

Abstract
BCL-2 family proteins are the regulators of apoptosis, but also have other functions. This family of interacting partners includes inhibitors and inducers of cell death. Together they regulate and mediate the process by which mitochondria contribute to cell death known as the intrinsic apoptosis pathway. This pathway is required for normal embryonic development and for preventing cancer. However, before apoptosis is induced, BCL-2 proteins have critical roles in normal cell physiology related to neuronal activity, autophagy, calcium handling, mitochondrial dynamics and energetics, and other processes of normal healthy cells. The relative importance of these physiological functions compared to their apoptosis functions in overall organismal physiology is difficult to decipher. Apoptotic and noncanonical functions of these proteins may be intertwined to link cell growth to cell death. Disentanglement of these functions may require delineation of biochemical activities inherent to the characteristic three-dimensional shape shared by distantly related viral and cellular BCL-2 family members.
WHAT ARE BCL-2 FAMILY PROTEINS AND HOW DO THEY WORK?
Human BCL-2 was discovered as the gene located near the junction at which chromosomes 18 and 14 (t14;18) are joined anomalously in the tumor cells of follicular lymphoma patients (Tsujimoto et al. 1984). This chromosome translocation leads to misregulation of the normal BCL-2 expression pattern to contribute to cancer (Tsujimoto et al. 1985; Nunez et al. 1989). Unlike previously identified oncogenes, BCL-2 was found to promote cell survival as opposed to promoting cell proliferation (Vaux et al. 1988; Tsujimoto 1989). That is, BCL-2 increases the total cell number by preventing cell death rather than by increasing cell division rate. Given that failure of these cells to die resulted in cancer, it was logical to assume that BCL-2 blocks aform of deliberate cell death. The term apoptosis (Gk: falling off, like a tree leaf ) had been coined some years earlier to refer to deliberate cell death, and thus was applied to the type of cell death blocked by BCL-2 (Kerr et al. 1972; Hockenbery et al. 1991).
Compelling genetic evidence that solidified and extended this model of apoptosis regulation came from simultaneous research on the worm Caenorhabditis elegans. The worm BCL-2 ortholog, CED-9, was identified as the gene responsible for preventing cell death during worm development (Hengartner et al. 1992; Hengartner and Horvitz 1994b). Further genetic studies revealed that CED-9 inhibits caspase-mediated cell death (Horvitz et al. 1983; Yuan and Horvitz 1990; Yuan et al. 1993). Although C. elegans has only one (multidomain) BCL-2 family member, eight additional homologs of BCL-2 ranging in size from 20 to 37 kDa (BCL-xL, MCL-1, BCL-w, BFL-1/A1, BCL-B, BAX, BAK, and BOK) plus five less related proteins sharing significant amino acid sequence similarity [BCL2L12, BCL-Rambo (BCL2L13), BCL-G (BCL2L14), BFK (BCL2L15), and BID] have been identified in the human genome (Fig. 1) (Blaineau and Aouacheria 2009). These proteins are thought to work on membranes of mitochondria and the endoplasmic reticulum (ER) facilitated by a hydrophobic membrane anchor/targeting domain near the carboxyl terminus of most BCL-2 homologs, and by a helical hairpin (helix 5 and 6 between BH1 and BH2) suggested to insert into membranes (Muchmore et al. 1996; Minn et al. 1997; Basanez and Hardwick 2008).
Figure 1.

Human, C. elegans, and Drosophila BCL-2 family members. BH motifs are numbered, BH4 (light gray, unnumbered box indicates traditional classification without verified sequence homology), BH3 (black), BH1-BH2 (white), transmembrane (dark gray), arrows (protease cleavage sites). Splice variants (isoforms) are shown as separate or partially overlapped diagrams. Scale bar at the bottom marks increments of 50 amino acids.
Although most BCL-2 homologs inhibit cell death, a subset is classified as proapoptotic (BAX, BAK, and BID). Proapoptotic BAX was first identified as an inhibitory binding partner of BCL-2 (Oltvai et al. 1993). The pro-death function of BAX is activated in response to a range of deleterious events inside or outside the cell, causing BAX to undergo conformational changes, membrane-insertion, and oligomerization to form a channel or other structure in the mitochondrial outer membrane. This is widely assumed to be the conduit through which cytochrome c exits mitochondria to trigger caspase activation and cell death (Cosulich et al. 1997; Kim et al. 1997; Jurgensmeier et al. 1998; Rosse et al. 1998; Kluck et al. 1999). The role of BCL-2-like antiapoptotic proteins is to inhibit their proapoptotic partners, leading to the original rheostat model in which the balance between counteracting anti- and proapoptotic BCL-2 family proteins determines cell fate (Korsmeyer et al. 1993). Although the ratios of anti- and pro-death family proteins indeed usually correlate with cell fate, this model is over-simplified in light of multiple subsequent discoveries, including the occasional interconversion of anti- and pro-death activities (Cheng et al. 1997a; Clem et al. 1998; Lewis et al. 1999), the existence of additional BCL-2-interacting proteins (Wang et al. 1996; Kelekar et al. 1997; Strasser et al. 2000; Puthalakath et al. 2001; Shamas-Din et al. 2011), the identification of BCL-2-like proteins unable to affect cell death (Bellows et al. 2002; Peterson et al. 2007; Galindo et al. 2009; Gonzalez and Esteban 2010), and the rapidly growing list of alternative nonapoptotic functions of BCL-2 family members that may have an important impact on cell survival.
The third functional subgroup of the BCL-2 family triangle is designated BH3-only because these proteins have only one of the four different BH (BCL-2 homology) motifs (Huang and Strasser 2000; Shamas-Din et al. 2011). BH motifs (numbered in order of discovery) are 10–20 amino acid regions of greatest amino acid sequence similarity across family members, though BH sequence identity can be low, and most BCL-2 homologs lack at least one BH motif (Fig. 1). The BH3 motif of proapoptotic family members is required for their pro-death activities. Eight BH3-only proteins (BID, BAD, BIK, BIM, BMF, HRK, NOXA, and PUMA) generally range in size from ~100 to 200 amino acids and are classified as BCL-2 family members based on their ability to bind and inhibit antiapoptotic BCL-2 proteins, though they lack significant overall amino acid sequence similarity (except BID) to BCL-2 proteins or to each other. BH3-only proteins promote apoptosis using one or both of two general strategies. They bind and directly activate BAX and BAK (e.g., tBID, BIM, and PUMA), or they promote death indirectly by inserting their BH3-containing helix into a hydrophobic groove on specific antiapoptotic BCL-2 proteins (Petros et al. 2000; Strasser 2005; Deng et al. 2007; Billen et al. 2008). This triangular model further explains that antiapoptotic BCL-2 proteins, of which BCL-xL is the best characterized, protect cells by binding and inhibiting the direct activator BH3-only proteins and the multi-BH proapoptotic proteins BAX and BAK. Other than the BH3 helix, three-dimensional structures of BH3-only proteins are unresolved except for BID, which adopts a BCL-2-like fold shared by both anti- and pro-death family members (Chou et al. 1999; McDonnell et al. 1999). Thus, BH3-only proteins are thought to be intrinsically disordered, possibly reflecting their dynamical functions and supported by extensive work (Hinds et al. 2007). Complete structures of partnered complexes would help distinguish this possibility from a case like the well ordered structures of Sgf11 and Sgf73 proteins that stretch across different domains of the SAGA deubiquitinating (DUBm) complex and would appear inherently unfolded in isolation (Samara et al. 2010).
The prevailing BCL-2 apoptosis model, in which antiapoptotic proteins are inhibited when their deep binding cleft is occupied by the BH3 helix of proapoptotic family members (e.g., BH3-only BAD), is strongly supported by the effects of a small molecule ABT-737, a BH3 mimetic designed to occupy the BCL-xL groove (Oltersdorf et al. 2005). ABT-737 derivatives are in clinical trials as anticancer agents with promising early results, and additional therapeutics specific to antiapoptotic MCL-1, which is not targeted by ABT-737, are being pursued (Tse et al. 2008; Yecies et al. 2010; Gandhi et al. 2011).
Considerable evidence now suggests that both pro- and antiapoptotic BCL-2 family proteins have additional functions required for normal physiology of healthy cells. These non-canonical functions are unlikely to be fully explained by classical apoptosis regulatory activities in which anti-death BCL-2 proteins directly bind and inhibit proapoptotic BCL-2 family proteins to control the release of cytochrome c from mitochondria in the intrinsic apoptosis pathway. In addition, there is a growing fourth class of BCL-2 family proteins that primarily lack apoptosis regulatory activities. Viruses encode many BCL-2-shaped proteins, but most of these appear to have functions distinct from regulating cell death (see below). Similarly, the cellular BCL-2 homologs of Drosophila may not regulate cell death in most cell types, and the role of cytochrome c in apoptosis is not uniformly conserved through evolution (Oberst et al. 2008; Galindo et al. 2009; Tanner et al. 2011; Bender et al. 2012). Noncanonical functions of BCL-2 family proteins include their ability to alter mitochondrial shape changes and energetics, to regulate autophagy, and to modulate innate immunity during virus infections (Stack et al. 2005; Hardwick et al. 2012). Furthermore, antiapoptotic BCL-2 proteins can become proapoptotic, whereas proapoptotic proteins can promote cell survival (Bellows et al. 2002; Peterson et al. 2007; Galindo et al. 2009; Gonzalez and Esteban 2010). A major unanswered question is the relative importance of “day-job” functions versus the apoptosis-related functions of BCL-2 proteins in determining cell fate, and if the biochemical details of these functions overlap.
