

Abstract
Sixteen FDA-approved drugs were investigated to elucidate their mechanisms of action (MOAs) and clinical functions by pathway analysis based on retrieved drug targets interacting with or affected by the investigated drugs. Protein and gene targets and associated pathways were obtained by data-mining of public databases including the MMDB, PubChem BioAssay, GEO DataSets, and the BioSystems databases. Entrez E-Utilities were applied and in-house Ruby scripts were developed for data retrieval and pathway analysis to identify and evaluate relevant pathways common to the retrieved drug targets. Pathways pertinent to clinical uses or MOAs were obtained for most drugs. Interestingly, some drugs identified pathways responsible for other diseases than their current therapeutic uses, and these pathways were verified retrospectively by in vitro, in vivo tests or clinical trials. The pathway enrichment analysis based on drug target information from public databases could provide a novel approach for elucidating drug MOAs and repositioning, therefore benefiting the discovery of new therapeutic treatments for diseases.
INTRODUCTION
Understanding the mechanisms of action (MOAs) of drugs is critical for drug development. The identification of drug MOAs has been primarily based on pharmacological experiments, but bioinformatics study by data-mining of public databases1–9 has become an alternative and important approach. Previous bioinformatics efforts have been focused on the establishment of compound-target associations based on drug similarity that could be used for the prediction of drug targets responsible for MOAs2, 3, 8, but recent studies have extended to the identification and analysis of biological pathways retrieved based on drug bioactivity and targets1, 4, 5, 9. A biological pathway involving a drug and its targets may point to the MOA, the biological function, or a disease associated with this drug. Identification of biological pathways may be helpful for the elucidation of the MOAs underlying drug effects. For instance, Covell et al explored the drug-gene-pathway relationships by the data mining of drug profiles from the NCI60 anticancer screen4, 5, 9 and even discussed MOAs indicated by the pathways with coexistence of gene expression change upon drug treatments4. However, these studies were either limited to certain data sets with gene expression data4, 5, 9 or focused on the overall relationship among drugs, their targets and their involved pathways1. The application of pathway analysis in identifying drug MOAs by integrating large-scale biomedical databases with drug target information is still lacking.
While MOA is essential for the development of new drugs, drug repurposing or repositioning, i.e., finding novel uses of existing drugs, has become an important strategy to exploit new treatments for diseases10–14. Computational methods, e.g. virtual screening of commercially-available drug databases based on small-molecule similarity and Structure-Activity Relationship (SAR) models have been employed for predicting new uses of existing drugs15–18. The most widely discussed bioinformatics approach for drug repositioning is to integrate the network among drugs, genes, and diseases, mostly by taking advantage of the high throughput gene expression data11–14, 19. For example, Dudley et al predicted the new uses of existing drugs based on drug-disease pairs identified by comparing gene expression signatures of drugs with those in Gene Expression Omnibus (GEO)12, 13, 20. Biological pathways involving drug targets may point to diseases not currently treated by a drug and therefore possibly indicate its new clinical uses. Although pathway enrichment analysis was recently applied in combination with gene expression profiles to explore new clinical uses among existing drugs21, 22, its application in drug repurposing based on large-scale biomedical database mining is still yet to explore.
The vast biological and biomedical information available at public databases have greatly facilitated the bioinformatics studies in recent years. In this work, sixteen FDA-approved drugs obtained from the DrugBank database were investigated to elucidate their MOAs and clinical functions with pathway analysis based on the drug target information retrieved from various public databases available at NCBI, including the MMDB, GEO, and PubChem Compound and BioAssay databases. Biological pathways involving drug molecules and their targets were identified by querying the NCBI’s BioSystems database. Most drugs retrieved biological pathways that were shared by their drug targets and pertinent to their clinical uses or MOAs, indicating identifying relevant pathways could aid the elucidation of the clinical functions and MOAs of drugs; some drugs have obtained pathways responsible for other diseases than those they are currently used for, indicating identifying biological pathways could provide an alternative approach for exploiting drug repurposing or repositioning. The insights into the pharmacology of individual drugs have been discussed, demonstrating how pathway analysis would facilitate the elucidation of MOAs and reveal new clinical functions of existing drugs, therefore benefiting the discovery of new therapeutic treatments for diseases.
