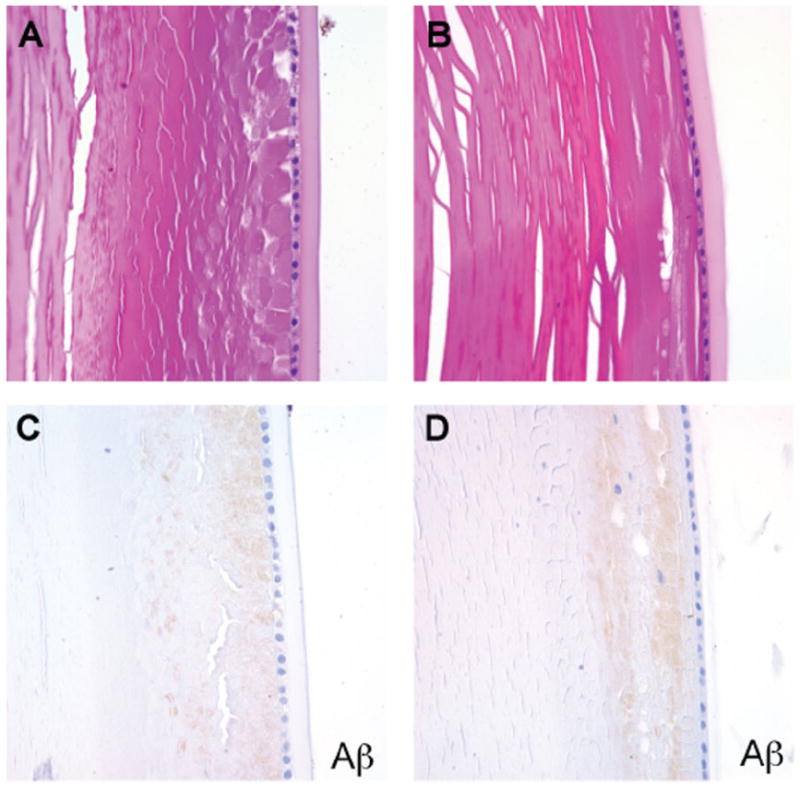
A. AD lens, H&E. B. Control lens, H&E. C. AD lens, Aβ immunostain. D. Control lens, Aβ immunostain. Faint granular Aβ staining was seen in the subcapsular region in both AD and control cases.
Our analysis failed to reveal congophilic amyloid or Aβ immunoreactivity in the retinal pigment epithelium or retina of either AD patients or controls (Figure 3). We also stained four autopsy eyes from patients with Down syndrome for whom corresponding brain tissue was not available, but detected no birefringent congophilic material in the lens or retinas of these cases. Of note, we did observe congophilic amyloid angiopathy in retinal sections from patients with cerebral amyloid angiopathy, which served as an additional internal positive control for the Congo red stain. Finally, no abnormal tau accumulation was seen in the lenses or retinas of AD cases, or the single case with incidental tau pathology in the brain by phospho-tau immunostains (Figure 4). We did not attempt to quantify ganglion cell density or nerve fiber layer thickness in our study, but did not appreciate any major differences between AD cases and control eyes in the representative OP nerve sections examined.
Figure 3. No extracellular or cytoplasmic amyloid deposits were detected in the retina of AD patients.