TURNING A NEW LEAF ON APOPTOSIS
Impressive research progress over the past 25 years has successfully driven home the fact that a subset of cells must die for an embryo to develop properly, even for the severed Planarian to regrow a new head (Gonzalez-Estevez and Salo 2010). But now it is time to let go of a few engrained assumptions to include a broader perspective. For example, we can no longer assume verbatim that the mere presence of caspase activity correlates with cell death, as caspases are also required for synaptic activity, cell growth, and inhibition of necrosis (Peter 2011; Li and Sheng 2012). We also no longer can assume that BID, BAX, and BAD are promoting cell death just because they are expressed (Fannjiang et al. 2003; Seo et al. 2004; Danial et al. 2010; Gimenez-Cassina et al. 2012). Even more complicated is the difficult task of experimentally distinguishing between noncanonical day-jobs versus apoptosis functions. For example, simply evaluating the degree of cell death will not reveal whether the disrupted function of BCL-2 was an essential day-job or its direct role in apoptosis, or both. One matter seems clear, most un-dead cells are detrimental to essentially all forms of life, and teleological reasoning suggests there is no better way to link fundamental cell functions to cell death than to use the same molecules for both processes.
The original assumption that antiapoptotic BCL-2/BCL-xL must bind to proapoptotic BAX/BAK to inhibit cell death was first challenged by point mutants of BCL-xL (e.g., BCL-xL F131V/D133A, BCL-xL Y101K) that retain significant anti-death activity but fail to interact with BAX or BAK (Cheng et al. 1996; Minn et al. 1999). However, these mutants have been reported to retain their second mode of action, the ability to bind specific BH3-only proteins, supporting the dual-strategy hypothesis whereby antiapoptotic BCL-2 proteins can suppress cell death by interfering with both subcategories of pro-death BCL-2 family proteins, the multi-domain and the BH3-only (direct activator) proteins (Cheng et al. 2001; Billen et al. 2008). Elegant biochemical and computational studies have rigorously probed the classic apoptosis mechanisms with purified components and provide compelling evidence for BCL-xL-inhibited, tBID-activated, BAX-mediated cytochrome c release. However, these findings cannot fully explain the protective effects of BCL-xL (Billen et al. 2008). These studies however do not directly test alternative noncanonical functions of BCL-2 family proteins, as both the F131V/D133A and Y101K BCL-xL mutants retain the ability to bind additional factors unrelated to BCL-2 family proteins that are not present or not being evaluated in these assays (Fig. 2). For example, wild type and F131V/D133A BCL-xL can promote cell survival by binding to Aven, a regulator of both Apaf1 (apoptosome) and of ATM kinase at the G2/M checkpoint (Chau et al. 2000; Guo et al. 2008; Roelofs and Hardwick 2011; Zou et al. 2011). BCL-2 proteins also bind to the mitochondrial metabolite channel VDAC (Cheng et al. 2003), autophagy regulator Beclin 1 (Pattingre et al. 2005), the mitochondrial fission and fusion factors Drp1 and Mfn1/2 (Rolland et al. 2009), and others (Chipuk et al. 2004). At present it is difficult to incorporate all of these and other reported interactions of BCL-xL into a unifying model for the physiological function of BCL-xL. The field currently lacks tools equivalent to the elegant reconstituted BCL-xL-tBID-BAX assays to probe these critical functions, though progress has been made (Alavian et al. 2011). We simply have insufficient knowledge at present to fully accomplish this task.
Figure 2.

Map of key BCL-xL mutants. Amino acid positions and single amino acid changes are indicated. tm, transmembrane region.
DEATH AND SURVIVAL FUNCTIONS OF PROAPOPTOTIC BCL-2 PROTEINS
BAX and BAK
Long before the discovery of BCL-2, Rita Levi-Montalcini and her colleagues were among the first to recognize programmed cell death while observing neurons in the developing chick embryo. With other colleagues, she later discovered the factor required to prevent this death of sympathetic and sensory neurons in the peripheral nervous system, nerve growth factor (NGF) (Levi-Montalcini and Angeletti 1968). Withdrawal of NGF leads to neuronal death that is mediated by the proapoptotic BCL-2 family protein BAX (Deckwerth et al. 1996). As a result, the use of BAX knockout mice has become a standard in the field for studying the functions of NGF and other factors because NGF-deprived bax−/− neurons can be evaluated without the complication of death of the subject under study (Glebova and Ginty 2004). In this scenario, BAX is thought to act downstream of NGF withdrawal by classical apoptosis mechanisms (see below).
As expected, BAX promotes cell death in the in the developing central nervous system, in which it is estimated that over half of the neurons born will die in a BAX-dependent manner, although this slow process can only be fully appreciated when evaluated over the long term in BAX-deficient animals (Sun et al. 2004). A role for caspase-dependent apoptosis in this process is evident from the enormous brains first observed in mice lacking caspase-3, caspase-9, or Apaf1 (Kuida et al. 1996, 1998; Yoshida et al. 1998; Zheng et al. 2000). These brain phenotypes are different from the BAX/BAK double knockout, which has an obvious increase in the number of neuroprogenitors in the periventricular zone of the brain (Lindsten et al. 2000). This increase in survival of neuroprogenitors is much more pronounced in the double knockout than in the BAX single knockout and is nearly undetectable in the BAK single knockout (Lindsten et al. 2000). BAX also suppresses neurogenesis in the hippocampus and the cerebellum of adult brains (Sun et al. 2004; Garcia et al. 2012). Adult-born neurons also survive in much greater numbers in the absence of BAX even when continued neurogenesis is ablated (Sahay et al. 2011). These undead neurons do not significantly impact spatial learning and memory. However, these extra neurons apparently can confer significantly improved contextual discrimination learning, a function that normally declines with age, raising the possibility that BAX suppression could delay premature diminution of neuronal function by allowing more neurons to survive and fulfill their functions (Sahay et al. 2011).
Contrary to the pro-death developmental functions of BAX and BAK, BAX-deficiency does not rescue the death of some neuron subtypes under pathological situations (Lindsten et al. 2000; Whitmore et al. 2003; Glebova and Ginty 2004). In fact, endogenous or exogenous BAX or BAK can even protect against cell death induced by infection with Sindbis virus, which primarily infects neurons of the brain and spinal cord and causes encephalitis in mice (Lewis et al. 1999; Fannjiang et al. 2003). In an extensive analysis of BAK knockout mice, BAK was found to either inhibit or enhance neuronal death depending on the developmental stage, death stimulus, and brain region (Fannjiang et al. 2003). For example, BAK promotes death of neurons of the cortex in a stroke model, but protects hippocampal neurons following kainate-induced seizures. However, the protection by BAK in a kainate-induced seizure model appears not to be because of classical antiapoptotic function, because the degree of neuronal cell death simply correlates with more severe seizures. That is, the cell death that occurred several days later is triggered by a different process (Fannjiang et al. 2003). Because BAK knockout mice showed more seizure behaviors within minutes after kainate injection, long before neuronal death, alternative functions of BAK are implicated, such as changes in neuronal activity that give rise to a seizure. This is supported by the altered electrophysiological recordings of acute brain slices prepared from BAK knockout and control mice (Fannjiang et al. 2003).
BAK also shows bipolar effects in the spinal cord. BAK inhibits the death of virus-infected spinal cord motor neurons in young mice, but promotes motor neuron death in more mature animals (Fannjiang et al. 2003). In this scenario, BAK is expected to kill by conventional apoptosis mechanisms, but it is also possible that BAK promotes death indirectly by an alternative mechanism, such as altered neuronal activity analogous to excitotoxicity (Fannjiang et al. 2003). Perhaps this apparent switch in function is analogous to the developmental switch in excitatory to inhibitory effects of the GABA neurotransmitter (Marty and Llano 2005). Thus, BAK and BAX could contribute to both survival and death of neurons by alternative nonapoptotic mechanisms, or potentially a combination of nonapoptotic and apoptotic mechanisms. Consistent with this possibility, it is intriguing that BAX inhibits neuronal death in brain slices in which neuro-connections are preserved, but kills when these same neurons are dissociated in a culture dish. For validation of genuine anti-death activity of BAX and BAK, targeted reconstitution of BAX or BAK into the neurons of their respective knockout mice by infecting these animals with Sindbis virus encoding a copy of BAX or BAK, dramatically rescues knockout mice from Sindbis virus-induced neuronal death and mortality (Lewis et al. 1999; Fannjiang et al. 2003). This is likely not a fluke of the model system, because the same model confirmed the anti-death activity of BCL-2 and the pro-death activity of BIMS (Seo et al. 2004).
BH3-Only Proteins
BAD is known to promote cell death by antagonizing anti-death BCL-2 proteins, but a number of studies clearly show that BAD has a normal physiological role in healthy cells. Endogenous and exogenous BAD strongly protect mice and their derived cells from Sindbis virus-and NMDA-induced neuronal death (Seo et al. 2004). The antiapoptotic functions of BAD are not limited to neurons. Overexpressed BAD in transfected cell lines can inhibit cell death similar to BCL-xL if the caspase cleavage sites in BAD are mutated to render BAD uncleavable (Condorelli et al. 2001; Kim et al. 2002; Seo et al. 2004). Interestingly, different death stimuli use distinct caspase cleavage sites to inactivate BAD. For example, mutation of the caspase cleavage site at Asp56 is required to block death following IL-3 withdrawal, whereas Asp61 must be mutated to protect against staurosporine and γ-irradiation (Seo et al. 2004). However, even proteolytically cleaved BAD (tBAD) cannot kill immature neurons in the brain, in which a further step apparently involving dephosphorylation of Ser residues (Ser112, S136, and S155) is required to turn off anti-death function and activate the death function of BAD in neonatal mice (Seo et al. 2004). This is consistent with the evidence that dephosphorylated BAD binds and inactivates antiapoptotic BCL-2 family proteins (Datta et al. 1997).
The mechanisms by which BAD inhibits cell death in these model systems is not known, but the findings are consistent with the classical view that dephosphorylation of BAD releases BAD from 14-3-3 to engage and inhibit BCL-2-like antiapoptotic proteins. However, this classical explanation of sequestering away proapoptotic BAD when phosphorylated is rather unsatisfying when attempting to explain how overexpression of phosphomimetic mutants of BAD show a gain of anti-death activity (Datta et al. 1997; Seo et al. 2004). Thus, BAD likely increases cell survival through its “day-job” mechanisms, which may involve its roles in glucose metabolism, autophagy, or cell cycle progression (Roy et al. 2009). The BH3 motif of BAD is critical for killing cells, as expected, but its BH3 is also required to promote health and well-being by activating glucokinase and increasing glucose metabolism (Danial et al. 2003, 2008). Similarly, phosphorylation is required both for its effects on glucose metabolism and for suppression of its proapoptotic activity. Hence, the cell survival and cell death functions of BAD appear to be intricately linked.