METHODS
Overall methodology
Pathways comprising component genes and proteins may account for biological processes or diseases that could be affected by a drug interacting with its pharmacological targets. Therefore, the pharmacological or clinical effects of a drug may be elucidated by analyzing the pathways enriched by drug targets with affinities or being affected by the investigated drug. In this study, pathway enrichment analysis was applied to elucidate drug MOAs and potential repositioning based on the integration of protein and gene expression information from public databases. There are three steps of this process: 1) Drug selection. 2) Drug target retrieval: protein targets with small molecule bioactivity data and crystal structures in complex with drugs were obtained; gene targets with gene expression level affected upon drug treatments were also retrieved. 3) Pathway retrieval and enrichment analysis: pathways with co-occurrence of identified drug targets were then obtained and ranked according to the number of identified drug targets involved in the pathways. The P-values associated with each pathway were calculated. A number of in-house scripts have been developed for data retrieval and analysis.
Public databases used for data retrieval
Several public databases at NCBI were utilized for retrieving the various types of data used in this work. These databases were selected due to the comprehensive and cross-linking information among them. The MMDB23 contains resolved protein structure data from the Protein Data Bank (PDB)24, added with useful information on small molecules (i.e. ligands in resolved structures), domains, and sequence similarity. PubChem25, a suite of three Entrez26 databases, i.e., Compound, Substance and BioAssay, is a public repository for chemical structures and biological properties of small molecules and RNAi reagents. With drug bioactivity and target information obtained from multiple depositors, PubChem BioAssay27 is a useful resource for biomedical and pharmaceutical research. The GEO20, 28 of NCBI is a repository of functional genomics data generated by high-throughput gene expression and genomic hybridization experiments. As one of the GEO databases, GEO DataSets20 contains entire experiments of gene expression measurements for all involved genes in certain conditions, such as upon small molecule or drug treatment in a sample. BioSystems database29 contains genes, proteins, and small molecules that are involved in biological systems, such as biological pathways and diseases. Currently it comprises records from several external databases, including Kyoto Encyclopedia of Genes and Genomes (KEGG)30, BioCyc31, Reactome32, the Pathway Interaction database33, Wikipathways34, 35, and Gene Ontology36, which are the major resources containing biological pathway data. Therefore, the BioSystems database is an informative and integrated platform to explore combinations of genes and proteins that are related by biological systems. All the above databases belong to Entrez26, an integrated retrieval system supported by NCBI with access to 37 databases, covering a wide variety of categories including literature search, DNA and RNA, proteins, genes and expression, genomes, genetics and medicine, chemicals and bioactivities, and domains and structures. Furthermore, the whole Entrez system is comprehensively integrated and powered by the cross-links among records available at various databases26.
Drug selection and target retrieval
Drugs were selected from the DrugBank database37, 38 and initially searched among the drugs with clear MOAs. The information about targets and MOAs responsible for drugs’ clinical functions was obtained from DrugBank and Wikipedia (http://en.wikipedia.org/wiki). Two categories of target were defined in this work: the primary target that is responsible for the clinical effects of a drug and other targets (or secondary targets denoted in this paper) that have shown biological affinity or effects with a drug. The drugs were further narrowed down by the availability of protein target information in the MMDB23 and PubChem BioAssay25, 27 databases. Each drug was searched in the PubChem Compound database25 for their compound CID (PubChem chemical structure accession) number. MMDB contains resolved protein structures derived from the Protein Data Bank (PDB)24. Structures in complex with each drug were retrieved with cross-links to MMDB in the PubChem Compound database; more protein targets were obtained within PubChem BioAssay by using the CID number of each drug. Only protein targets with the taxonomy of Homo sapiens retrieved from MMDB and BioAssay remained for further study. Drugs without any target information obtained from MMDB and BioAssay were excluded from further consideration.
