

Abstract
Pharmaceuticals and industrial chemicals, both in the environment and in research settings, commonly interact with aquatic vertebrates. Due to their short life-cycles and the traits that can be generalized to other organisms, fish and amphibians are attractive models for the evaluation of toxicity caused by endocrine disrupting chemicals (EDCs) and adverse drug reactions. EDCs, such as pharmaceuticals or plasticizers, alter the normal function of the endocrine system and pose a significant hazard to human health and the environment.
The selection of suitable animal models for toxicity testing is often reliant on high sequence identity between the human proteins and their animal orthologs. Herein, we compare in silico the ligand-binding sites of 28 human `side-effect' targets to their corresponding orthologs in Danio rerio, Pimephales promelas, Takifugu rubripes, Xenopus laevis, and Xenopus tropicalis, as well as sub-pockets involved in protein interactions with specific chemicals. We found that the ligand-binding pockets had much higher conservation than the full proteins, while the peroxisome proliferator-activated receptor γ and corticotropin-releasing factor receptor 1, were notable exceptions. Furthermore, we demonstrated that the conservation of sub-pockets may vary dramatically. Finally, we identified the aquatic model(s) with the highest binding site similarity, compared to the corresponding human toxicity target.
Introduction
Aquatic vertebrates are targeted by pharmaceutical and industrial chemicals, both intentionally and unintentionally, in a variety of research and environmental contexts. In the wild, these animals are exposed to the pharmaceuticals and industrial chemicals present in the surface waters. In research settings, aquatic vertebrates may be used to evaluate novel chemicals for toxicity, including the early identification of adverse drug reaction (ADR) or endocrine disruption (ED) potential of pharmaceutical candidates and industrial chemicals.
Lower order vertebrates, such as amphibians and fish, are being increasingly viewed as a replacement for rodent models. They are convenient and cost-effective model organisms due to their short life-cycles and the presence of traits that can be generalized to other organisms.1 Species that are commonly used for toxicological evaluations include Danio rerio (zebrafish), Pimephales promelas (fathead minnow), Takifugu rubripes (Japanese pufferfish), Xenopus laevis (African clawed frog), and Xenopus tropicalis (Western clawed frog).1–4 Specifically, D. rerio has been widely used to study ADRs that include reproductive toxicity, cardiotoxicity, hepatotoxicity and neurotoxicity,5 as well as the evaluation of potential endocrine disrupting chemicals (EDCs; reviewed in1). P. promelas has been used to predict the aquatic toxicity of environmental chemicals,2 and T. rubripes has been used to evaluate EDCs.6,7 Amphibians are known to be good models for studying EDCs that interact with thyroid hormone receptors8 and X. laevis has been used to study ADRs related to membrane transporters.9
Toxicity, for chemicals with low concentrations in the target organisms, is most frequently caused by their specificity to particular proteins in the organism. Comparing the protein sequences and structures of human toxicity targets to their orthologs in aquatic species can assist in the identification of the most similar ortholog.
For the reliable prediction of pharmaceutical or environmental toxicity, robust animal models are required whose proteins are highly similar to the orthologous human ADR and toxicity targets. Additionally, in the wild, these species are more vulnerable than others to pharmaceuticals present in the environment that have been specifically designed for high-affinity interactions with the designated proteins.10
Typically in toxicity studies, one rodent model and one non-rodent model are employed.11 However, depending on the target and the class of chemicals in question, some animal models may be more relevant than others. The ever-increasing number of species with fully sequenced genomes has begun to allow for druggable genome and proteome comparisons. Recently, the genomes of eight relevant toxicological species were compared to the human genome.12 Target similarity has been assessed at the level of protein sequence, with the degree of conservation of specific drug targets in humans and model organisms evaluated by performing sequence-by-sequence alignments,10 and limited studies have been conducted on the domain conservation for the androgen receptor (AR) and estrogen receptor α (ERα).13
Yet the levels of conservation between orthologous sequences usually vary throughout the sequence (Figure 1). Thus, it is important to focus on the similarity of sections of the sequence that are most relevant to chemical interactions. The conservation of residues directly involved in ligand binding is a more relevant parameter for evaluation of aquatic species models than full sequence similarity. Inter-species variations in the amino-acid composition of the binding-pocket can sometimes have dramatic effects on the utility of species in pharmacological assays. For example, in the serotonin 6 receptor (5-HT6R), two residues in the ligand-binding pocket were found to significant change the pharmacology of the mouse 5-HT6R (resulting in a systematic one log unit shift of the 5-HT6R ligands), compared to the human and rat 5-HT R,15,16 6 making the mouse model an unfavorable choice for testing 5-HT6R-targeting pharmaceuticals, whilst the rat 5-HT6R binding pocket is identical to humans. Similarly, a two (out of 13) minor amino-acid substitutions (Thr to Ala and Ala to Val) in the binding pocket of the rat and mouse histamine H3 receptors (H3R), compared to the human H3R, leads to a systematic compound potency measurement error and limits both of their utility in H3-related studies.17
Figure 1.
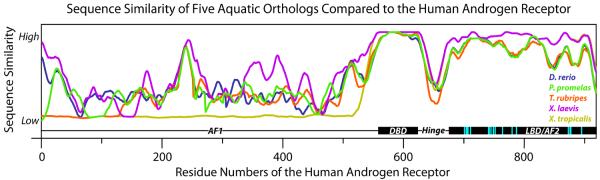
Variations in sequence conservation across sequence of the AR for D. rerio, P. promelas, T. rubripes, X. laevis, and X. tropicalis compared to the human AR (binding site residues highlighted in cyan). All sequences were window averaged across 25 residues. Abbreviations: AF1/2, activation function 1/2; DBD, DNA binding domain; LBD, ligand binding domain.14
Because orthologous proteins in different species typically bind the same or similar endogenous ligands,8 the conservation of the binding pockets far exceeds the full length sequence conservation. They are also likely to bind the same exogenous chemicals. The aim of this research was to identify the aquatic organisms (from the set of D. rerio, P. promelas, T. rubripes, X. laevis, and X. tropicalis) that share the highest binding pocket similarity with humans in each of the 28 best-characterized toxicity targets. X-ray crystal structures were used to identify the amino-acid residues constituting the ligand-binding pockets, which were extrapolated to the aquatic orthologs. Sequence similarity and identity were calculated for the ligand-binding sites and the most similar orthologs to the 28 human toxicity targets were identified.