An intriguing nonapoptotic role for BAD in the control of potassium-ATP channels through its effects on glucose metabolism is also regulated by BAD phosphorylation (Gimenez-Cassina et al. 2012). In this manner, BAD links metabolism to the control of seizure activity measured both as behavioral and electrographic seizures. Recently, BAD and BAX have also been shown to exert nonapoptotic functions in long-term depression (LTD) of synaptic transmission in CA1 hippocampal neurons (Jiao and Li 2011). BAD and BAX-mediated activation of limited caspase-3 activity is required for NMDA receptor-dependent LTD but not for mGluR-LTD. Activation of this pathway is sufficient to induce synaptic depression and is inhibited by both BAD and BAX siRNAs or knockout. Activation of BAD by dephosphorylation is limited, and apparently BAX does not translocate to mitochondria in this model (Jiao and Li 2011).
BID has a key role in crosstalk between the extrinsic (extracellular ligand binding to cell surface death receptors) and intrinsic apoptosis pathways. Cleavage of BID by caspase-8 in the extrinsic pathway generates amino-terminally deleted BID known as truncated BID (tBID) (Li et al. 1998; Luo et al. 1998; Gross et al. 1999). tBID then activates the intrinsic pathway by well-studied mechanisms in which tBID transiently binds and induces BAX activation on mitochondrial membranes in vitro and in cells (Kuwana et al. 2002; Billen et al. 2008; Lovell et al. 2008). The pro-death function of the activator BH3-only proteins BID, BIM, and PUMA are apparent from studies of triple knockout mice that fail to activate BAX/BAK-dependent apoptosis in neurons and lymphocytes (Ren et al. 2010).
Although cleavage of full-length BID near its amino terminus (Fig. 1) to expose its killer BH3 domain could be analogous to the cleavage and removal of inhibitory prodomains of proteases for example, it is also possible that full-length BID has a “day-job” in healthy cells. Indeed, in addition to its apoptotic function BID also has a nonapoptotic role in regulation of the DNA damage response (Zinkel et al. 2005). It has been shown that DNA damage induces translocation of BID to the nucleus in which it is phosphorylated by ATM and regulates an intra S-phase checkpoint. More recently, BID was also shown to mediate the ATR-directed DNA damage response to replicative stress by interacting with the Atrip/RPA complex (Liu et al. 2011). Interestingly, BID was recently identified in a screen for factors that facilitate innate immune responses in the gut. BID appears to be required for activation of host defense mechanisms to control bacterial infections, but may also exacerbate immune-mediated inflammatory bowel disease (Yeretssian et al. 2011). This function of BID apparently does not involve its classical apoptosis mechanisms, as knockin mice with uncleavable BID are competent for immune signaling. Conversely, mutations in the amino-terminal region of BID (not found in tBID) interfere with binding to NOD1 (member of a large family of host-defense proteins also found in plants), which forms a complex with RIP2 through their mutual CARD domains and activate IKK to mediate NF-κB activation.
NOXA, which is best known for antagonizing the anti-death function of MCL-1, has dual roles in apoptosis and metabolism, which became apparent by studying posttranslational modifications of NOXA (Lowman et al. 2010). Phosphorylation of NOXA appears to increase glucose metabolism through the pentose phosphate shunt, and this requires an intact BH3 domain, but not for binding to MCL-1 and inducing apoptosis.
MITOCHONDRIAL MEMBRANE STRUCTURE AND INTRAMITOCHONDRIAL FUNCTIONS OF BCL-2 PROTEINS
A major theme has emerged in recent years, the involvement of BCL-2 family proteins in mitochondrial shape-changes and organelle localization (Fig. 3) (Frank 2006; Detmer and Chan 2007; Knott et al. 2008). Some yet undetermined but fundamental underlying function of BCL-2 proteins, possibly in mitochondrial energetics (Vander Heiden et al. 2001), could easily influence mitochondrial morphology and function indirectly. Many studies now suggest close interactions between BCL-2 family proteins and the dynamin-like GTPases Drp1 and Mfn1/2 that physically mediate mitochondrial outer membrane fission and fusion, respectively (Li et al. 2008; Rolland et al. 2009). Here too, BCL-2 family proteins also appear to have polar opposite effects, but with a twist. Both anti- and pro-death BCL-2 family proteins functionally interact with both fission and fusion factors, but the connections between specific protein players and cell death/survival outcomes are far from clear (Jagasia et al. 2005; Cheng et al. 2006; Delivani et al. 2006; Parone et al. 2006; Li et al. 2008; Tan et al. 2008; Breckenridge et al. 2009). Nevertheless, these mechanisms may be conserved across species even when apoptosis functions of BCL-2 family proteins appear not to be conserved (Delivani et al. 2006). Genetic studies in C. elegans and biochemical strategies indicate that the fission and death functions of Drp1 are separable, and that both involve BCL-2 family members (Cassidy-Stone et al. 2008; Abdelwahid et al. 2010). A role for BAX in fusion has also been connected to the permeability transition pore opening on the mitochondrial inner membrane, leading to necrosis (Whelan et al. 2012).
Figure 3.
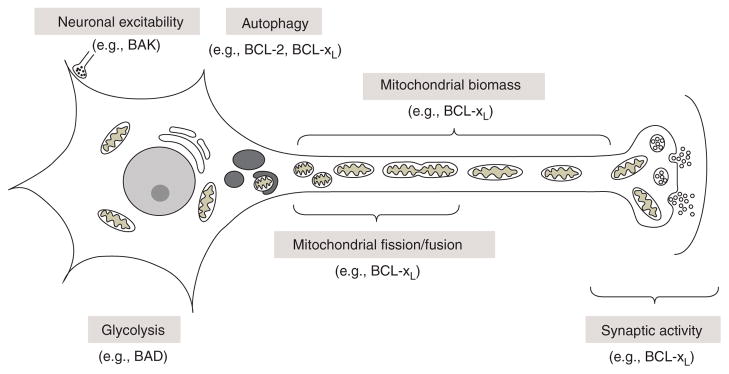
Neuronal functions of BCL-2 family proteins in healthy cells.
The link between BCL-2 family proteins and mitochondrial dynamics was first uncovered when Drp1 (homolog of yeast Dnm1) was found to promote BAX-induced mitochondrial fission and cell death (Frank et al. 2001; Karbowski et al. 2002). In contrast to dying cells, BAX promotes mitochondrial fusion in healthy cells (Karbowski et al. 2006). This dichotomy is shared by the C. elegans BCL-2 homolog CED-9, which is an essential inhibitor of cell death but can show pro-death functions (Hengartner and Horvitz 1994a). Although CED-9 interacts with FZO-1 (human Mfn1/2) to stimulate mitochondrial fusion in healthy cells, CED-9 also is required for DRP1-dependent fragmentation of mitochondria during developmental cell death (arguably both a death- and day-job) (Jagasia et al. 2005). Similarly, human BCL-xL can bind and induce Drp1-dependent mitochondrial fission, but this appears to be a day-job rather than a cell death function, because BCL-xL expression increases the number of neuronal synapses and the number of mitochondria localized to synapses, which is at least partially dependent on Drp1 (Berman et al. 2008, 2009; Li et al. 2008) (Fig. 3). These changes induced by BCL-xL are associated with dramatically increased spontaneous synaptic activity (Li et al. 2008). More direct evidence that BCL-2 family proteins can alter synaptic activity comes from studies using the squid giant synapse. Microinjection of recombinant BCL-xL protein into the squid nerve cell terminals dramatically increases synaptic activity within ~15 min (Jonas et al. 2003). Interestingly, BCL-xL can have opposite effects on neuronal activity in this squid model depending on the stimulus. Microinjected BCL-xL delays hypoxia-induced synaptic decline, but also enhances the detrimental effects of excessive neuronal activity (Jonas et al. 2004, 2005b; Hickman et al. 2008b). These findings were confirmed using the tailor-fit small molecular inhibitor of BCL-xL ABT-737. The effects of BCL-2 family proteins on synaptic activity correlates well with their ability to form large versus small channels on patch-clamped intracellular mitochondria, in which recombinant proteins are delivered through the inner patch pipette (Jonas et al. 2003, 2004, 2005a). The ability of caspase-cleaved BCL-xL to promote synaptic run-down in squid under some conditions is supported by the ability of ABT-737 to suppress this effect of BCL-xL (Hickman et al. 2008a; Ofengeim et al. 2012). This is in striking contrast to the death-promoting effects of ABT-737 in treating cancer.
Merging of day-job and apoptotic mechanisms may be most apparent in the retraction and growth of new neuronal projections and particularly dendritic spines that can be rapidly remodeled within minutes. Thus, it is conceivable that BCL-2 family proteins carry out their classical apoptotic functions except that instead of death of the entire cell, only a tiny appendage of an enormous neuron is effectively “killed” as part of normal synaptic plasticity. However, it is unlikely to be this simple. Nevertheless, recent analysis of a caspase-resistant BCL-xL knockin mouse is consistent with this hypothesis (Ofengeim et al. 2012). A mouse in which the two caspase cleavage sites in BCL-xL were mutated to render BCL-xL uncleavable is strikingly resistant to transient ischemic injury. Fitting with a pro- death role of cleaved BCL-xL, and not simply preservation of uncleaved full-length BCL-xL, ABT-737 protects wild-type animals from neuronal loss in the hippocampus following an is-chemic event, again opposite to its expected role in killing tumor cells (Ofengeim et al. 2012). However, these effects of BCL-xL appear not to involve BAX and BAK, but may involve non-apoptotic functions of caspases.