Drug target information was also retrieved according to affected gene expression data upon drug administration. For drugs that failed to obtain relevant pathways with just protein target information from the MMDB and PubChem BioAssay databases, additional gene targets were obtained by checking PubMed literature cross-linked with GEO DataSets20, 28 records associated with each drug.
Pathway retrieval and enrichment analysis
The Entrez programming utilities (E-Utilities)26, 39 is an Application Programming Interface (API) that is used to search, link, and download data from the databases available at NCBI (http://www.ncbi.nlm.nih.gov/books/NBK25501/). The Gene IDs associated with each drug target were obtained from the Gene40 database of NCBI. Human biological pathways involving every drug target were retrieved from the BioSystems database by using the Gene IDs via the ESearch of E-Utilities. The title of each pathway was obtained by using ESummary of E-Utilities, with the BioSystems IDs (BSIDs) of pathways as queries. The significance of retrieved pathways was evaluated with the enrichment statistics P values as calculated in a similar way to DAVID41 based on the modified Fisher’s Exact Test compared to the background with all human pathways available in the BioSystems database. In-house Ruby scripts were developed for large-scale retrieval of drug target Gene IDs and pathways by using the E-Utilities, for identification of pathways common to the retrieved drug targets, and for calculation of P values. All public databases were accessed in June-July, 2013. Extra gene targets for celecoxib were obtained by accessing PubMed in Dec, 2013.
RESULTS
Drug selection and target retrieval
Drugs were searched in the order of their DrugBank primary accession numbers (Drugbank IDs) until a certain number of drugs were reached. In detail, the drugs were initially selected among 240 drugs with DrugBank ID prior to DB00860. Antibiotics, antifungals, and other drugs targeting non-human organisms were removed. The remaining drugs were then selected based on the availability of clear primary targets and MOAs, and retrieved protein and gene targets that makes the pathway analysis feasible. At last, sixteen drugs with various targets and clinical functions were obtained for subsequent pathway analysis (Table 1). Primary targets were mainly obtained from DrugBank, meanwhile secondary targets were obtained from the MMDB, BioAssay, and GEO databases. All drug targets retrieved to search the biological pathways are shown in Supporting Information, Table S1. The drug CID numbers and links used for obtaining the protein targets available at the MMDB and BioAssay databases are shown in Supporting Information, Table S2. Seven drugs obtained crystal structures of target proteins from MMDB, some of which are the primary targets. The corresponding gene symbols and PDB IDs of these protein structure complexes are shown in Supporting Information, Table S3. The numbers of retrieved protein and gene targets via the MMDB, BioAssay and GEO databases ranged from 0 to 7, 2 to 68 and 0 to 433, respectively, which are summarized in Table 1 together with the drug therapeutic information.
Table 1.
Profiles of drugs and retrieved proteins, genes, and relevant pathways.
| id | Drug | Therapeutic category | Primary target(s) |
No. targets | ||
|---|---|---|---|---|---|---|
| MMDBa | BioAssayb | GEOc | ||||
| 1 | Actinomycin | cancer | DNA | 0 | 2 | 14 |
| 2 | Adenosine | antiarrhythmic | ADORs | 7* | 6 | NAd |
| 3 | Bortezomib | relapsed multiple myeloma Mantle cell lymphoma | protease | 0 | 11 | 28 |
| 4 | Celecoxib | anti-inflammatory | PTGS2 | 1 | 19 | 56 |
| 5 | Disulfiram | chronic alcoholism addiction | ALDHs | 0 | 59 | 0 |
| 6 | Doxorubicin | cancer | DNA | 0 | 9 | 28 |
| 7 | Fluorouracil | cancer | TYMS | 1 | 5 | 80 |
| 8 | Gefitinib | cancer | EGFR | 1* | 68 | NA |
| 9 | Mifepristone | abortifacient | PGR | 1 | 14 | 433 |
| 10 | Propofol | hypnotic | GABRs | 0 | 11 | 88 |
| 11 | Rosiglitazone | antidiabetic | PPARs | 0 | 5 | 14 |
| 12 | Tamoxifen | breast cancer | ESR1 | 0 | 57 | NA |
| 13 | Tretinoin | topical retinoid | RARs | 2* | 38 | 89 |
| 14 | Triiodothyronine | multiple functions | THRs | 3* | 11 | 275 |
| 15 | Valproic Acid | anticonvulsant, mood-stabilizing | ABAT | 0 | 3 | 196 |
| 16 | Vincristine | Cancer | tubulin | 0 | 2 | 11 |
Number of target proteins retrieved from the links to protein structures at the PubChem Compound database.