Materials and Methods
Selection of human EDC and ADR targets
An initial set of 85 unique human proteins that have been previously characterized as side-effect and toxicity targets were compiled from the 73 protein assays listed in the Novartis in vitro safety panels (Table S1), eleven targets from the VirtualToxLab,18,19 and the Constitutive Androstane Receptor (CAR; NR1I3). All 85 proteins were used for sequence analyses. For binding pocket similarity analyses, the 85 targets were matched against the Pocketome encyclopedia (http://pocketome.org),20 a collated set of annotated, binding pocket structure ensembles from the Protein Data Bank (PDB).21 At the time of this study, 28 out of the 85 targets had Pocketome entries for their ligand-binding pockets available (Table S1) that contained at least one co-crystalized ligand making it possible to precisely identify the binding site residues. These 28 targets were used for binding site similarity and identity comparisons.
Identification of orthologs of human EDC and ADR targets in the aquatic species
The complete proteomes of D. rerio, Mus musculus (mouse), P. promelas, Rattus norvegicus (rat), T. rubripes, X. laevis, and X. tropicalis were downloaded in FASTA format from the UniProt Knowledgebase.22 The M. musculus and R. norvegicus results have been included in all Supporting Information for comparison purposes. For each of the files, BLAST search index was generated using the bioinformatics module of the Internal Coordinate Mechanics (ICM) software version 3.7–3a (Molsoft L.L.C., La Jolla, CA).23,24 A BLAST search25 was performed to identify orthologs of the 85 human proteins in the corresponding aquatic species. One hit per target per species was retained using the following prioritization rules: (i) manually annotated orthologs of the toxicity and side-effect targets were retained with the highest priority; (ii) for automatically annotated analogues, orthologs with the same gene name as the human protein and the highest probability score to the human protein were kept; (iii) if only sequence fragments were available, the longest fragment was retained.
Sequence alignment and analysis
Pairwise alignments were constructed between the full sequence of human protein and the corresponding orthologs and pairwise sequence scores were calculated with the Needleman and Wunsch algorithm26 modified for the zero end-gap penalties (the ZEGA algorithm27) as implemented in the ICM program. We used gap opening and gap extension penalties of 2.4 and 0.15, respectively. Sequence identity was represented by the number of identical residues over the total number of aligned residues. Sequence similarity was calculated using the GONNET residue substitution comparison matrix.28
Binding site definition and classification using ligand contact strength fingerprints
For each ligand in the pocketome entry and each non-hydrogen atom in the protein, distance-dependent contact strengths were calculated using the parameters developed in context of GPCR Dock 2010 evaluation.29,30 The per-atom contact strengths were aggregated into per-residue contact strength values by taking the sum over all non-hydrogen atoms in the residue side-chain. Only residue side-chains were included in the calculation because, except for proline, ligand contacts with backbone atoms may not be affected by residue substitutions between species. If a ligand was co-crystalized in multiple structures, the vectors of per-residue contact strengths were averaged. To reduce noise and binding site definition artifacts associated with increased conformational variability of individual residues, the contact strength vector components were multiplied by a factor ranging from 0 to 1 and inversely proportional to the observed conformational variability of the corresponding residue in the Pocketome ensemble.
Each unique ligand Li was characterized by a vector FPi of per-residue numbers ranging from 0 (no contact) to 32 (extensive close contact with Phe168 in the adenosine A2A receptor (A2AR); Table S1). Normalized fingerprint distance between ligands Li and Lj was calculated as where Min(FPi,FPj) and (FPi+FPj)/2 are vectors of element-wise minima and element-wise averages between vectors FPi and FPj, respectively.30 When defined that way, ligand fingerprint distances range from 0 (for identical fingerprints) to 1 (for non-overlapping fingerprints). Ligand interaction fingerprints were clustered at the distance cutoff of D = 0.35 to identify classes of ligand occupying distinct areas in the binding site. The cutoff of 0.35 was found to be the optimal tradeoff between the excessive number of clusters and the unwanted aggregation of substantially different ligand chemotypes in multiple targets. This cutoff indicates that the ligands will be classified as belonging to different clusters if their fingerprints vary by one-third (or more) of the contacts.
Next, clusters of unique crystallographic ligands were ordered by their size, starting with the most populated one and ending with singletons (i.e. clusters containing only a single ligand). Top clusters containing 80% of the ligands were combined to define the set of residues interacting with the majority of the ligands. The remaining 20% were disregarded in the pocket definition to ensure that it is not affected by occasional or spurious contacts.
Binding pocket sequence identity and similarity calculations
For each sub-pocket in the binding site, as determined by ligand contact strength fingerprint clustering, a sub-alignment was extracted by projecting the full sequence alignment between human and ortholog sequences onto the corresponding residue selection. Binding pocket/sub-pocket identity and similarity were calculated from these sub-alignments using the same parameters as the full sequence alignments. The same was done for the set of residues forming the interaction site(s) for at least 80% of the ligands, as described above, and thus representing the aggregation of the consistently populated regions of the pocket. The comparison of complete pockets (including interaction fingerprints of all crystallographic ligands) is available in Supporting Information.