Currently, it is not clear if the opposite effects of individual mammalian and worm (and perhaps Drosophila) BCL-2 family proteins on mitochondrial shape changes require any shared biochemical mechanisms. However, recent in vitro studies argue even stronger that BCL-2family proteins may have an intimate role in membrane fusion reactions. Biochemical approaches suggest that Drp1 induces a hemifusion state in the mitochondrial outer membrane (Montessuit et al. 2010; Landes and Martinou 2011). In this manner, Drp1 creates a local lipid topology that promotes BAX oligomerization, consistent with colocalization of Drp1 and BAX in spots found at sites of mitochondrial fission (Karbowski et al. 2002; Montessuit et al. 2010). This function of Drp1 does not require its GTPase activity, which is required for mitochondrial fission. If BAX oligomerizes at stalled fission junctions, it is conceivable that outer membrane permeability is the result of a defective, excessive, or incomplete fission process (Montessuit et al. 2010).
On the day-job side, development of a powerful in vitro fusion assay has served to show a role for BAX in mitochondrial fusion in vitro (Meeusen and Nunnari 2007; Hoppins et al. 2011). By mixing together two populations of mitochondria decorated with different fusion protein components unique to the cells from which they were isolated, the effects of these protein components can be rigorously evaluated. These studies reveal that BAX and BCL-xL (separately) can promote Mfn2-dependent fusion of isolated mitochondrial organelles in vitro, fitting with their functions in healthy mitochondria. These mammalian studies are built on earlier work in yeast, Drosophila, and C. elegans that first identified the mitochondrial fission, fusion, and maintenance factors and continue to significantly advance knowledge that would not otherwise be possible (Labrousse et al. 1999; Shaw and Nunnari 2002; Tan et al. 2008; Rolland et al. 2009).
Although some of the effects on mitochondrial structure are exerted at the OMM, recent findings indicate that BCL-2 proteins can also affect mitochondrial structure and function by acting intramitochondrially (Hardwick et al. 2012). Although the long-standing dogma indicates that the apoptotic functions of BCL-2 family proteins are exerted exclusively at the OMM, this topic was recently revisited as several studies have shown that antiapoptotic BCL-2 proteins can be imported into mitochondria and likely associate with the inner mitochondrial membrane in which they are suggested to carry out nonapoptotic functions (Huang and Yang-Yen 2010; Vento et al. 2010; Alavian et al. 2011; Chen et al. 2011; Warr et al. 2011; Perciavalle et al. 2012). These studies follow earlier reports of inner mitochondrial membrane localization of BCL-2 (Hardwick et al. 2012). Intramitochondrially localized BCL-xL is suggested to regulate mitochondrial ATP production by interacting with the F1F0 ATP synthase complex and by stabilizing the inner membrane potential, thereby providing significant energy conservation (Alavian et al. 2011; Chen et al. 2011). MCL-1 has been shown to possess a bonafide mitochondrial presequence that mediates ΔΨ-dependent MCL-1 import into the mitochondrial matrix. Matrix-localized MCL-1, while devoid of antiapoptotic activity, has been shown to regulate the structure of mitochondrial cristae and the ATP synthase to alter ATP production as well (Perciavalle et al. 2012).
Non-BCL-2familyapoptosisregulatorshave also been found to be imported into mitochondria, suggesting much broader roles for apoptosis regulators at this unexpected location. Recently, p53 was reported to accumulate in the mitochondrial matrix and trigger mitochondrial permeability transition pore (PTP) opening by interaction with the PTP regulator cyclophilin D (CypD) in response to oxidative stress (Vaseva et al. 2012). Interestingly, BCL-2, which in some studies localizes preferentially at the inner mitochondrial membrane (Gotow et al. 2000) and not OMM, was show recently to also interact with matrix-localized Cyp D (Eliseev et al. 2009). Discrepancies between studies regarding the subcellular localization of different BCL-2 family proteins may reflect different cell types and energetic states, but may also reflect hereto unappreciated conformation-dependent epitopes recognized by different antibodies. The availability of genetic knockouts/knockdowns will help clarify these issues.
REGULATORS OF CALCIUM HOMEOSTASIS
Although mitochondria are considered the primary site of action of BCL-2 family proteins, many of these proteins also localize at the ER and recent studies have shown that pro-and anti-apoptotic family members exert opposing effects on ER Ca2+ handling. Although early studies noted that overexpression of BCL-2 can affect Ca2+ signaling and redistribution of Ca2+ from ER to mitochondria (Baffy et al. 1993), it was subsequently found that BCL-2 reduces basal ER Ca2+ levels specifically through increasing Ca2+ leak into the cytosol (Palmer et al. 2004). BCL-xL was later found to exert a similar effect on enhancing Ca2+ leak from ER and maintaining a low basal ER calcium concentration (White et al. 2005).
Although it was originally hypothesized that BCL-2 (and BAX) alter ER calcium levels through their channel-forming ability, it was later shown that this effect does not depend on their putative pore-forming domains (Chami et al. 2004). Subsequent studies suggested instead that regulation of ER calcium levels by BCL-2 proteins occurs through direct or indirect modulation of ER calcium channels. BCL-2 was found to bind and inactivate the calcium pump, SERCA (Dremina et al. 2004), and to induce a decline in SERCA2b levels following overexpression (Vanden Abeele et al. 2002). More recent studies pointed to a role of BCL-2 family proteins in regulating the IP3R function, although the mechanisms involved are still debated (Pinton and Rizzuto 2006). Several groups have shown that the prosurvival effect of BCL-2 and BCL-xL at the ER is promoted by an increase in ER Ca2+ leak leading to low basal ER Ca2+ concentrations and thus to a reduction in stress-induced ER Ca2+ release (Oakes et al. 2005; White et al. 2005). BCL-2 and BCL-xL were shown to directly bind IP3R and modulate its Ca2+ conductance, which in the case of BCL-2 appears to involve an effect on IP3R phosphorylation state. MCL-1 has been recently shown to bind IP3R as well and function in a similar manner at the ER (Eckenrode et al. 2010). These effects of BCL-2 proteins appear to involve activation of IP3R channel gating by an allosteric mechanism that sensitizes the channel to low inositol 1,4,5-tris-phosphate concentrations and accounts for the reduced steady-state ER Ca2+ levels (White et al. 2005; Eckenrode et al. 2010). At least for BCL-2, ER calcium regulation appears to be modulated by phosphorylation as phosphorylated BCL-2, which resides primarily at the ER, cannot reduce basal ER Ca2+ levels (Bassik et al. 2004; Oakes et al. 2006).
Other studies have found instead that interaction between IP3R and BCL-2 results in inhibition of IP3R and consequently a reduction in stress-induced IP3R-mediated Ca2+ release, elevation of cytosolic calcium and mitochondrial calcium overload. Consistent with this alternate model a peptide derived from IP3R has been shown to disrupt the BCL-2/IP3R interaction and reverse the inhibitory effect of BCL-2 on IP3R (Rong et al. 2008). The inhibitory effect of BCL-2 was attributed to the BH4 domain of BCL-2 that binds the regulatory and coupling domain of IP3R (Rong et al. 2009). The interaction of BCL-2 proteins with the IP3R likely involves multiple binding sites, as amino-terminal truncation of MCL-1 still binds efficiently to the IP3R (Eckenrode et al. 2010). Although this second model is also supported by substantial evidence, it does not provide an explanation for the observed effects of BCL-2 and BCL-xL on resting ER calcium levels.
Regarding the proapoptotic BCL-2 proteins it has been found that cells from BAX and BAK double knockout (DKO) mice also have lower resting ER calcium levels and are protected from apoptotic stimuli that signal through calcium (Scorrano et al. 2003). Based on these findings it has been proposed that in contrast to BCL-2 and BCL-xL, BAX and BAK elevate ER Ca2+ concentration and trigger ER Ca2+ release and its uptake by the mitochondria following stress (Scorrano et al. 2003; Oakes et al. 2005). Although BAX and BAK have been shown to regulate IP3R1 and Ca2+ leak, no direct interaction between BAX/BAK and IP3R has been observed and these effects may be mediated through modulation of BCL-2/IP3R1 interaction and IP3R1 phosphorylation state (Oakes et al. 2005). Although overexpression of BCL-2 or BCL-xL also results in a decreased capacitative Ca2+ entry, reduction of calreticulin and SERCA2 levels, as well as altered IP3R levels, none of these effects were noted in BAX/BAK DKO cells (Scorrano et al. 2002, 2003).
A role in regulating ER calcium release has also been reported for several BH3-only proteins, including BIK/NBK, PUMA, and NIX/BNIP3. ER-localized BIK has been shown to be required for BAX/BAK-dependent ER Ca2+ release and cytochrome c release in response to genotoxic stress (Mathai et al. 2005). Similarly, PUMA has been shown to contribute to ERCa2+ depletion-induced apoptosis by modulating BAX activity (Luo et al. 2005). Another study showed that ER-localized NIX/BNIP3 was required to induce Ca2+-mediated PTP opening and loss of ΔΨm in cardiomyocytes, although the mechanism by which NIX modulates ER Ca2+ levels is unknown (Diwan et al. 2008).
The release of calcium from the ER has been reported to be a control point for initiation of apoptosis in response to several stimuli, such as arachidonic acid, ceramide, and H2O2 (Scorrano et al. 2003). Subsequent studies have shown, however, that ER calcium regulation is also involved in nonapoptotic functions of these proteins (i.e., mitochondrial energy metabolism and T-cell activation [Jones et al. 2007]), suggesting that ER Ca2+ regulation represents another day-job function of these proteins. Such nonapoptotic functions of BCL-2 proteins appear to be conserved as the zebrafish BCL-2 homolog Nrz has been recently shown to control the cytoskeletal dynamics during zebrafish development by regulating ER Ca2+ release through direct interaction with the IP3R1 (Popgeorgiev et al. 2011).