Numbers with * indicate that the crystal structure of the primary target in complex with the drug is included.
Number of total target proteins retrieved from PubChem BioAssay (excluding the targets with crystal structures).
Number of total target genes retrieved via GEO DataSets (excluding the targets with crystal structures and obtained from BioAssay).
Drug targets were not retrieved via GEO because there were relevant pathways identified based on only MMDB and PubChem BioAssay.
Pathway enrichment analysis: identified pathways with participating drug targets
Ruby scripts were developed for pathway retrieval and data processing. The scripts are attached as Supporting Information. There were usually multiple pathways retrieved for each drug target. We hypothesize that biological pathways common to the primary and secondary targets may indicate significant biological systems that are important for the therapeutic functions of drugs. In this study, all drugs have found biological pathways with coexistence of primary and secondary drug targets. The list of all these pathways for each drug is shown in Supporting Information, Table S4. The obtained pathways were ranked in terms of the number of retrieved drug targets involved in the pathways. The official full names of participating genes in the identified pathways are shown in Supporting Information, Table S5.
Most drugs have identified pathways related to the MOAs, biological functions, or clinical uses (denoted as relevant pathways in the paper) shared by the primary and secondary targets. Examples of these pathways are shown in Table 2. Four investigated drugs, i.e., adenosine, doxorubicin, gefitinib, and tamoxifen found relevant pathways to their clinical uses or MOAs common to at least four protein targets (including the primary target) retrieved from MMDB and BioAssay. For the remaining drugs, additional gene targets were obtained via GEO DataSets records with gene information indicated by PubMed. Seven drugs, i.e., actinomycin, bortezomib, celecoxib, fluorouracil, rosiglitazone, tretinoin, and vincristine, have found relevant pathways among at least four target proteins and genes. Interestingly, four drugs, i.e., gefitinib, celecoxib, tamoxifen, and tretinoin, identified biological pathways responsible for diseases other than those currently treated by the drugs, with the majority of them supported by previous in vitro, in vivo tests or clinical trials. See Supporting Information for detailed discussions of the relevant pathways identified for each drug.
Table 2.
Relevant pathways shared by multiple drug targets.
| ida | drug | Primary target(s) |
Relevant Pathways | Pathway BSID |
No. of targets | P-valuee | ||
|---|---|---|---|---|---|---|---|---|
| Mb | Bc | Gd | ||||||
| 1 | Actinomycin | DNA | Integrated Pancreatic Cancer Pathway | 711360 | NA f | 0 | 8 | 2.42e-10 |