Results
Orthologs of human EDC and ADR targets in aquatic vertebrates
Five fish and amphibians frequently used in toxicological evaluations were used in this study: D. rerio, P. promelas, T. rubripes, X. laevis, and X. tropicalis. In their proteomes, we identified the orthologs of the known human side-effect and environmental target proteins. In some cases, orthologs could not be found: 89% of the toxicity targets were identified in D. rerio, 20% in P. promelas, 84% in T. rubripes, 51% in X. laevis, and 85% in X. tropicalis (Table S1). This may be explained by the fact that only the genomes of D. rerio,31,32T. rubripes33 and X. tropicalis34 have been fully sequenced, while the remaining two genomes (P. promelas and X. laevis), and thus proteomes, are incomplete. Additionally, in some cases, only protein fragments of the toxicity target orthologs have been identified. The sequences of the human and orthologous toxicity proteins were aligned and the full sequence similarity was calculated (Figure 2a, Table S1).
Figure 2.

Sequence similarity (percentage and color) and sequence identity (number of identical residues/number of aligned residues is shown in parenthesis) for the 28 toxicity target proteins of a. the full sequence and b. the ligand-contact residues conserved for 80% of the co-crystalized ligands. White spaces indicate that no ortholog was identified (often due to an incomplete proteome).
Full sequence similarity between human EDC/ADR targets and their orthologs in aquatic vertebrates
The relevance of a model organism for prediction of toxicity in humans has previously been evaluated using the amino acid conservation across entire protein sequences, e.g. ref.10. In the present study, the majority of the human toxicity targets displayed 60 to 70% sequence similarity with their aquatic vertebrate orthologs (Figure 2a). The average full sequence similarity between the human proteins and the aquatic orthologs was 69% for D. rerio, 63% for P. promelas, 70% for T. rubripes, 71% for X. laevis, and 72% for X. tropicalis (Figure S1). In some cases, the overall sequence similarity was relatively high. For example, X. tropicalis had the highest full sequence similarity for the androgen receptor (AR, 88%). However, the protein sequence for X. tropicalis was only a fragment of the full sequence that lacked the N-terminal domain of the protein compared to the other species, giving artificially higher sequence similarity. The corticotropin-releasing factor receptor 1 (CRF1R) is highly conserved in four species (~85% sequence similarity). The interspecies variations in full sequence similarity were more informative for the estrogen receptors α and β (ERα and ERβ, respectively), and the glucocorticoid receptor (GR), where the full sequences were similar in length. X. laevis and X. tropicalis shared higher conservation of these receptors with human (9–24% higher sequence similarity) than D. rerio, P. promelas and T. rubripes. The impact of the variability of the sequence length on the full sequence similarity demonstrates the difficulties with using the full protein sequence (or longest available sequence) in these calculations.
Ligand-binding pocket similarity between human EDC/ADR targets and their orthologs in aquatic vertebrates
As expected, the ligand-binding pockets of the orthologous proteins generally shared higher sequence conservation with the human toxicity targets than the full protein sequences (Figure 2b). For example, the ligand-binding site of human AR shared ~98% sequence similarity with all five species, whereas the full sequence similarity was only 47–88%. Likewise, the binding sites of ERα, ERβ and GR are 92–100% conserved in all five aquatic species, while the highest full sequence conservation observed in X. laevis and X. tropicalis did not exceed 70–76%. The relative ranking of species by the full sequence similarity to humans often varies from that by binding pocket similarity. For example, based on full sequence similarity, one would choose X. laevis or X. tropicalis as the most relevant model for testing ERα-targeting chemicals; however, our pocket similarity analysis indicates that all five species are almost equally good, with the fish species having a slight advantage over the frogs. Similarly, despite being most similar to human in terms of full β2 adrenergic receptor (β2AR) sequence, X. tropicalis is probably the least accurate of the five models for evaluation of β2AR ligand pharmacology, as it has as many as 5 residue substitutions in the binding pocket (Figure S3).
Surprisingly, two targets had lower sequence conservation in the binding site as compared to the full sequence. These were the obesity- and stress-related targets, peroxisome proliferator-activated receptor γ (PPARγ) and CRF1R. PPARγ displayed lower binding-site similarity (56–85%) than full sequence similarity (74–89%). CRF1R displayed higher sequence similarity across the full protein sequence (~85%) than in the peptide-binding site in its extracellular domain (46–78%). However, GPCRs often have a greater degree of sequence variability in the extracellular domains, hence the lower sequence similarity in the peptide-binding site of CRF1R is consistent with the nature of this receptor.
Ligand-binding pockets in ADR/EDC targets: one size does not fit all
On closer inspection of the ligand-binding interactions in the X-ray crystal structures of the human EDC and ADR targets, there were often noticeably different residue interaction fingerprints for different ligand chemotypes. In some cases, different chemotypes can bind to distinct ligand-binding pockets, or “sub-pockets” of the proteins.
This is exemplified by the identification of three different sub-pockets of the adenosine A2A receptor (A2AR). Promisingly, the three sub-pockets identified for A2AR (Figure 3a) correspond to an agonist-bound structure (Figure 3b), the endogenous agonist-bound structure (Figure 3c) and the antagonist-bound structures (Figure 3d), respectively. All sub-pockets were fully conserved in X. laevis and X. tropicalis. Additionally, significant variations in the conservation of sub-pockets can be observed for the ortholog of β2AR in X. tropicalis (Figure S3), where sub-pocket 1 displays 75% conservation, yet sub-pocket 2 has only 48% sequence similarity.
Figure 3.
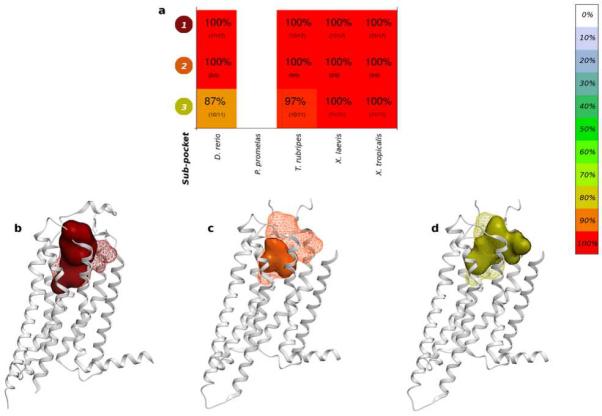
a. Sequence similarity (percentage and color) and sequence identity (number of identical residues/number of aligned residues is shown in parenthesis) for the three A2AR sub-pockets (white spaces indicate that no ortholog was identified). A2AR crystal structures (grey ribbons), all co-crystalized ligands (mesh) and sub-pocket (solid surface); b. sub-pocket 1 (agonist-bound structures), c. sub-pocket 2 (the endogenous agonist-bound structure), d. sub-pocket 3 (antagonist-bound structures).