VIRAL BCL-2 PROTEINS: AN UNEXPECTED MIXED BAG
Many viruses encode proteins that localize to mitochondria, for example, the RNA viruses HIV and influenza virus, but the functions of these proteins in virus infection and virus-host cell interactions are only partially delineated (Boya et al. 2004; Gocnikova and Russ 2007). Although BCL-2-like proteins have not been identified in the genomes of RNA viruses or small DNA viruses, in which coding capacities are preciously conserved, all three large DNA virus families infecting mammals, herpesviridae, adenoviridae, and poxviridae, encodeproteins that are included in the BCL-2 family by at least one criterion. However, many of these viral BCL-2-like factors differ greatly in sequence or function from each other and from cellular BCL-2, but all appear to maintain a BCL-2-like three-dimensional structure. The open reading frames of viral BCL-2-like genes are unspliced and generally located in variable regions of their respective viral genomes, suggesting that they were acquired from their host cell to successfully establish a stable virus–host relationship. This assumption challenges some theories suggesting precellular origins for at least some viral genomes. Like the viral oncogenes of avian and other retroviruses that differ from their homologs in host cells (proto-oncogene), viral BCL-2-like proteins appear to be resistant to cellular regulatory mechanisms relative to their cellular counterparts (Bellows et al. 2000; Irusta et al. 2003).
Antiapoptotic activity has been confirmed for several unrelated poxvirus BCL-2-like proteins, including vaccinia virus F1L (Wasilenko et al. 2003) and N1L (Cooray et al. 2007), virus M11L (Su et al. 2006), parapoxvirus ORF virus ORFV125 (Westphal et al. 2007), and fowlpox virus FPV039 (Banadyga et al. 2007), plus the obvious homologs of these proteins encoded by related poxviruses (Fig. 4). Similarly, obvious BCL-2 homologs with antiapoptotic activity are found in essentially all γ herpesviruses (Henderson et al. 1993; Cheng et al. 1997b; Nava et al. 1997). Furthermore, one or both of the BCL-2 homologs of γ herpesvirus Epstein–Barr virus (BHRF1 and BALF1) is required for this virus to immortalize B cells and inhibit cell death and perhaps other functions (Altmann and Hammerschmidt 2005; Seto et al. 2010), although BALF1 lacks obvious antiapoptotic activity (Bellows et al. 2002). It has long been assumed that the antiapoptotic activity of viral BCL-2 homologs of γ herpesviruses contribute importantly to the B-cell lymphomas and many other cancers associated with these viruses. It was this transformation-promoting function of adenovirus E1B-19K that first revealed the importance of anti-death activity in E1A-induced transformation as well as mammalian virus pathogenesis (Rao et al. 1992). However, adenoviruses are not associated with any known human cancers, implying that E1B-19K serves to combat virus-induced apoptosis that is activated as a host defense-response (Degenhardt et al. 2000). The mechanisms by which viral BCL-2-like proteins inhibit BAX/BAK-dependent cell death is best characterized for E1B-19K (Perez and White 2000; Cuconati and White 2002; Shimazu et al. 2007).
Figure 4.

Viral BCL-2 family proteins with and without significant amino acid sequence similarity. BCL-2 family assignment of viral proteins are based on three-dimensional structure determinations or predicted structures. These proteins are found in the three large DNA virus families of mammals as indicated, although some lack detectable activities related to apoptosis (yellow boxes). See Figure 1 legend for BH motif key.
Functional analyses and three-dimensional structure determination of a number of poxvirus proteins has revealed an unexpected plethora of diverse BCL-2-like proteins. Some of these proteins show antiapoptotic activity, but contain limited sequence similarity to the BH motifs that define the cellular BCL-2 family (Fig. 4; FPV039, ORFV125) (Taylor et al. 2006; Banadyga et al. 2009), whereas others have no amino acid sequence similarity to other known BCL-2 proteins, yet adopt a BCL-2-like helical structure (Fig. 4; e.g., F1L, N1L) (Taylor et al. 2006).
Three-dimensional structure determination has uncovered many other BCL-2-like proteins encoded by vaccinia virus, and thereby inferred for other related Orthopox viruses. In addition to the characterized antiapoptotic F1L and N1L proteins, there are seven additional genes in the common laboratory strain Western Reserve and 12 genes in the related Copenhagen vaccine strain (Fig. 4) that also appear to adopt a BCL-2-like fold despite the lack of any obvious sequence similarity to cellular BCL-2 proteins (Graham et al. 2008; Kalverda et al. 2009). Three-dimensional structures have been solved for a subset of these proteins referred to as the A46 subfamily (A46, A52, B14, and K7). Unlike F1L and N1L, the A46 protein family lacks detectable antiapoptotic function despite obvious BCL-2-like structural similarity. Rather, functions assigned to these factors include inhibition of TLR signaling and suppression of host immune responses (Gonzalez and Esteban 2010). Consistent with the lack of antiapoptotic activity, the three-dimensional structures of A52, B14, and K7 reveal that the BH3-binding hydrophobic groove common to antiapoptotic viral and cellular BCL-2-like proteins is occluded and unable to bind BH3-peptides. In contrast, the antiapoptotic F1L and N1L vaccinia virus proteins (and M11L of myxoma virus) have an open groove and bind with high affinity to BH3 peptides of proapoptotic proteins (Aoyagi et al. 2007; Douglas et al. 2007; Kvansakul et al. 2008). The remaining 4–5 putative BCL-2-like proteins of vaccinia viruses are more distantly related to the A46 subgroup, but sequence analysis and secondary structure predictions suggest that several members of the C6 and C16/B22 groups, as well as N2 and C1 proteins also have an all-α helical structure compatible with the 3D structural fold of BCL-2 despite lacking sequence homology to other BCL-2 proteins. Similarly, they are suggested to function in innate immunity (Gonzalez and Esteban 2010). Unlike antiapoptotic F1L, M11L, and FPV039, which are single copy genes, multiple A46-related genes are present in a single genome, raising the possibility that many more yet unidentified BCL-2-like proteins may be present in other viral and cellular genomes.
AUTOPHAGY REGULATION BY VIRAL AND CELLULAR BCL-2 PROTEINS
Recent studies indicate that in addition to their apoptotic roles, members of BCL-2 family proteins also regulate autophagy (Levine et al. 2008, 2011; He and Levine 2010; Kang et al. 2011). Antiapoptotic BCL-2 proteins including BCL-2 (Pattingre et al. 2005), BCL-xL (Maiuri et al. 2007b), BCL-w (Erlich et al. 2007), and MCL-1 (Germain and Slack 2011; Maliket al. 2011) have been shown to interact with the autophagy regulator Beclin 1, the homolog of yeast Atg6, and to inhibit autophagy. Inhibition of autophagy and binding to Beclin 1 has also been reported for herpesvirus BCL-2 homologs, including KsBCL-2 (Pattingre et al. 2005), and M11, the latter binding Beclin 1 with higher affinity and being a stronger suppressor of autophagy than cellular BCL-2 (Ku et al. 2008). Interestingly, suppression of autophagy but not apoptosis by M11 is required for chronic infection by γHerpesvirus 68 as a virus encoding a mutant M11 that cannot bind Beclin 1 but still binds proapoptotic BCL-2 proteins is impaired in establishing chronic infection (E et al. 2009). Structural and mutational studies show that Beclin 1 contains a functional BH3 motif that mediates binding to antiapoptotic proteins (Maiuri et al. 2007b; Oberstein et al. 2007). This is supported by structures of the Beclin 1 BH3 motif inserted into the hydrophobic groove on BCL-xL in a manner similar to other BH3 domains of proapoptotic proteins (Maiuri et al. 2007b; Oberstein et al. 2007; Sinha et al. 2008). This finding raises the possibility that autophagy could also be modulated by other BH3-containing proteins as well. Indeed BH3-only proteins including BAD and BIK have been reported to disrupt BCL-xL-Beclin and BCL-2-Beclin interaction, respectively, to induce autophagy (Maiuri et al. 2007a; Chang et al. 2010). The role of BH3-only proteins in autophagy regulation appears to be evolutionarily conserved as a gain-of-function mutation of EGL-1 increases autophagy whereas EGL-1 deletion results in impaired autophagy in the nematode C. elegans (Maiuri et al. 2007b). Similarly, the small molecule BH3-mimetic ABT-737 competitively disrupts the interaction between BCL-2/xL and Beclin-1 to stimulate autophagy (Maiuri et al. 2007a). However, the involvement of multidomain proapoptotic BAX and BAK in autophagy is unclear. Although cells deficient in both BAX and BAK have an increased rate of autophagy suggesting that BAX/BAK directly or indirectly inhibit autophagy (Moretti et al. 2007), other studies have found that BAX expression induces mitochondrial autophagy (Yee et al. 2009). Perhaps this is not surprising given that endogenous BAX promotes mitochondrial fusion in healthy cells, which could be expected to slow mitophagy, but overexpressed BAX induces mitochondrial fission in apoptotic cells, fitting with evidence that fission serves to produce small mitochondria destined for destruction (Twig et al. 2008).
BCL-2 and other family members have been reported to reside on ER membranes and to be involved in calcium homeostasis at this site. Evidence suggests that only ER-localized BCL-2, and not mitochondrial BCL-2, negatively regulates Beclin 1-dependent autophagy at an early step by disrupting Beclin 1 interaction with Vps34 (Pattingre et al. 2005). Consistent with this model, the ER-localized protein NAF-1 (nutrient-deprivation autophagy factor-1) enhances the interaction of BCL-2 with Beclin 1 to suppress autophagy, and further promotes this cell-survival process by binding and inhibiting BH3-only protein BIK (Chang et al. 2010). In apparent contrast with the above findings, the mitochondrial pool of BCL-2 is also able to inhibit autophagy by interacting with and sequestering the Beclin 1-binding, positive regulator of autophagy, AMBRA1 (Strappazzon et al. 2011). These are early days in the quest for delineating the role of BCL-2 proteins in autophagy, further hampered by the lack of biochemical details. In addition, none of these proposed mechanisms are mutually exclusive.