| Apoptosis | 198797 | NA | 0 | 7 | 2.48e-11 | |||
| 105648 | NA | 0 | 5 | 5.35e-06 | ||||
| 83060 | NA | 0 | 5 | 1.14e-07 | ||||
| Integrated Breast Cancer Pathway | 219801 | NA | 0 | 7 | 9.04e-08 | |||
| Pathways in Cancer | 83105 | NA | 0 | 7 | 3.62e-07 | |||
| Acute Myeloid Leukemia | 83117 | NA | 0 | 6 | 4.36e-08 | |||
| Apoptosis Modulation and Signaling | 198822 | NA | 0 | 5 | 1.14e-07 | |||
| Prostate Cancer | 755440 | NA | 0 | 5 | 6.04e-07 | |||
| Integrated Cancer Pathway | 672450 | NA | 0 | 3 | 2.15e-05 | |||
| Pancreatic Cancer | 83108 | NA | 0 | 3 | 8.92e-03 | |||
| Small Cell Lung Cancer | 83118 | NA | 0 | 2 | 8.11e-03 | |||
| Prostate Cancer | 83111 | NA | 0 | 2 | 8.67e-03 | |||
| Endometrial Cancer | 83109 | NA | 0 | 2 | 3.04e-03 | |||
| Colorectal Cancer | 83106 | NA | 0 | 2 | 4.30e-03 | |||
| 2 | Adenosine | ADORs | Vascular Smooth Muscle Contraction | 96530 | 1* | 3 | NA | 3.77e-04 |
| 3 | Bortezomib | protease | Protein Processing in Endoplasmic Reticulum | 167325 | NA | 2 | 4 | 2.08e-06 |
| Parkin-Ubiquitin Proteasomal System pathway | 700638 | NA | 1 | 4 | 1.72e-03 | |||
| 4 | Celecoxib | PTGS2 | Pathways in Cancer | 83105 | 0 | 1 | 11 | 1.87e-05 |
| Small Cell Lung Cancer | 83118 | 0 | 1 | 8 | 1.58e-07 | |||
| Integrated Pancreatic Cancer Pathway | 711360 | 0 | 1 | 7 | 4.82e-04 | |||
| Eicosanoid Synthesis | 198888 | 0 | 2 | 2 | 4.24e-04 | |||
| Arachidonic AcidMetabolism | 82991 | 0 | 2 | 2 | 1.07e-02 | |||
| 685553 | 0 | 2 | 2 | 6.02e-03 | ||||
| 6 | Doxorubicin | DNA | Integrated Pancreatic Cancer Pathway | 711360 | NA | 4 | 14 | 6.57e-22 |
| Pathways in Cancer | 83105 | NA | 3 | 14 | 2.06e-16 | |||
| Apoptosis | 198797 | NA | 2 | 14 | 7.89e-25 | |||
| 105648 | NA | 2 | 11 | 1.15e-14 | ||||
| 83060 | NA | 2 | 11 | 1.12e-18 | ||||
| Apoptosis Modulation and Signaling | 198822 | NA | 2 | 11 | 3.45e-19 | |||
| Prostate Cancer | 755440 | NA | 1 | 11 | 1.74e-15 | |||
| Integrated Breast Cancer Pathway | 219801 | NA | 1 | 10 | 3.50e-12 | |||
| Pancreatic Cancer | 83108 | NA | 2 | 6 | 5.97e-11 | |||
| Prostate Cancer | 83111 | NA | 2 | 6 | 6.92e-10 | |||
| Small Cell Lung Cancer | 83118 | NA | 2 | 5 | 1.81e-08 | |||
| Integrated Cancer Pathway | 672450 | NA | 1 | 6 | 2.58e-11 | |||
| Endometrial Cancer | 83109 | NA | 1 | 6 | 4.92e-10 | |||
| Colorectal Cancer | 83106 | NA | 1 | 6 | 1.76e-09 | |||
| Chronic myeloid leukemia | 83116 | NA | 1 | 6 | 1.76e-09 | |||
| Acute Myeloid Leukemia | 83117 | NA | 0 | 6 | 4.36e-08 | |||
| Bladder Cancer | 83115 | NA | 1 | 4 | 2.04e-07 | |||
| Glioma | 83110 | NA | 0 | 4 | 8.00e-05 | |||
| Melanoma | 83114 | NA | 1 | 2 | 2.03e-03 | |||
| Gastric cancer network 2 | 760637 | NA | 1 | 2 | 2.33e-04 | |||
| 7 | Fluorouracil | TYMS | Cell Cycle | 530733 | 0 | 2 | 19 | 1.01e-09 |
| Cell Cycle, Mitotic | 105765 | 0 | 1 | 17 | 7.80e-09 | |||