Because the likelihood of a chemical interacting with an aquatic species ortholog of its target protein largely depends on the conservation of specific interacting residues and not the entire binding site, we sought to identify the individual sub-pockets in each of the target pockets and to separately evaluate their similarity to the corresponding sub-pockets in the studied aquatic organisms. Sub-pockets were identified by the clustering of contact-strength fingerprints (see Methods).
GPCR sub-pocket sequence conservation
GPCRs are a superfamily of membrane bound proteins characterized by seven transmembrane (TM) helices and many have been implicated in ADRs, endocrine disruption and reproductive toxicity.35 The A2AR is implicated in a number of ADRs such as palpitations and angina.18 ADRs for the β2 adrenergic receptor (β2AR) include tremor, cardiac failure, angina18; it has also been implicated in ED in aquatic vertebrates.4,36 The serotonin 2B receptor (5-HT2BR) is linked to valvular heart disease,37 the histamine H1 receptor (H1R) is involved in sedation and the human M2 muscarinic acetylcholine receptor (M2R) is associated with constipation.18 The dopamine D3 receptor (D3R) is implicated in dyskinesia and Parkinsonism18 and shown to bind the known endocrine disruptor BPA.38 Two class B GPCRs were also evaluated; CRF1R, which is implicated in stress-related disorders,39,40 and the gastric inhibitory polypeptide receptor (GIPR), which is implicated in diabetes and obesity.41
Two sub-pockets were identified for β2AR (Figure S3), the classical orthosteric site (sub-pocket 1) and the orthosteric site with some additional residues from the less conserved TM1/TM2/TM7 region (sub-pocket 2). Generally, X. tropicalis displayed poor ligand-binding pocket conservation to the human β2AR (75% and 48%, sub-pockets 1 and 2, respectively). Due to the scarcity of multiple crystal structures for many GPCRs, sub-pockets were unable to be explored for the 5-HT2BR, D3R H1R, κ opioid receptor (κOR) and M2R, however the binding pockets were generally well conserved (69–100%; Figure 2b and Figure S4.
At the time of this study, crystal structures were only available for the extracellular domains of the GPCRs CRF1R and GIPR, which contain the peptide-binding sites. These peptide-binding sites were expected to have lower levels of conservation because it is well established that the extracellular domains of GPCRs have a large degree of sequence variability. Only X. tropicalis had a moderately conserved ortholog for GIPR (61%, Figure S4), indicating that alternate animal models should also be investigated. X. laevis and X. tropicalis displayed higher ligand-binding pocket similarity across both sub-pockets (60–78%, Figure S4). However, it is unlikely that peptides in the environment would result in endocrine disruption via the peptide-binding site of CRF1R and GIPR in either humans or the fish and amphibians evaluated in this study, as potential ED peptides are unlikely to be readily absorbed. Consequently, this technique should also be applied to the small molecule binding site of GIPR when a structure becomes available, and to the recently released structure of CRF1R.42
Nuclear receptor sub-pocket conservation
Nuclear receptors are a superfamily of proteins that regulate development, growth and homeostasis and they are commonly implicated in endocrine disruption. Some classic examples of ED that occur via nuclear receptors include the weak agonistic activity of the plasticizer bisphenol A (BPA) against the ERα;43 the feminization of fish by 17α-ethinylestradiol (EE2), a synthetic estrogen in human contraceptives;44 and modulation of PPARγ by EDCs, which is implicated in obesity.45
ERα sub-pockets were generally highly conserved across the aquatic species (94–100%, Figure S5), with the exception of T. rubripes for sub-pocket 8, which is bound to a large estradiol metal chelate ligand (88%). The binding pocket of ERβ across the five species, compared to the human ERβ, was generally highly conserved (92–100%, Figure S4). However, the across all the sub-pockets T. rubripes was slightly less conserved (92–95% vs. 98–100%). ERR1 has only been co-crystalized with two unique ligands in two unique sub-pockets (Figure S4), with sub-pocket 2, co-crystalized with a thiazolidinedione, having higher sequence conservation (82–89% vs. 54–60%). The sub-pockets of the glucocorticoid receptor (GCR) were generally well conserved with the human receptor (91–98%, Figure S4). The binding sites of the progesterone receptor (PR) for X. laevis and X. tropicalis shared slightly higher pocket conservation with the human receptor (98–100%, Figure S4). The sub-pockets of the androgen receptor (AR) were highly conserved (96–100%, Figure S6) and the sub-pockets of the thyroid hormone receptor β (TRβ) were fully conserved (100%, Figure S4). Unlike TRβ, the thyroid hormone receptor α (TRα) did not show full sequence conservation across all species (86–100%; Figure S4). All sub-pockets across all species (except for P. promelas for which no ortholog was identified) were fully conserved for the Liver X Receptor (LXR; Figure S4). Whilst no sub-pockets were identified for the mineralocorticoid receptor (MCR; Figure S4), X. tropicalis had lowest LBD similarity (81%). Of the five aquatic species, T. rubripes consistently displayed higher homology to the human Pregnane X receptor (PXR; 54–64%, Figure S7). Despite this, the overall pocket similarity was relatively low (maximum 64%), indicating that PXR is not well conserved in these aquatic vertebrates and that other animal models with higher binding site conservation should also be investigated. Similarly, low binding-pocket conservation was observed for the Constitutive Androstane Receptor (CAR; 35–43%; Figure S4). In 15 out of the 16 sub-pockets, X. tropicalis had the highest ligand-binding pocket sequence similarity to the human PPARγ (81–100%; Figure S8). Interestingly X. laevis, a close relative of X. tropicalis, had significantly lower ligand-binding pocket sequence similarity (50–80%).