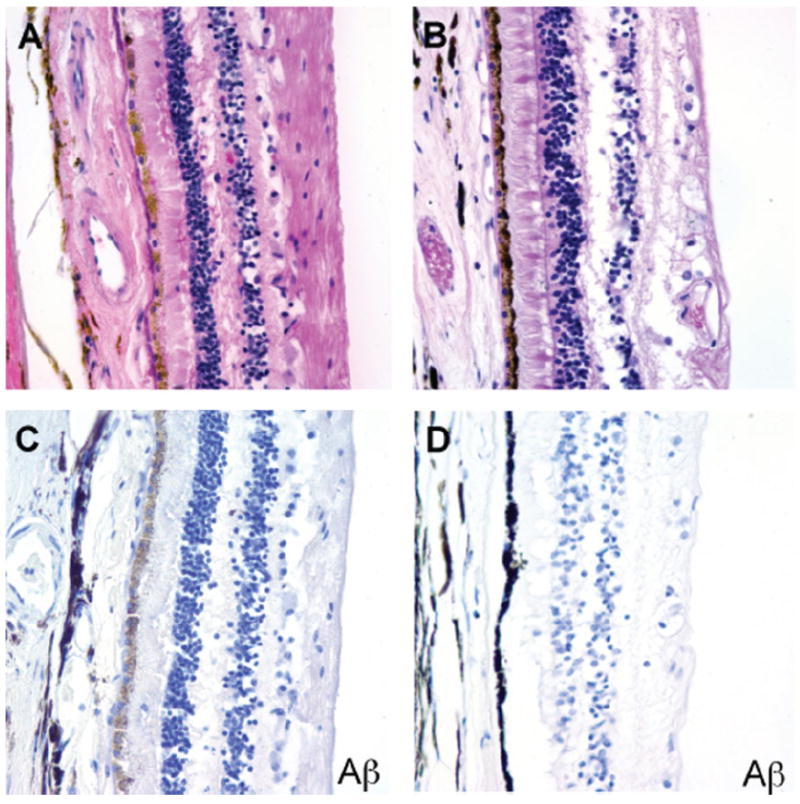
A. AD retina, H&E. B. Control retina, H&E. C. AD retina, Aβ immunostain. D. Control retina, Aβ immunostain.
Figure 4. No neurofibrillary tangles or abnormal tau aggregates were detected in the retina of AD patients on phospho-tau immunostains.
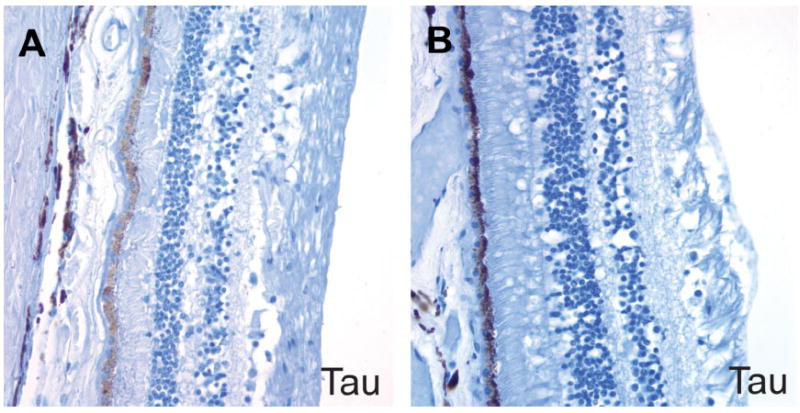
A. AD retina. B. Control retina.
Definite LBs and Lewy neurites are lacking in PD and AD eyes
On H&E sections, the PD eyes also did not show any specific pathology, with no discernable inner retinal layer thinning as compared to controls. α-synuclein immunohistochemical studies demonstrated scattered-to-frequent cells in the ganglion cell layer and inner nuclear layer of retina with diffuse cytoplasmic immunostaining (Figure 5). The α-synuclein immunoreactivity was detected in 7 of 11 (64%) AD cases, 4 of 6 (67%) PD cases and 3 of 6 (50%) control cases. However, no morphologically appropriate LBs or Lewy neurites were identified in the lenses or retinas. The retinal α-synuclein positivity also did not correlate with the presence or extent of the LB pathology in the brain.
Figure 5. Diffuse cytoplasmic α-synuclein immunoreactivity in the ganglion cell layer and inner nuclear layer of retina.
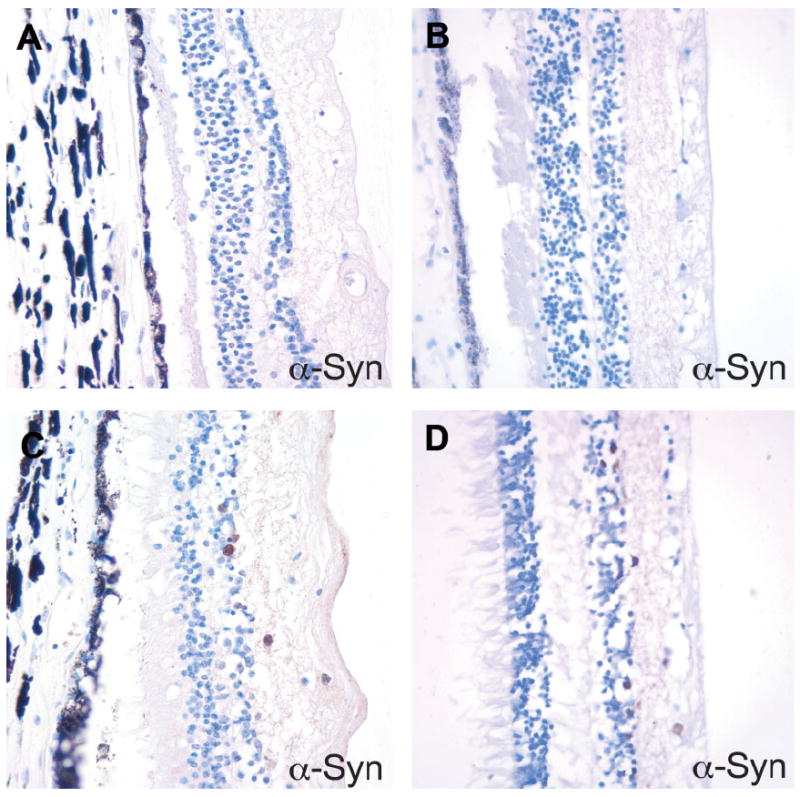
A. Control and B. PD retina with no α-synuclein immunoreactivity. C. Control and D. PD retina with frequent α-synuclein-positive cells. The α-synuclein staining pattern was diffuse cytoplasmic and did not have the morphology of a Lewy body.