| Integrated Pancreatic Cancer Pathway | 711360 | 0 | 2 | 16 | 2.10e-13 | |||
| Mitotic G1-G1/S Phases | 160941 | 0 | 1 | 11 | 1.96e-09 | |||
| G1/S Transition | 105769 | 0 | 1 | 8 | 4.50e-07 | |||
| Pyrimidine Metabolism | 82946 | 1 | 1 | 4 | 1.57e-03 | |||
| Pyrimidine Deoxyribonucleotides de novo Biosynthesis I | 782380 | 0 | 1 | 3 | 2.16e-03 | |||
| Pyrimidine Metabolism | 106281 | 1 | 1 | 2 | 9.12e-04 | |||
| 8 | Gefitinib | EGFR | Integrated Pancreatic Cancer Pathway | 711360 | 1* | 8 | NA | 3.67e-05 |
| Pathways in Cancer | 83105 | 1* | 7 | NA | 5.11e-03 | |||
| Signaling by FGFR in disease | 645274 | 1* | 6 | NA | 1.13e-03 | |||
| Signaling by FGFR | 106343 | 1* | 6 | NA | 7.00e-04 | |||
| Integrated Breast Cancer Pathway | 219801 | 1* | 4 | NA | 1.76e-02 | |||
| Signaling Pathways in Glioblastoma | 672458 | 1* | 4 | NA | 1.77e-03 | |||
| Pancreatic Cancer | 83108 | 1* | 3 | NA | 8.93e-03 | |||
| Bladder Cancer | 83115 | 1* | 2 | NA | 2.51e-02 | |||
| 11 | Rosiglitazone | PPARs | PPAR Signaling Pathway | 83042 | NA | 1 | 4 | 5.83e-06 |
| Insulin Signaling Pathway | 83090 | NA | 1 | 3 | 1.89e-03 | |||
| 12 | Tamoxifen | ESR1 | Integrated Pancreatic Cancer Pathway | 711360 | NA | 6 | NA | 3.88e-03 |
| Integrated Breast Cancer Pathway | 219801 | NA | 4 | NA | 4.93e-02 | |||
| 13 | Tretinoin | RARs | Pathways in Cancer | 83105 | 0 | 7 | 4 | 6.36e-03 |
| Transcriptional Misregulation in Cancer | 523016 | 0 | 4 | 7 | 6.16e-05 | |||
| Integrated Pancreatic Cancer Pathway | 711360 | 0 | 6 | 2 | 6.36e-03 | |||
| Retinoic Acid Receptors-mediated Signaling | 138039 | 0 | 5 | 0 | 2.14e-04 | |||
| Non-small Cell Lung Cancer | 83119 | 0 | 5 | 0 | 4.65e-03 | |||
| Small Cell Lung Cancer | 83118 | 0 | 4 | 1 | 2.04e-02 | |||
| Acute Myeloid Leukemia | 83117 | 0 | 3 | 2 | 4.96e-03 | |||
| 16 | Vincristine | tubulin | Cell Cycle | 530733 | NA | 2 | 2 | 4.25e-03 |
| Cell Cycle, Mitotic | 105765 | NA | 2 | 1 | 1.74e-02 | |||
Numbers are consistent with those in Table 1.
Number of target proteins retrieved from the links to the protein structures at the PubChem Compound database (short cut to MMDB).
Numbers with * indicate that the crystal structure of the primary target in complex with the drug is included.
Number of target proteins retrieved from PubChem BioAssay (excluding the targets with crystal structures) involved in the particular pathway.
Number of target genes retrieved via GEO DataSets (excluding the targets with crystal structures and obtained from BioAssay) involved in the particular pathway.
The enrichment statistics P values were calculated based on a modified Fisher’s Exact Test compared to a background with all human pathways in the BioSystems database. Only pathways with P < 0.05 are shown as being enriched for investigated drugs in the table.
Not available. No targets retrieved via PubChem Compound (link to MMDB) or GEO.