Cytochrome P450 sub-pocket sequence conservation
Cytochrome P450s (CYPs) are a superfamily of enzymes that catalyze the oxidation of a diverse range of organic compounds and are commonly involved in the metabolism of xenobiotic compounds. CYPs typically have large and conformationally flexible binding sites in order to accommodate a wide range of chemically dissimilar compounds,46,47 which is supported by the diverse array of sub-pockets identified. There were closely related orthologs to the human CYP1A2, with D. rerio having the highest pocket similarity (96%, Figure S4). Both D. rerio and P. promelas had closely related orthologs of CYP3A4 across five out of six sub-pockets (89–100%, Figure S9). X. laevis and X. tropicalis had the highest sub-pocket similarities for CYP2C9 (60–78%), however the ligand-binding pocket conservation was moderate (Figure S4). Orthologs of CYP2D6 were only identified in X. laevis and X. tropicalis, which displayed good conservation to the human protein (Figure S4).
Sub-pocket sequence conservation of other enzymes
Monoamine oxidase A (MAO-A) is involved in the catabolism of neurotransmitters and dietary amines and inhibition can lead to neuroendocrine disruption48 and it is implicated in ADRs including psychosis and hypertensive crisis.49ADRs associated with cAMP-specific 3',5'-cyclic phosphodiesterase 4D (PDE4D) include diarrhea and nausea,50 and due to its role in the endocrine system PDE4D may also be a target for EDCs.51 The binding site of PDE4D was fully conserved across the identified ortholog binding sites (100%, Figure S4). The sub-pockets for MAO-A, however, displayed higher sequence similarity for D. rerio and T. rubripes (95%, Figure S4).
Discussion
The present study performs a comparison of 28 human toxicity targets to their orthologs in five aquatic species, with the goal of identifying the aquatic organisms with the highest ligand-binding pocket sequence similarity to the human toxicity target. The comparison was performed not only at the level of full protein sequences but also, more relevantly, at the level of the ligand-binding sites. By using the X-ray crystal structures of human toxicity targets, residue-level interaction fingerprints were calculated for each unique co-crystalized ligand, binding pockets and spatially distinct sub-pockets were identified, with each residue selection extrapolated onto the orthologous proteins in the five aquatic vertebrates. In some cases, the contact fingerprints could also separate the toxicity target crystal structures based on the mode of action of the co-crystalized ligands (such as A2AR; Figure 3), providing a basis for the understanding the sub-pocket sequence conservation.
We identified the aquatic vertebrate(s) that share the highest sequence similarity for the ligand-binding pockets (Table 1), compared to the human toxicity targets, as well as the determining the sequence similarity of the spatially distinct sub-pockets. X. tropicalis, had the largest number of orthologs that shared the highest conservation with the human toxicity targets (out of the five aquatic species), having the highest ligand-binding site similarity for 21 out of the 28 toxicity targets, closely followed by D. rerio (19), T. rubripes (19) and X. laevis (18). P. promelas had the lowest number of highly conserved ligand-binding pockets with only 7 ligand-binding sites with high similarity, which can be partially attributed to an incomplete genome.
Table 1.
Identification of the aquatic vertebrate model(s) with the highest ligand-binding pocket similarity (denoted by X) compared to the corresponding human toxicity target (*indicates targets where other species should be investigated due to orthologs with only low or moderate pocket similarity).
| Aquatic vertebrate model(s) with the highest ligand-binding pocket similarity | |||||
|---|---|---|---|---|---|
| Receptor | D. rerio | P. promelas | T. rubripes | X. laevis | X. tropicalis |
| 5-HT2BR | X | X | |||
| A2AR | X | X | X | X | |
| M2R | X | X | X | ||
| β2AR | X | X | X | X | |
| AR | X | X | X | X | X |
| MAO-A | X | X | |||
| CYP1A2 | X | X | |||
| CYP2C9* | X | X | |||
| CYP2D6 | X | X | |||
| CYP3A4 | X | X | |||
| CRF1R | X | X | |||
| D3R | X | X | |||
| ERR1 | X | X | X | X | |
| ERα | X | X | X | X | X |
| ERβ | X | X | X | X | X |
| GCR | X | X | X | X | X |
| GIPR | X | ||||
| H1R | X | X | X | ||
| MCR | X | X | X | ||
| LXR | X | X | X | X | |
| PXR* | X | X | X | ||
| CAR* | X | X | |||
| κOR | X | X | X | ||
| PDE4D | X | X | X | ||
| PPARγ | X | ||||
| PR | X | X | |||
| TRα | X | X | X | ||
| TRβ | X | X | X | X | X |
In this study, we demonstrated that the major difficulty faced when using the full sequence similarity for the comparison of toxicity target orthologs to human proteins is due to variations in the length of the amino acid sequences. For example, whilst X. tropicalis has the highest full sequence similarity for AR (88%), the longest available sequence of the AR of X. tropicalis was actually incomplete, lacking the N-terminal of the protein including the DNA binding domain (393 residues vs. > 729 residues), thus giving artificially higher sequence similarity. This also occurred for some of the aquatic orthologs of MCR, PDE4D, PPARγ, PR, TRα and TRβ. Additionally, we have shown that high full sequence similarity does not always correlate with high ligand-binding site conservation. For example, the sequence similarity for the extracellular domains of CRF1R for all species is high (~85%), yet the peptide-binding sites have lower conservation (46–78%). Generally, we have demonstrated that the ligand-binding sites share higher conservation between orthologs, compared to the full sequences (Figure 2). Consequently, we also have shown that ligand-binding site similarity is the preferred method for the identification of the most conserved orthologs, because it is more informative than the full sequence similarity and it is not influenced by variations in the length of the longest available amino acid sequence of an ortholog. Additionally, if full sequence similarity alone is to be considered, variations in the length of the full (or longest available sequence) should also be incorporated into these assessments.