No significant pathological findings were identified in the optic nerve of AD or PD cases, except for one AD case with mild optic nerve atrophy with reactive astrocytosis. Pronounced nerve fiber thinning or loss was not noted in our H&E stained sections, but no special studies to quantify axon number or size were performed. NFTs and Lewy neurites were absent in the optic nerve. Congo red stains and immunostains for Aβ, tau and α-synuclein were negative in the optic nerve of AD and PD cases as well as controls.
DISCUSSION
AD and PD are two prevalent and devastating neurodegenerative disorders. Although a clinical diagnosis can be made based on the medical history, neuropsychological assessment and neurological examination, definitive diagnosis still requires postmortem histopathologic examination. Recent research efforts have focused on early detection of the conditions even before symptoms occur, so treatment intervention can start at the earliest stages. Neuroimaging is potentially a powerful tool for the differential diagnosis of neurodegenerative diseases and for monitoring disease course, but generally uses positron emission tomography (PET) tracers that are costly and can be difficult to generate (8, 31). Given the direct connection of many ocular tissues to the brain, and our increasingly sophisticated ability to directly image its contents, the eye would be an attractive site for early diagnosis and monitoring of neurodegenerative disease.
We were not able to demonstrate selective deposition of abnormal proteins in the retinas of patient with AD. Using the same procedures to examine the brain and the eye, 11 AD cases were examined, along with six age-matched controls. While plaques with strong Aβ staining were present in the brains of the affected subjects, no immunoreactivity was identified in the retinas of the AD or control patients. Congo red stains also failed to highlight NPs or other structures in any retinas. Our findings suggest that abnormal Aβ does not accumulate in the retina to the same degree as the brain in AD patients.
These findings are consistent with some studies, but not with others. Hinton and colleagues failed to identify NPs, NFTs or amyloid in the retinas of four AD patients, but their special studies were limited to light microscopy, ultrastructural analysis and thioflavin S staining (3, 9). Loffler et al and Leger et al studied 24 and 19 eyes, respectively, from mostly older patients, with the latter study including two AD patients. The first group found no signs of intraretinal amyloid using Congo red stains and Aβ immunohistochemistry, while the second utilized only Aβ immunohistochemistry which was also negative (16, 17). Most recently, Schon and colleagues could detect one form of hyperphosphorylated tau in retinas from AD patients, but did not observe any fibrillar tau or Aβ aggregates, and concluded that measuring amyloid or tau in the retina was unlikely to be of diagnostic value (28).
In contrast, Koronyo-Hamaoui and colleagues examined eight postmortem eyes from AD patients, five suspected early-stage cases and five controls, and found Aβ plaques selectively in affected individuals using curcumin, thioflavin-S and Aβ immunostaining (14). This study focused mainly on immunofluorescent analysis of frozen material, which may be more sensitive in detecting amyloid than the analyses of formalin-fixed paraffin-embedded tissue we used. In addition, the curcumin used in some of their studies has been shown to bind tau (15) and potentially other proteins with oligomer-forming ability, thus its specificity for amyloid is not clear. The potentially most important difference was the fact that Koronyo-Hamaoui and colleagues looked at retinal whole mounts, which allow deposits scattered across the entire retina to be observed. In contrast, we examined representative cross-sections of the retina, which include much less tissue.