Case A: drugs with pathways indicating potential new therapeutic uses
Four drugs identified relevant pathways that might indicate new drug uses (Table 2). Gefitinib (or iressa) is a known selective inhibitor of the tyrosine kinase domain of epidermal growth factor receptor (EGFR) and is used for the treatment of lung, breast and other cancers42–44. The crystal structure of the primary target of human EGFR in complex with gefitinib was retrieved from MMDB (PDB ID: 2ITY); sixty eight more protein targets (most of them are tyrosine kinases) with moderate affinity to gefitinib were retrieved from the BioAssay database (Table 1). Pathways pertinent to the EGFR signaling were identified, such as 1) the Signaling by FGFR in Disease (BSID: 645274, shared by EGFR, ERBB2-4, LCK, MKNK1, SLK, and SRC); 2) Signaling by FGFR (BSID: 106343, shared by EGFR, ERBB2-4, LCK, MKNK1, and SRC). A few cancer-related pathways were identified, including 1) Integrated Breast Cancer Pathway (BSID: 219801); 2) Pancreatic Cancer Pathways (BSID: 711360 and 83108); 3) Pathways in Cancer (BSID: 83105); 4) Signaling Pathways in Glioblastoma (BSID: 672458); 5) Bladder cancer (BSID: 83115). The retrieved breast cancer pathway confirmed gefitinib as a chemotherapy agent for this disease; other pathways, such as those about pancreatic and ladder cancers as well as glioblastoma were supported retrospectively by previous in vivo or in vitro studies on the potential uses of gefitinib against these cancers. For instance, gefitinib was found to prevent progression of pancreatic carcinoma in mouse model45 and inhibit pancreatic cancer cell growth and invasion in vitro46; gefitinib was able to inhibit tumor cell migration in EFGR-overexpressed human glioblastoma47; gefitinib also suppressed bladder cancer cell growth48 and decreased occurrence of bladder tumors in murine models49, 50. Misregulation of protein kinases especially EGFR is widely believed as a cause of uncontrolled cell proliferation51–55. Therefore, EGFR has become a popular target for anti-cancer therapy56, 57. Figure 1(A) demonstrates the relevant pathways identified for gefitinib, indicating the primary target EGFR and the various corresponding EFGR signaling pathways may account for the current or potential clinical uses of gefitinib as an anti-cancer agent.
Figure 1.
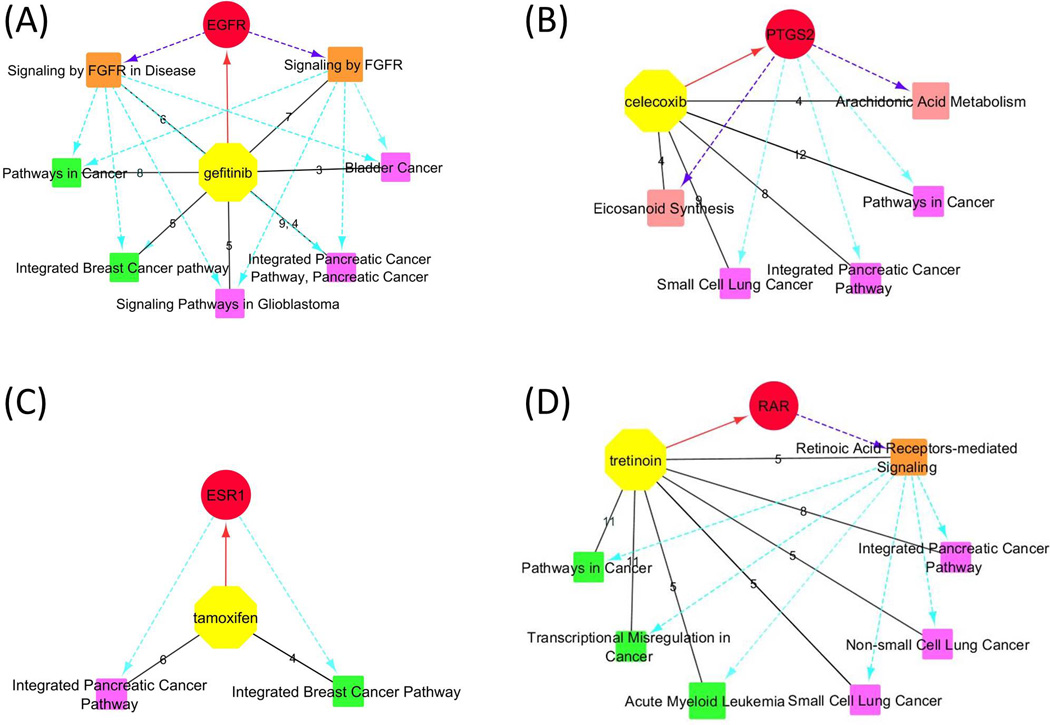
Relevant pathways identified for gefitinib(A), celecoxib(B), tamoxifen(C), and tretinoin(D). Yellow octagons and red circles represent the drugs and their primary targets. The orange, rose, green, and magenta squares represent relevant pathways responsible for the MOAs, biological functions, current and potential clinical uses of drugs, respectively. Black solid lines are between a drug and the retrieved relevant pathways. Edge labels indicate numbers of target proteins and genes shared by each pathway. Red solid lines with an arrow represent a drug and its primary target. Purple dashed lines with an arrow represent that the primary targets are responsible for the MOAs or biological functions indicated by pathways. Cyan dashed lines with an arrow represent the primary targets or pathways of MOAs are responsible for those indicating current or potential clinical uses.