There are a few caveats that need to be taken into consideration when using orthologous sequence comparisons to aid in the selection of animal models for the evaluation of toxicity. Firstly, the provided principles only suggest toxicity target orthologs in aquatic species based on sequence similarity, without attention to possible variations in the protein function or the downstream pathways.10,52 This method unfortunately does not provide any detail regarding the signaling pathways for orthologous protein and will, of course, require a certain level of understanding of the animal model. Binding pocket similarity may be a necessary but not a sufficient condition for model utility, as exemplified by the pair of human and rat ARs: a large-scale study of inter-species variations in binding affinity of chemicals17 identified, this pair as having systematic one log unit differences in potency of multiple diverse chemicals, despite the fact that not only the binding pockets, but also the entire ligand binding domain of AR is strictly conserved between human and rat. Secondly, our method is reliant on the availability of the proteome of the organisms or, at the very least, the availability of sequences of the orthologs of the toxicity targets. And thirdly, calculating ligand-binding site conservation requires X-ray crystal structures of the human toxicity targets, preferably in complex with a diverse range of chemicals. Both of the problems regarding the availability of the full proteomes and crystal structures can be addressed in future studies, due to the increasing availability of these data. Thus, this study could be expanded to a wider range of toxicity targets and species, including toxicity targets that lack crystal structures, by using crystal structures of highly homologous proteins.
By calculating the amino acid similarity in the ligand-binding pockets, we have successfully avoided the problem of full sequence length variability in sequence similarity calculations, to determine the aquatic orthologs with the most similar ligand-binding pockets for 28 human toxicity targets. This method also allows for the calculation of binding site similarity for sub-pockets that are involved in the specific chemical-protein interactions. We believe that this study will be a useful tool when designing target-specific assays for the assessment of ADRs and ED potential of chemicals.
Supplementary Material
Acknowledgements
This work was partially supported by NIH grants R01 GM071872, U01 GM094612, and U54 GM094618. V.S. was a recipient of a Genentech Foundation research scholarship. The authors gratefully acknowledge Prof. Bryan W. Brooks for useful discussions and sharing his expertise, as well as Clarisse Ricci for useful discussions and for critically reading the manuscript.
Footnotes
Conflict of Interest Disclosure The authors declare no competing financial interest.
Supporting Information Supporting Table S1 includes the full sequence similarity for D. rerio, P. promelas, T. rubripes, X. laevis, and X. tropicalis, as well as M. musculus and R. norvegicus against all 85 toxicity targets, including the details of the 28 Pocketome entries. Figures S1 and S2 show the average sequence similarity of orthologs compared to the corresponding human toxicity target, Figures S3, S5–S9 display selected heat-maps of sequence similarity and crystal structures for selected toxicity targets and Figure S4 shows the heat-maps for the remaining toxicity targets. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Segner H. Zebrafish (Danio rerio) as a model organism for investigating endocrine disruption. Comp. Biochem. Phys. C. 2009;149(2):187–195. doi: 10.1016/j.cbpc.2008.10.099. [DOI] [PubMed] [Google Scholar]
- 2.Ankley GT, Villeneuve DL. The fathead minnow in aquatic toxicology: Past, present and future. Aquat. Toxicol. 2006;78(1):91–102. doi: 10.1016/j.aquatox.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 3.Berg C, Gyllenhammar I, Kvarnryd M. Xenopus tropicalis as a Test System for Developmental and Reproductive Toxicity. J. Toxicol. Env. Health A. 2009;72(3–4):219–225. doi: 10.1080/15287390802539079. [DOI] [PubMed] [Google Scholar]
- 4.Massarsky A, Trudeau VL, Moon TW. β-blockers as endocrine disruptors: the potential effects of human β-blockers on aquatic organisms. J. Exp. Zool. Part A. 2011;315A(5):251–265. doi: 10.1002/jez.672. [DOI] [PubMed] [Google Scholar]
- 5.McGrath P, Li C-Q. Zebrafish: a predictive model for assessing drug-induced toxicity. Drug Discov. Today. 2008;13(9–10):394–401. doi: 10.1016/j.drudis.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Milnes MR, Garcia A, Grossman E, Grün F, Jason S, Tabb MM, Kawashima Y, Katsu Y, Watanabe H, Iguchi T, Blumberg B. Activation of Steroid and Xenobiotic Receptor (SXR, NR1I2) and Its Orthologs in Laboratory, Toxicologic, and Genome Model Species. Environ. Health Persp. 2008;116(7):880–885. doi: 10.1289/ehp.10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oba Y, Yamauchi A, Hashiguchi Y, Satone H, Miki S, Nassef M, Shimasaki Y, Kitano T, Nakao M, Kawabata S.-i., Honjo T, Oshima Y. Purification and characterization of tributyltin-binding protein of tiger puffer, Takifugu rubripes. Comp. Biochem. Phys. C. 2011;153(1):17–23. doi: 10.1016/j.cbpc.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 8.Kloas W, Urbatzka R, Opitz R, Würtz S, Behrends T, Hermelink B, Hofmann F, Jagnytsch O, Kroupova H, Lorenz C, Neumann N, Pietsch C, Trubiroha A, Van Ballegooy C, Wiedemann C, Lutz I. Endocrine Disruption in Aquatic Vertebrates. Ann. N.Y. Acad. Sci. 2009;1163(1):187–200. doi: 10.1111/j.1749-6632.2009.04453.x. [DOI] [PubMed] [Google Scholar]