CONCLUDING REMARKS
Considerable evidence now indicates that pro-and antiapoptotic BCL-2 family proteins have functions other than regulating apoptosis and that these functions are important for normal physiology of healthy cells. These noncanonical functions of BCL-2 family proteins are unlikely to be fully explained by classical apoptosis regulatory activities in which anti-death BCL-2 proteins directly bind and inhibit proapoptotic BCL-2 family proteins to control the release of cytochrome c from mitochondria in the intrinsic apoptosis pathway. The identification of multiple BCL-2-shaped proteins encoded by viruses, which either regulate apoptosis or have nonapoptotic functions, raises interesting questions about the underlying biochemical function inherent to BCL-2-shaped proteins, including those bacterial toxins that also share structural similarity to BCL-2 family proteins (e.g., Diphtheria toxin), but lack significant amino acid sequence similarity. Despite remarkable progress, currently there is no known biochemical function that explains the three-dimensional protein structure shared by BCL-2 family members. Thus, both anti- and pro-apoptotic BCL-2 proteins potentially harbor a common underlying biochemical function not yet discovered. Compelling evidence further indicates that the characteristic shape of BCL-2 proteins in solution will differ greatly from their membrane-inserted structures, but the details of these alternative structures remain elusive. Perhaps resolution of these novel structures will help answer another major unanswered question in the field about the relative importance of noncanonical versus the apoptosis functions of BCL-2 proteins in determining cell fate.
References
- Abdelwahid E, Rolland S, Teng X, Conradt B, Hardwick JM, White K. Mitochondrial involvement in cell death of non-mammalian eukaryotes. Biochim Biophys Acta. 2010;1813:597–607. doi: 10.1016/j.bbamcr.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1F0 ATP synthase. Nat Cell Biol. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann M, Hammerschmidt W. Epstein–Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005;3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi M, Zhai D, Jin C, Aleshin AE, Stec B, Reed JC, Liddington RC. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 2007;16:118–124. doi: 10.1110/ps.062454707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffy G, Miyashita T, Williamson JR, Reed JC. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hematopoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem. 1993;268:6511–6519. [PubMed] [Google Scholar]
- Banadyga L, Gerig J, Stewart T, Barry M. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the pro-apoptotic protein Bak. J Virol. 2007;81:11032–11045. doi: 10.1128/JVI.00734-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banadyga L, Veugelers K, Campbell S, Barry M. The fowlpox virus BCL-2 homologue, FPV039, interacts with activated Bax and a discrete subset of BH3-only proteins to inhibit apoptosis. J Virol. 2009;83:7085–7098. doi: 10.1128/JVI.00437-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basanez G, Hardwick JM. Unravelling the bcl-2 apoptosis code with a simple model system. PLoS Biol. 2008;6:e154. doi: 10.1371/journal.pbio.0060154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004;23:1207–1216. doi: 10.1038/sj.emboj.7600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows DS, Chau BN, Lee P, Lazebnik Y, Burns WH, Hardwick JM. Antiapoptotic herpesvirus Bcl-2 homologs escape caspase-mediated conversion to proapoptotic proteins. J Virol. 2000;74:5024–5031. doi: 10.1128/jvi.74.11.5024-5031.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM. Epstein–Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J Virol. 2002;76:2469–2479. doi: 10.1128/jvi.76.5.2469-2479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender CE, Fitzgerald P, Tait SW, Llambi F, McStay GP, Tupper DO, Pellettieri J, Alvarado AS, Salvesen GS, Green DR. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci. 2012;109:4904–4909. doi: 10.1073/pnas.1120680109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SB, Pineda FJ, Hardwick JM. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008;15:1147–1152. doi: 10.1038/cdd.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, et al. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-xL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaineau SV, Aouacheria A. BCL2DB: Moving “helix-bundled” BCL-2 family members to their database. Apoptosis. 2009;14:923–925. doi: 10.1007/s10495-009-0376-0. [DOI] [PubMed] [Google Scholar]
- Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G. Viral proteins targeting mitochondria: Controlling cell death. Biochim Biophys Acta. 2004;1659:178–189. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Kang BH, Xue D. Bcl-2 Proteins EGL-1 and CED-9 do not regulate mitochondrial fission or fusion in Caenorhabditis elegans. Curr Biol. 2009;19:768–773. doi: 10.1016/j.cub.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, Rizzuto R. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem. 2004;279:54581–54589. doi: 10.1074/jbc.M409663200. [DOI] [PubMed] [Google Scholar]
- Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010;29:606–618. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau BN, Cheng EH, Kerr DA, Hardwick JM. Aven, a novel inhibitor of caspase activation, binds Bcl-xL and Apaf-1. Mol Cell. 2000;6:31–40. [PubMed] [Google Scholar]
- Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011;195:263–276. doi: 10.1083/jcb.201108059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Levine B, Boise LH, Thompson CB, Hard-wick JM. Bax-independent inhibition of apoptosis by Bcl-xL. Nature. 1996;379:554–556. doi: 10.1038/379554a0. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science. 1997a;278:1966–1968. doi: 10.1126/science.278.5345.1966. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS, Hardwick JM. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci. 1997b;94:690–694. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-xL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- Cheng WC, Berman SB, Ivanovska I, Jonas EA, Lee SJ, Chen Y, Kaczmarek LK, Pineda F, Hardwick JM. Mitochondrial factors with dual roles in death and survival. Oncogene. 2006;25:4697–4705. doi: 10.1038/sj.onc.1209596. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. doi: 10.1016/s0092-8674(00)80572-3. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Cheng EH, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, et al. Modulation of cell death by Bcl-xL through caspase interaction. Proc Natl Acad Sci. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli F, Salomoni P, Cotteret S, Cesi V, Srinivasula SM, Alnemri ES, Calabretta B. Caspase cleavage enhances the apoptosis-inducing effects of BAD. Mol Cell Biol. 2001;21:3025–3036. doi: 10.1128/MCB.21.9.3025-3036.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooray S, Bahar MW, Abrescia NG, McVey CE, Bartlett NW, Chen RA, Stuart DI, Grimes JM, Smith GL. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J Gen Virol. 2007;88:1656–1666. doi: 10.1099/vir.0.82772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr Biol. 1997;7:913–920. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- Cuconati A, White E. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes Dev. 2002;16:2465–2478. doi: 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJ, Datta SR, Pitter KL, Bird GH, Wikstrom JD, et al. Dual role of proapoptotic BAD in insulin secretion and β cell survival. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gimenez-Cassina A, Tondera D. Homeostatic functions of BCL-2 proteins beyond apoptosis. Adv Exp Med Biol. 2010;687:1–32. doi: 10.1007/978-1-4419-6706-0_1. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Perez D, White E. Pathways used by adenovirus E1B 19K to inhibit apoptosis. Symp Soc Exp Biol. 2000;52:241–251. [PubMed] [Google Scholar]
- Delivani P, Adrain C, Taylor RC, Duriez PJ, Martin SJ. Role for CED-9 and Egl-1 as regulators of mitochondrial fission and fusion dynamics. Mol Cell. 2006;21:761–773. doi: 10.1016/j.molcel.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Diwan A, Koesters AG, Capella D, Geiger H, Kalfa TA, Dorn GW., II Targeting erythroblast-specific apoptosis in experimental anemia. Apoptosis. 2008;13:1022–1030. doi: 10.1007/s10495-008-0236-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AE, Corbett KD, Berger JM, McFadden G, Handel TM. Structure of M11L: A myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. 2007;16:695–703. doi: 10.1110/ps.062720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dremina ES, Sharov VS, Kumar K, Zaidi A, Michaelis EK, Schoneich C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) Biochem J. 2004;383:361–370. doi: 10.1042/BJ20040187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EX, Hwang S, Oh S, Lee JS, Jeong JH, Gwack Y, Kowalik TF, Sun R, Jung JU, Liang C. Viral Bcl-2-mediated evasion of autophagy aids chronic infection of γherpesvirus 68. PLoS Pathog. 2009;5:e1000609. doi: 10.1371/journal.ppat.1000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010;285:13678–13684. doi: 10.1074/jbc.M109.096040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliseev RA, Malecki J, Lester T, Zhang Y, Humphrey J, Gunter TE. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J Biol Chem. 2009;284:9692–9699. doi: 10.1074/jbc.M808750200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007;3:561–568. doi: 10.4161/auto.4713. [DOI] [PubMed] [Google Scholar]
- Fannjiang Y, Kim CH, Huganir RL, Zou S, Lindsten T, Thompson CB, Mito T, Traystman RJ, Larsen T, Griffin DE, et al. BAK alters neuronal excitability and can switch from anti- to pro-death function during postnatal development. Dev Cell. 2003;4:575–585. doi: 10.1016/s1534-5807(03)00091-1. [DOI] [PubMed] [Google Scholar]
- Frank S. Dysregulation of mitochondrial fusion and fission: An emerging concept in neurodegeneration. Acta Neuropathol. 2006;111:93–100. doi: 10.1007/s00401-005-0002-3. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Galindo KA, Lu WJ, Park JH, Abrams JM. The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development (Cambridge, England) 2009;136:275–283. doi: 10.1242/dev.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011;29:909–916. doi: 10.1200/JCO.2010.31.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia I, Crowther AJ, Gama V, Ryan Miller C, Deshmukh M, Gershon TR. Bax deficiency prolongs cerebellar neurogenesis, accelerates medulloblastoma formation and paradoxically increases both malignancy and differentiation. Oncogene. 2012 doi: 10.1038/onc.2012.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M, Slack RS. MCL-1 regulates the balance between autophagy and apoptosis. Autophagy. 2011;7:549–551. doi: 10.4161/auto.7.5.15098. [DOI] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Martinez-Francois JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012;74:719–730. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Heterogeneous requirement of NGF for sympathetic target innervation in vivo. J Neurosci. 2004;24:743–751. doi: 10.1523/JNEUROSCI.4523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocnikova H, Russ G. Influenza a virus PB1-F2 protein. Acta Virol. 2007;51:101–108. [PubMed] [Google Scholar]