The lens is another ocular tissue in which investigators have previously demonstrated the presence of amyloid plaques, raising the possibility of using optical imaging for diagnosis of AD. In 2003, Goldstein and colleagues identified amyloid precursor protein as well as Aβ1–40 and Aβ1–42 peptides in the lenses of AD patients using Western blot and mass spectrometry. They also identified supranuclear cataracts in the AD patients, and found Aβ deposits using immunohistochemistry (7). Recently, the same group extended their observations to include Down syndrome patients with early-onset AD (23). The superficial cortical region of the lenses from four of our AD patients and three controls demonstrated a faint granular staining pattern, and Congo red stains showed patchy orange-red color in scattered lens fibers, but all cases were negative for the green birefringent material seen in true amyloid. Because the very weak Aβ staining was nonspecific, and present in both AD and control cases, it seems most likely to be unrelated to AD pathology. We also failed to identify birefringent congophilic material in the lenses or retinas of four additional eyes from patients with Down syndrome for whom corresponding brains were not available, although three of these patients were children. Our findings are similar to those recently reported by Michael and colleagues, who examined lenses obtained postmortem from 21 patients with AD and found some orange staining but no birefringent material diagnostic of amyloid (21).
Relatively little is known about the presence of PD-associated proteins in the eye. We performed α-synuclein immunostains on all 23 cases in our study, and found immunoreactivity in the retinas of the majority of the cases. Compared to previous studies that showed cytoplasmic α-synuclein aggregates in the inner nuclear layer of the retina (15, 30), the α-synuclein staining pattern we saw was diffuse and cytoplasmic. Maurage and colleagues reported pale inclusions in the outer plexiform layer of a patient with dementia with LBs, but the structures did not stain on α-synuclein immunostains (19). Given the fact that no LBs or neurites were detected, and the random α-synuclein positivity among AD, PD and control cases, we do not believe that the α-synuclein staining is associated with LB formation. Instead, it most likely reflects the abundant nature of this protein in neural tissue. It is possible that the use of phospho-specific antibodies in retina would assist in detecting forms of the protein associated with neurodegenerative pathology.
In conclusion, when eyes and brains are processed and examined using the diagnostic stains in current standard use at our institution, abnormal protein aggregation characteristic of AD and PD is not present in the retinas or lenses of affected patients. It is not entirely clear why our results differ from some prior studies, but the use of different assays, frozen tissues and whole mount preparations that allow more of the retina to be examined may account for some of these discrepancies. Nevertheless, our data suggest that if present, AD and PD pathology is much smaller in magnitude or extent in the eye than in the brain.
References
- 1.Alexandrov PN, Pogue A, Bhattacharjee S, Lukiw WJ. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. Neuroreport. 2011;22:623–627. doi: 10.1097/WNR.0b013e3283497334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bitel CL, Kasinathan C, Kaswala RH, Klein WL, Frederikse PH. Amyloid-β and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimers Dis. 2012;32:291–305. doi: 10.3233/JAD-2012-120571. [DOI] [PubMed] [Google Scholar]
- 3.Blanks JC, Hinton DR, Sadun AA, Miller CA. Retinal ganglion cell degeneration in Alzheimer’s disease. Brain Res. 1989;501:364–372. doi: 10.1016/0006-8993(89)90653-7. [DOI] [PubMed] [Google Scholar]
- 4.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer’s disease. Graefes Arch Clin Exp Ophthalmol. 2009;247:1213–1221. doi: 10.1007/s00417-009-1060-3. [DOI] [PubMed] [Google Scholar]
- 6.Frost S, Martins RN, Kanagasingam Y. Ocular biomarkers for early detection of Alzheimer’s disease. Int J Alzheimers Dis. 2010;22:1–16. doi: 10.3233/JAD-2010-100819. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang X. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- 8.Herholz K, Ebmeier K. Clinical amyloid imaging in Alzheimer’s disease. Lancet Neurol. 2011;10:667–670. doi: 10.1016/S1474-4422(11)70123-5. [DOI] [PubMed] [Google Scholar]
- 9.Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic-nerve degeneration in Alzheimer’s disease. N Engl J Med. 1986;315:485–487. doi: 10.1056/NEJM198608213150804. [DOI] [PubMed] [Google Scholar]