- 9.Giacomini KM, Huang S-M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L, The International Transporter Consortium Membrane transporters in drug development. Nat. Rev. Drug. Discov. 2010;9(3):215–236. doi: 10.1038/nrd3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunnarsson L, Jauhiainen A, Kristiansson E, Nerman O, Larsson DGJ. Evolutionary Conservation of Human Drug Targets in Organisms used for Environmental Risk Assessments. Environ. Sci. Technol. 2008;42(15):5807–5813. doi: 10.1021/es8005173. [DOI] [PubMed] [Google Scholar]
- 11.Dixit R, Boelsterli UA. Healthy animals and animal models of human disease(s) in safety assessment of human pharmaceuticals, including therapeutic antibodies. Drug Discov. Today. 2007;12(7–8):336–342. doi: 10.1016/j.drudis.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Vamathevan JJ, Hall MD, Hasan S, Woollard PM, Xu M, Yang Y, Li X, Wang X, Kenny S, Brown JR, Huxley-Jones J, Lyon J, Haselden J, Min J, Sanseau P. Minipig and beagle animal model genomes aid species selection in pharmaceutical discovery and development. Toxicol. Appl. Pharmacol. 2013;270(2):149–157. doi: 10.1016/j.taap.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Kohno S, Katsu Y, Iguchi T, Guillette LJ. Novel approaches for the study of vertebrate steroid hormone receptors. Integr. Comp. Biol. 2008;48(4):527–534. doi: 10.1093/icb/icn080. [DOI] [PubMed] [Google Scholar]
- 14.Ho CKM, Habib FK. Estrogen and androgen signaling in the pathogenesis of BPH. Nat. Rev. Urol. 2011;8(1):29–41. doi: 10.1038/nrurol.2010.207. [DOI] [PubMed] [Google Scholar]
- 15.Setola V, Roth BL. Why Mice Are Neither Miniature Humans nor Small Rats: A Cautionary Tale Involving 5-Hydroxytryptamine-6 Serotonin Receptor Species Variants. Mol. Pharmacol. 2003;64(6):1277–1278. doi: 10.1124/mol.64.6.1277. [DOI] [PubMed] [Google Scholar]
- 16.Hirst WD, Abrahamsen B, Blaney FE, Calver AR, Aloj L, Price GW, Medhurst AD. Differences in the Central Nervous System Distribution and Pharmacology of the Mouse 5-Hydroxytryptamine-6 Receptor Compared with Rat and Human Receptors Investigated by Radioligand Binding, Site-Directed Mutagenesis, and Molecular Modeling. Mol. Pharmacol. 2003;64(6):1295–1308. doi: 10.1124/mol.64.6.1295. [DOI] [PubMed] [Google Scholar]
- 17.Kruger FA, Overington JP. Global Analysis of Small Molecule Binding to Related Protein Targets. PLoS Comput. Biol. 2012;8(1):e1002333. doi: 10.1371/journal.pcbi.1002333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Cote S, Shoichet BK, Urban L. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486(7403):361–367. doi: 10.1038/nature11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vedani A, Dobler M, Smiesko M. VirtualToxLab - A platform for estimating the toxic potential of drugs, chemicals and natural products. Toxicol. Appl. Pharmacol. 2012;261(2):142–153. doi: 10.1016/j.taap.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 20.Kufareva I, Ilatovskiy AV, Abagyan R. Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012;40(Database Issue):D535–40. doi: 10.1093/nar/gkr825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The UniProt Consortium Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40(D1):D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.ICM. version 3.7-a Molsoft L.L.C.: La Jolla; 2012. [Google Scholar]
- 24.Abagyan R, Totrov M. Biased Probability Monte Carlo Conformational Searches and Electrostatic Calculations for Peptides and Proteins. J. Mol. Biol. 1994;235(3):983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 25.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48(3):443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 27.Abagyan RA, Batalov S. Do aligned sequences share the same fold? J. Mol. Biol. 1997;273(1):355–368. doi: 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- 28.Gonnet G, Cohen M, Benner S. Exhaustive matching of the entire protein sequence database. Science. 1992;256(5062):1443–1445. doi: 10.1126/science.1604319. [DOI] [PubMed] [Google Scholar]
- 29.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Status of GPCR Modeling and Docking as Reflected by Community-wide GPCR Dock 2010 Assessment. Structure. 2011;19(8):1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kufareva I, Abagyan R. Methods of Protein Structure Comparison. In: Orry AJW, Abagyan R, editors. Homology Modeling. Vol. 857. Humana Press; 2012. pp. 231–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Postlethwait JH, Yan Y-L, Gates MA, Horne S, Amores A, Brownlie A, Donovan A, Egan ES, Force A, Gong Z, Goutel C, Fritz A, Kelsh R, Knapik E, Liao E, Paw B, Ransom D, Singer A, Thomson M, Abduljabbar TS, Yelick P, Beier D, Joly JS, Larhammar D, Rosa F, Westerfield M, Zon LI, Johnson SL, Talbot WS. Vertebrate genome evolution and the zebrafish gene map. Nat. Genet. 1998;18(4):345–349. doi: 10.1038/ng0498-345. [DOI] [PubMed] [Google Scholar]
- 32.Amores A, Force A, Yan Y-L, Joly L, Amemiya C, Fritz A, Ho RK, Langeland J, Prince V, Wang Y-L, Westerfield M, Ekker M, Postlethwait JH. Zebrafish hox Clusters and Vertebrate Genome Evolution. Science. 1998;282(5394):1711–1714. doi: 10.1126/science.282.5394.1711. [DOI] [PubMed] [Google Scholar]