- Gonzalez JM, Esteban M. A poxvirus Bcl-2-like gene family involved in regulation of host immune response: Sequence similarity and evolutionary history. Virol J. 2010;7:59. doi: 10.1186/1743-422X-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Estevez C, Salo E. Autophagy and apoptosis in planarians. Apoptosis. 2010;15:279–292. doi: 10.1007/s10495-009-0445-4. [DOI] [PubMed] [Google Scholar]
- Gotow T, Shibata M, Kanamori S, Tokuno O, Ohsawa Y, Sato N, Isahara K, Yayoi Y, Watanabe T, Leterrier JF, et al. Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell Death Differ. 2000;7:666–674. doi: 10.1038/sj.cdd.4400694. [DOI] [PubMed] [Google Scholar]
- Graham SC, Bahar MW, Cooray S, Chen RA, Whalen DM, Abrescia NG, Alderton D, Owens RJ, Stuart DI, Smith GL, et al. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog. 2008;4:e1000128. doi: 10.1371/journal.ppat.1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-xL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274:1156–1163. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- Guo JY, Yamada A, Kajino T, Wu JQ, Tang W, Freel CD, Feng J, Chau BN, Wang MZ, Margolis SS, et al. Aven-dependent activation of ATM following DNA damage. Curr Biol. 2008;18:933–942. doi: 10.1016/j.cub.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick JM, Chen YB, Jonas EA. Multipolar functions of BCL-2 proteins link energetics to apoptosis. Trends Cell Biol. 2012;22:318–328. doi: 10.1016/j.tcb.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson S, Huen D, Rowe M, Dawson C, Johnson G, Rickinson A. Epstein–Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci. 1993;90:8479–8483. doi: 10.1073/pnas.90.18.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature. 1994a;369:318–320. doi: 10.1038/369318a0. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994b;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA. Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J Neurophysiol. 2008a;99:1515–1522. doi: 10.1152/jn.00598.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman JA, Hardwick JM, Kaczmarek LK, Jonas EA. Bcl-xL inhibitor ABT-737 reveals a dual role for Bcl-xL in synaptic transmission. J Neurophysiol. 2008b;99:1515–1522. doi: 10.1152/jn.00598.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds MG, Smits C, Fredericks-Short R, Risk JM, Bailey M, Huang DC, Day CL. Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2 protein is topographically restricted in tissues characterized by apoptotic cell death. Proc Natl Acad Sci. 1991;88:6961–6965. doi: 10.1073/pnas.88.16.6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Edlich F, Cleland MM, Banerjee S, Mc-Caffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41:150–160. doi: 10.1016/j.molcel.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR, Sternberg PW, Greenwald IS, Fixsen W, Ellis HM. Mutations that affect neural cell lineages and cell fates during the development of the nematode Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol. 1983;48:453–463. doi: 10.1101/sqb.1983.048.01.050. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Huang CR, Yang-Yen HF. The fast-mobility isoform of mouse Mcl-1 is a mitochondrial matrix-localized protein with attenuated anti-apoptotic activity. FEBS Lett. 2010;584:3323–3330. doi: 10.1016/j.febslet.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Irusta PM, Chen YB, Hardwick JM. Viral modulators of cell death provide new links to old pathways. Curr Opin Cell Biol. 2003;15:700–705. doi: 10.1016/j.ceb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Jagasia R, Grote P, Westermann B, Conradt B. DRP-1-mediated mitochondrial fragmentation during EGL-1-induced cell death in C. elegans. Nature. 2005;433:754–760. doi: 10.1038/nature03316. [DOI] [PubMed] [Google Scholar]
- Jiao S, Li Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron. 2011;70:758–772. doi: 10.1016/j.neuron.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hoit D, Hickman JA, Brandt TA, Polster BM, Fannjiang Y, McCarthy E, Montanez MK, Hardwick JM, Kaczmarek LK. Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J Neurosci. 2003;23:8423–8431. doi: 10.1523/JNEUROSCI.23-23-08423.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Chachar M, Polster BM, Brandt TA, Fannjiang Y, Ivanovska I, Basanez G, Kinnally KW, Zimmerberg J, et al. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc Natl Acad Sci. 2004;101:13590–13595. doi: 10.1073/pnas.0401372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas EA, Hardwick JM, Kaczmarek LK. Actions of BAX on mitochondrial channel activity and on synaptic transmission. Antioxid Redox Signal. 2005a;7:1092–1100. doi: 10.1089/ars.2005.7.1092. [DOI] [PubMed] [Google Scholar]
- Jonas EA, Hickman JA, Hardwick JM, Kaczmarek LK. Exposure to hypoxia rapidly induces mitochondrial channel activity within a living synapse. J Biol Chem. 2005b;280:4491–4497. doi: 10.1074/jbc.M410661200. [DOI] [PubMed] [Google Scholar]
- Jones RG, Bui T, White C, Madesh M, Krawczyk CM, Lindsten T, Hawkins BJ, Kubek S, Frauwirth KA, Wang YL, et al. The proapoptotic factors Bax and Bak regulate T cell proliferation through control of endoplasmic reticulum Ca2+ homeostasis. Immunity. 2007;27:268–280. doi: 10.1016/j.immuni.2007.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalverda AP, Thompson GS, Vogel A, Schroder M, Bowie AG, Khan AR, Homans SW. Poxvirus K7 protein adopts a Bcl-2 fold: Biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J Mol Biol. 2009;385:843–853. doi: 10.1016/j.jmb.2008.09.048. [DOI] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Chang BS, Harlan JE, Fesik SW, Thompson CB. Bad is a BH3 domain-containing protein that forms an inactivating dimer with Bcl-xL. Mol Cell Biol. 1997;17:7040–7046. doi: 10.1128/mcb.17.12.7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CN, Wang X, Huang Y, Ibrado AM, Liu L, Fang G, Bhalla K. Overexpression of Bcl-xL inhibits Ara- C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res. 1997;57:3115–3120. [PubMed] [Google Scholar]
- Kim BC, Mamura M, Choi KS, Calabretta B, Kim SJ. Transforming growth factor β1 induces apoptosis through cleavage of BAD in a Smad3-dependent mechanism in FaO hepatoma cells. Mol Cell Biol. 2002;22:1369–1378. doi: 10.1128/mcb.22.5.1369-1378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck RM, Esposti MD, Perkins G, Renken C, Kuwana T, Bossy-Wetzel E, Goldberg M, Allen T, Barber MJ, Green DR, et al. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/Bax: A rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol. 1993;4:327–332. [PubMed] [Google Scholar]
- Ku B, Woo JS, Liang C, Lee KH, Hong HSEX, Kim KS, Jung JU, Oh BH. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine γ-herpesvirus 68. PLoS Pathog. 2008;4:e25. doi: 10.1371/journal.ppat.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA, Huang DC, Colman PM. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 2008;15:1564–1571. doi: 10.1038/cdd.2008.83. [DOI] [PubMed] [Google Scholar]
- Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- Landes T, Martinou JC. Mitochondrial outer membrane permeabilization during apoptosis: The role of mitochondrial fission. Biochim Biophys Acta. 2011;1813:540–545. doi: 10.1016/j.bbamcr.2011.01.021. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Angeletti PU. Nerve growth factor. Phys Rev. 1968;48:534–569. doi: 10.1152/physrev.1968.48.3.534. [DOI] [PubMed] [Google Scholar]
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, Korsmeyer SJ, Zou S, Hardwick JM. Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med. 1999;5:832–835. doi: 10.1038/10556. [DOI] [PubMed] [Google Scholar]
- Li Z, Sheng M. Caspases in synaptic plasticity. Mol Brain. 2012;5:15. doi: 10.1186/1756-6606-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, et al. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci. 2008;105:2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Vaithiyalingam S, Shi Q, Chazin WJ, Zinkel SS. BID binds to replication protein A and stimulates ATR function following replicative stress. Mol Cell Biol. 2011;31:4298–4309. doi: 10.1128/MCB.05737-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B, Andrews DW. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell. 2008;135:1074–1084. doi: 10.1016/j.cell.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Lowman XH, McDonnell MA, Kosloske A, Odumade OA, Jenness C, Karim CB, Jemmerson R, Kelekar A. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol Cell. 2010;40:823–833. doi: 10.1016/j.molcel.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Luo X, He Q, Huang Y, Sheikh MS. Transcriptional upregulation of PUMA modulates endoplasmic reticulum calcium pool depletion-induced apoptosis via Bax activation. Cell Death Differ. 2005;12:1310–1318. doi: 10.1038/sj.cdd.4401659. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-xL. Autophagy. 2007a;3:374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-xL and a BH3-like domain in Beclin-1. EMBO J. 2007b;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik SA, Shen S, Marino G, Benyounes A, Maiuri MC, Kroemer G. BH3 mimetics reveal the network properties of autophagy-regulatory signaling cascades. Autophagy. 2011;7 doi: 10.4161/auto.7.8.15785. [DOI] [PubMed] [Google Scholar]
- Marty A, Llano I. Excitatory effects of GABA in established brain networks. Trend Neurosci. 2005;28:284–289. doi: 10.1016/j.tins.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Shore GC. BH3-only BIK regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–23836. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ, Cowburn D. Solution structure of the proapoptotic molecule BID: A structural basis for apoptotic agonists and antagonists. Cell. 1999;96:625–634. doi: 10.1016/s0092-8674(00)80573-5. [DOI] [PubMed] [Google Scholar]