- 10.Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol. 2009;256(Suppl 3):S270–S279. doi: 10.1007/s00415-009-5243-y. [DOI] [PubMed] [Google Scholar]
- 11.Kesler A, Vakhapova V, Korczyn AD, Naftaliev E, Neudorfer M. Retinal thickness in patients with mild cognitive impairment and Alzheimer’s disease. Clin Neurol Neurosurg. 2011;113:523–526. doi: 10.1016/j.clineuro.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–24384. [PubMed] [Google Scholar]
- 13.Koronyo Y, Salumbides BC, Black KL, Koronyo-Hamaoui M. Alzheimer’s disease in the retina: imaging retinal aβ plaques for early diagnosis and therapy assessment. Neurodegener Dis. 2012;10:285–293. doi: 10.1159/000335154. [DOI] [PubMed] [Google Scholar]
- 14.Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, et al. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage. 2011;54(Suppl 1):S204–S217. doi: 10.1016/j.neuroimage.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landau M, Sawaya MR, Faull KF, Laganowsky A, Jiang L, Sievers SA, et al. Towards a pharmacophore for amyloid. Plos Biol. 2011;9:1–13. doi: 10.1371/journal.pbio.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leger F, Fernagut PO, Canron MH, Léoni S, Vital C, Tison F. Protein aggregation in the aging retina. J Neuropathol Exp Neurol. 2011;70:63–68. doi: 10.1097/NEN.0b013e31820376cc. [DOI] [PubMed] [Google Scholar]
- 17.Loffler KU, Edward DP, Tso MO. Immunoreactivity against tau, amyloid precursor protein, and beta-amyloid in the human retina. Invest Ophthalmol Vis Sci. 1995;36:24–31. [PubMed] [Google Scholar]
- 18.Lu Y, Li Z, Zhang X, Ming B, Jia J, Wang R, Ma D. Retinal nerve fiber layer structural abnormalities in early Alzheimer’s disease: evidence in optical coherence tomography. Neurosci Lett. 2010;480:69–72. doi: 10.1016/j.neulet.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Maurage CA, Ruchoux MM, de Vos R, Surguchov A, Destee A. Retinal involvement in dementia with Lewy bodies: a clue to hallucinations? Ann Neurol. 2003;54:542–547. doi: 10.1002/ana.10730. [DOI] [PubMed] [Google Scholar]
- 20.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 21.Michael R, Rosandić J, Montenegro GA, Lobato E, Tresserra F, Barraquer RI, Vrensen GF. Absence of beta-amyloid in cortical cataracts of donors with or without Alzheimer’s disease. Exp Eye Res. 2013;106:5–13. doi: 10.1016/j.exer.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 22.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II: standardization of neuropathological assessment of Alzheimer’s disease. Neurology. 2005;65:1863–1872. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 23.Moncaster JA, Pineda R, Moir RD, Lu S, Burton MA, Ghosh JG, et al. Alzheimer’s disease amyloid-beta links lens and brain pathology in Down syndrome. PLoS ONE. 2010;5:e10659. doi: 10.1371/journal.pone.0010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moschos MM, Markopoulos I, Chatziralli I, Rouvas A, Papageorgiou SG, Ladas I, Vassilopoulos D. Structural and functional impairment of the retina and optic nerve in Alzheimer’s disease. Curr Alzheimer Res. 2012;9:782–788. doi: 10.2174/156720512802455340. [DOI] [PubMed] [Google Scholar]
- 25.Muchowski PJ, Ramsden R, Nguyen Q, Arnett EE, Greiling TM, Anderson SK, Clark JI. Noninvasive measurement of protein aggregation by mutant huntingtin fragments or alpha-synuclein in the lens. J Biol Chem. 2008;283:6330–6336. doi: 10.1074/jbc.M709678200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nussbaum RL, Ellis CE. Alzheimer’s disease and Parkinson’s disease. N Engl J Med. 2003;348:1356–1364. doi: 10.1056/NEJM2003ra020003. [DOI] [PubMed] [Google Scholar]
- 27.Parnell M, Guo L, Abdi M, Cordeiro MF. Ocular manifestations of Alzheimer’s disease in animal models. Int J Alzheimers Dis. 2012;2012:786494. doi: 10.1155/2012/786494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schon C, Hoffmann NA, Ochs SM, Burgold S, Filser S, Steinbach S. Long-term in vivo imaging of fibrillary tau in the retina of P301S transgenic mice. PLoS ONE. 2012;7:e53547. doi: 10.1371/journal.pone.0053547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Selkoe DJ. Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. doi: 10.1146/annurev.cb.10.110194.002105. [DOI] [PubMed] [Google Scholar]
- 30.Surguchov A, McMahan B, Masliah E, Surgucheva I. Synucleins in ocular tissues. J Neurosci Res. 2001;65:68–77. doi: 10.1002/jnr.1129. [DOI] [PubMed] [Google Scholar]
- 31.Thompson PM, Vinters HV. Pathologic lesions in neurodegenerative diseases. Prog Mol Biol Transl Sci. 2012;107:1–40. doi: 10.1016/B978-0-12-385883-2.00009-6. [DOI] [PubMed] [Google Scholar]