- 33.Aparicio S, Chapman J, Stupka E, Putnam N, Chia J.-m., Dehal P, Christoffels A, Rash S, Hoon S, Smit A, Gelpke MDS, Roach J, Oh T, Ho IY, Wong M, Detter C, Verhoef F, Predki P, Tay A, Lucas S, Richardson P, Smith SF, Clark MS, Edwards YJK, Doggett N, Zharkikh A, Tavtigian SV, Pruss D, Barnstead M, Evans C, Baden H, Powell J, Glusman G, Rowen L, Hood L, Tan YH, Elgar G, Hawkins T, Venkatesh B, Rokhsar D, Brenner S. Whole-Genome Shotgun Assembly and Analysis of the Genome of Fugu rubripes. Science. 2002;297(5585):1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 34.Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, Ovcharenko I, Putnam NH, Shu S, Taher L, Blitz IL, Blumberg B, Dichmann DS, Dubchak I, Amaya E, Detter JC, Fletcher R, Gerhard DS, Goodstein D, Graves T, Grigoriev IV, Grimwood J, Kawashima T, Lindquist E, Lucas SM, Mead PE, Mitros T, Ogino H, Ohta Y, Poliakov AV, Pollet N, Robert J, Salamov A, Sater AK, Schmutz J, Terry A, Vize PD, Warren WC, Wells D, Wills A, Wilson RK, Zimmerman LB, Zorn AM, Grainger R, Grammer T, Khokha MK, Richardson PM, Rokhsar DS. The Genome of the Western Clawed Frog Xenopus tropicalis. Science. 2010;328(5978):633–636. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin MT, Knudsen TB, Reif DM, Houck KA, Judson RS, Kavlock RJ, Dix DJ. Predictive Model of Rat Reproductive Toxicity from ToxCast High Throughput Screening. Biol. Reprod. 2011;85(2):327–339. doi: 10.1095/biolreprod.111.090977. [DOI] [PubMed] [Google Scholar]
- 36.Owen SF, Giltrow E, Huggett DB, Hutchinson TH, Saye J, Winter MJ, Sumpter JP. Comparative physiology, pharmacology and toxicology of β-blockers: Mammals versus fish. Aquat. Toxicol. 2007;82(3):145–162. doi: 10.1016/j.aquatox.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 37.Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL. Evidence for Possible Involvement of 5-HT2B Receptors in the Cardiac Valvulopathy Associated With Fenfluramine and Other Serotonergic Medications. Circulation. 2000;102(23):2836–2841. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- 38.Mizuo K, Narita M, Yoshida T, Narita M, Suzuki T. Functional changes in dopamine D3 receptors by prenatal and neonatal exposure to an endocrine disruptor bisphenol-A in mice. Addict. Biol. 2004;9(1):19–25. doi: 10.1080/13556210410001674059. [DOI] [PubMed] [Google Scholar]
- 39.Overstreet DH, Knapp DJ, Breese GR. Can CRF1 receptor antagonists become antidepressant and/or anxiolytic agents? Drug Develop. Res. 2005;65(4):191–204. [Google Scholar]
- 40.Hauger RL, Grigoriadis DE, Dallman MF, Plotsky PM, Vale WW, Dautzenberg FM. International Union of Pharmacology. XXXVI. Current Status of the Nomenclature for Receptors for Corticotropin-Releasing Factor and Their Ligands. Pharmacol. Rev. 2003;55(1):21–26. doi: 10.1124/pr.55.1.3. [DOI] [PubMed] [Google Scholar]
- 41.Irwin N, Flatt PR. Therapeutic potential for GIP receptor agonists and antagonists. Best Pract. Res. Cl. En. 2009;23(4):499–512. doi: 10.1016/j.beem.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Hollenstein K, Kean J, Bortolato A, Cheng RKY, Dore AS, Jazayeri A, Cooke RM, Weir M, Marshall FH. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499(7459):438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- 43.Flint S, Markle T, Thompson S, Wallace E. Bisphenol A exposure, effects, and policy: A wildlife perspective. J. Environ. Manage. 2012;104:19–34. doi: 10.1016/j.jenvman.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 44.Parrott JL, Blunt BR. Life-cycle exposure of fathead minnows (Pimephales promelas) to an ethinylestradiol concentration below 1 ng/L reduces egg fertilization success and demasculinizes males. Environ. Toxicol. 2005;20(2):131–141. doi: 10.1002/tox.20087. [DOI] [PubMed] [Google Scholar]
- 45.Janesick A, Blumberg B. Minireview: PPARγ as the target of obesogens. J. Steroid Biochem. Mol. Biol. 2011;127(1–2):4–8. doi: 10.1016/j.jsbmb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pochapsky TC, Kazanis S, Dang M. Conformational Plasticity and Structure/Function Relationships in Cytochromes P450. Antioxid. Redox Sign. 2010;13(8):1273–1296. doi: 10.1089/ars.2010.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ekroos M, Sjögren T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Nat. Acad. Sci. U.S.A. 2006;103(37):13682–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milestone CB, Orrego R, Scott PD, Waye A, Kohli J, O‚ÄôConnor BI, Smith B, Engelhardt H, Servos MR, MacLatchy DL, Smith DS, Trudeau VL, Arnason JT, Kovacs T, Heid Furley T, Slade AH, Holdway DA, Hewitt LM. Evaluating the Potential of Effluents and Wood Feedstocks from Pulp and Paper Mills in Brazil, Canada, and New Zealand to Affect Fish Reproduction: Chemical Profiling and In Vitro Assessments. Environ. Sci. Technol. 2011;46(3):1849–1858. doi: 10.1021/es203382c. [DOI] [PubMed] [Google Scholar]
- 49.Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliver. Rev. 2008;60(13–14):1527–1533. doi: 10.1016/j.addr.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boswell-Smith V, Spina D. PDE4 inhibitors as potential therapeutic agents in the treatment of COPD-focus on roflumilast. Int. J. Chron. Obstruct. Pulmon. Dis. 2007;2:121–129. [PMC free article] [PubMed] [Google Scholar]
- 51.Vezzosi D, Bertherat J. Phosphodiesterases in endocrine physiology and disease. Eur. J. Endocrinol. 2011;165(2):177–188. doi: 10.1530/EJE-10-1123. [DOI] [PubMed] [Google Scholar]
- 52.Searls DB. Pharmacophylogenomics: genes, evolution and drug targets. Nat. Rev. Drug. Discov. 2003;2(8):613–623. doi: 10.1038/nrd1152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.