- Meeusen SL, Nunnari J. Mitochondrial fusion in vitro. Methods Mol Biol. 2007;372:461–466. doi: 10.1007/978-1-59745-365-3_32. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Velez P, Schendel SL, Liang H, Muchmore SW, Fesik SW, Fill M, Thompson CB. Bcl-xL forms an ion channel in synthetic lipid membranes. Nature. 1997;385:353–357. doi: 10.1038/385353a0. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Kettlun CS, Liang H, Kelekar A, Vander Heiden MG, Chang BS, Fesik SW, Fill M, Thompson CB. Bcl-xL regulates apoptosis by heterodimerization- dependent and -independent mechanisms. EMBO J. 1999;18:632–643. doi: 10.1093/emboj/18.3.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P, et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell. 2010;142:889–901. doi: 10.1016/j.cell.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti L, Attia A, Kim KW, Lu B. Crosstalk between Bak/Bax and mTOR signaling regulates radiation-induced autophagy. Autophagy. 2007;3:142–144. doi: 10.4161/auto.3607. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Nava VE, Cheng EH, Veliuona M, Zou S, Clem RJ, Mayer ML, Hardwick JM. Herpesvirus saimiri encodes a functional homolog of the human bcl-2 oncogene. J Virol. 1997;71:4118–4122. doi: 10.1128/jvi.71.5.4118-4122.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez G, Seto M, Seremetis S, Ferrero D, Grignani F, Korsmeyer SJ, Dalla-Favera R. Growth- and tumor-promoting effects of deregulated BCL2 in human B-lymphoblastoid cells. Proc Natl Acad Sci. 1989;86:4589–4593. doi: 10.1073/pnas.86.12.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SA, Lin SS, Bassik MC. The control of endoplasmic reticulum-initiated apoptosis by the BCL-2 family of proteins. Curr Mol Med. 2006;6:99–109. doi: 10.2174/156652406775574587. [DOI] [PubMed] [Google Scholar]
- Oberst A, Bender C, Green DR. Living with death: The evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 2008;15:1139–1146. doi: 10.1038/cdd.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-xL-Beclin 1 peptide complex: Beclin 1 is a novel BH3- only protein. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Chen YB, Miyawaki T, Li H, Sacchetti S, Flannery RJ, Alavian KN, Pontarelli F, Roelofs BA, Hickman JA, et al. N-terminally cleaved Bcl-xL mediates ischemia-induced neuronal death. Nat Neurosci. 2012;15:574–580. doi: 10.1038/nn.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 hetero-dimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parone PA, James DI, Da Cruz S, Mattenberger Y, Donze O, Barja F, Martinou JC. Inhibiting the mitochondrial fission machinery does not prevent Bax/Bak-dependent apoptosis. Mol Cell Biol. 2006;26:7397–7408. doi: 10.1128/MCB.02282-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez D, White E. TNF-α signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19K. Mol Cell. 2000;6:53–63. [PubMed] [Google Scholar]
- Peter ME. Programmed cell death: Apoptosis meets necrosis. Nature. 2011;471:310–312. doi: 10.1038/471310a. [DOI] [PubMed] [Google Scholar]
- Peterson JS, Bass BP, Jue D, Rodriguez A, Abrams JM, McCall K. Noncanonical cell death pathways act during Drosophila oogenesis. Genesis. 2007;45:396–404. doi: 10.1002/dvg.20306. [DOI] [PubMed] [Google Scholar]
- Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB, et al. Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 2000;9:2528–2534. doi: 10.1110/ps.9.12.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton P, Rizzuto R. Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ. 2006;13:1409–1418. doi: 10.1038/sj.cdd.4401960. [DOI] [PubMed] [Google Scholar]
- Popgeorgiev N, Bonneau B, Ferri KF, Prudent J, Thibaut J, Gillet G. The apoptotic regulator Nrz controls cytoskeletal dynamics via the regulation of Ca2+ trafficking in the zebrafish blastula. Dev Cell. 2011;20:663–676. doi: 10.1016/j.devcel.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Villunger A, O’Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A. Bmf: A proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–1832. doi: 10.1126/science.1062257. [DOI] [PubMed] [Google Scholar]
- Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc Natl Acad Sci. 1992;89:7742–7746. doi: 10.1073/pnas.89.16.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. BID, BIM, and PUMA are essential for activation of the BAX-and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs BA, Hardwick JM. Flying to a halt: Drosophila Aven arrests the cell cycle. Cell cycle. 2011;10:1351–1352. doi: 10.4161/cc.10.9.15436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland SG, Lu Y, David CN, Conradt B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J Cell Biol. 2009;186:525–540. doi: 10.1083/jcb.200905070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell. 2008;31:255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong YP, Barr P, Yee VC, Distelhorst CW. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim Biophys Acta. 2009;1793:971–978. doi: 10.1016/j.bbamcr.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- Roy SS, Madesh M, Davies E, Antonsson B, Danial N, Hajnoczky G. Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+-dependent cell survival and death. Mol Cell. 2009;33:377–388. doi: 10.1016/j.molcel.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samara NL, Datta AB, Berndsen CE, Zhang X, Yao T, Cohen RE, Wolberger C. Structural insights into the assembly and function of the SAGA deubiquitinating module. Science. 2010;328:1025–1029. doi: 10.1126/science.1190049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- Seo SY, Chen YB, Ivanovska I, Ranger AM, Hong SJ, Dawson VL, Korsmeyer SJ, Bellows DS, Fannjiang Y, Hardwick JM. BAD is a pro-survival factor prior to activation of its pro-apoptotic function. J Biol Chem. 2004;279:42240–42249. doi: 10.1074/jbc.M406775200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. Micro RNAs of Epstein–Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophysica Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trend Cell Biol. 2002;12:178–184. doi: 10.1016/s0962-8924(01)02246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Degenhardt K, Nur EKA, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M. NBK/BIK antagonizes MCL-1 and BCL-xL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev. 2007;21:929–941. doi: 10.1101/gad.1522007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Colbert CL, Becker N, Wei Y, Levine B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the γ-herpesvirus 68 Bcl-2 homolog M11. Autophagy. 2008;4:989–997. doi: 10.4161/auto.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AM-BRA1-induced autophagy. EMBO J. 2011;30:1195–1208. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A. The role of BH3-only proteins in the immune system. Nat Rev. 2005;5:189–200. doi: 10.1038/nri1568. [DOI] [PubMed] [Google Scholar]
- Strasser A, Puthalakath H, Bouillet P, Huang DC, O’Connor L, O’Reilly LA, Cullen L, Cory S, Adams JM. The role of bim, a proapoptotic BH3-only member of the Bcl-2 family in cell-death control. Ann NY Acad Sci. 2000;917:541–548. doi: 10.1111/j.1749-6632.2000.tb05419.x. [DOI] [PubMed] [Google Scholar]
- Su J, Wang G, Barrett JW, Irvine TS, Gao X, McFadden G. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J Virol. 2006;80:1140–1151. doi: 10.1128/JVI.80.3.1140-1151.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Winseck A, Vinsant S, Park OH, Kim H, Oppenheim RW. Programmed cell death of adult-generated hippocampal neurons is mediated by the pro-apoptotic gene Bax. J Neurosci. 2004;24:11205–11213. doi: 10.1523/JNEUROSCI.1436-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan FJ, Husain M, Manlandro CM, Koppenol M, Fire AZ, Hill RB. CED-9 and mitochondrial homeostasis in C. elegans muscle. J Cell Sci. 2008;121:3373–3382. doi: 10.1242/jcs.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner EA, Blute TA, Brachmann CB, McCall K. Bcl-2 proteins and autophagy regulate mitochondrial dynamics during programmed cell death in the Drosophila ovary. Development. 2011;138:327–338. doi: 10.1242/dev.057943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Quilty D, Banadyga L, Barry M. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J Biol Chem. 2006;281:39728–39739. doi: 10.1074/jbc.M607465200. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y. Overexpression of the human BCL-2 gene product results in growth enhancement of Ep-stein–Barr virus-immortalized B cells. Proc Natl Acad Sci. 1989;86:1958–1962. doi: 10.1073/pnas.86.6.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228:1440–1443. doi: 10.1126/science.3874430. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Abeele F, Skryma R, Shuba Y, Van Coppenolle F, Slomianny C, Roudbaraki M, Mauroy B, Wuytack F, Prevarskaya N. Bcl-2-dependent modulation of Ca2+ homeostasis and store-operated channels in prostate cancer cells. Cancer Cell. 2002;1:169–179. doi: 10.1016/s1535-6108(02)00034-x. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 Opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Vento MT, Zazzu V, Loffreda A, Cross JR, Downward J, Stoppelli MP, Iaccarino I. Praf2 is a novel Bcl-xL/Bcl-2 interacting protein with the ability to modulate survival of cancer cells. PLoS ONE. 2010;5:e15636. doi: 10.1371/journal.pone.0015636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: A novel BH3 domain-only death agonist. Genes Dev. 1996;10:2859–2869. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Warr MR, Mills JR, Nguyen M, Lemaire-Ewing S, Baardsnes J, Sun KL, Malina A, Young JC, Jeyaraju DV, O’Connor-McCourt M, et al. Mitochondrion-dependent N-terminal processing of outer membrane Mcl-1 protein removes an essential Mule/Lasu1 protein-binding site. J Biol Chem. 2011;286:25098–25107. doi: 10.1074/jbc.M111.218321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilenko ST, Stewart TL, Meyers AF, Barry M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc Natl Acad Sci. 2003;100:14345–14350. doi: 10.1073/pnas.2235583100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal D, Ledgerwood EC, Hibma MH, Fleming SB, Whelan EM, Mercer AA. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J Virol. 2007;81:7178–7188. doi: 10.1128/JVI.00404-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan RS, Konstantinidis K, Wei AC, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-xL modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH. PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ. 2009;16:1135–1145. doi: 10.1038/cdd.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeretssian G, Correa RG, Doiron K, Fitzgerald P, Dillon CP, Green DR, Reed JC, Saleh M. Non-apoptotic role of BID in inflammation and innate immunity. Nature. 2011;474:96–99. doi: 10.1038/nature09982. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- Zheng TS, Hunot S, Kuida K, Momoi T, Srinivasan A, Nicholson DW, Lazebnik Y, Flavell RA. Deficiency in caspase-9 or caspase-3 induces compensatory caspase activation. Nat Med. 2000;6:1241–1247. doi: 10.1038/81343. [DOI] [PubMed] [Google Scholar]
- Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A, Korsmeyer SJ. A role for proapoptotic BID in the DNA-damage response. Cell. 2005;122:579–591. doi: 10.1016/j.cell.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Zou S, Chang J, LaFever L, Tang W, Johnson EL, Hu J, Wilk R, Krause HM, Drummond-Barbosa D, Irusta PM. Identification of dAven, a Drosophila melanogaster ortholog of the cell cycle regulator Aven. Cell Cycle. 2011;10:989–998. doi: 10.4161/cc.10.6.15080. [DOI] [PMC free article] [PubMed] [Google Scholar]