Celecoxib is a well-known anti-inflammatory agent as a selective inhibitor of prostaglandin-endoperoxide synthase 2 (PTGS2 or COX2)58, 59, an enzyme that converts arachidonic acid (AA) to prostaglandin H2 (PGH2). The crystal structure of a target protein carbonic anhydrase II (CA2) bound with celecoxib (PDB ID: 1OQ5) was retrieved from MMDB. Nineteen additional protein targets and fifth six affected genes were retrieved. Two biological pathways relevant to its biological functions were retrieved, i.e. the pathways of Arachidonic Acid Metabolism (BSID: 82991 and 685553, shared by PTGS2, CYP2J2, GGT1, and PTGES) and Eicosanoid Synthesis (BSID: 198888, shared by PTGS2, GGT1, GGT2, and PTGES). Although celecoxib is not a chemotherapy drug, some cancer pathways were also identified, including the Pathways in Cancer (BSID: 83105), Integrated Pancreatic Cancer Pathway (BSID: 711360), and Small Cell Lung Cancer (BSID: 83118). These results are consistent with numerous publications of the efficacy to cancer cells by solely or combinatory use of celecoxib60–67. Particularly, celecoxib or its combination with other drugs prevented progression of pancreatic cancer cells in vitro and in vivo68–72. While celecoxib was mostly reported to have inhibitory effects on non-small cell lung cancer cells63, 73–76, clinical trials have shown that co-administration of celecoxib with chemotherapy agents improved treatment of small cell lung cancer77, 78. As discussed by previous studies, the effects of celecoxib on cancer cells may be attributed to its inhibition on COX-2, which in turn regulates multiple cancer pathways79–84. The relevant pathways are shown in Figure 1(B), indicating the primary target of celecoxib is responsible for its biological functions and its potential drug repositioning in cancer treatment.
Tamoxifen is an antagonist of the estrogen receptor 1 (ESR1) that is used for treatment of breast cancer85. No resolved structure was available for tamoxifen according to the link with MMDB. Fifty seven protein targets with affinity or inhibition with tamoxifen were retrieved from the BioAssay database. Among them, ESR1, AR, EGFR, and TP53 shared the common pathway of Integrated Breast Cancer Pathway (BSID: 219801) based on the search of BioSystems database, supporting the usage of tamoxifen as an anti-breast cancer drug. Another cancer pathway, the Integrated Pancreatic Cancer Pathway, was shared by ESR1, AR, EGFR, ERBB2, ESR2, and TP53, which is consistent with in vivo tests and clinical trials by sole or combinatory administration of tamoxifen and other chemotherapy drugs for the treatment of pancreatic cancer86–90. Figure 1(C) demonstrates the relevant pathways identified for tamoxifen that could be attributed to its primary target ESR1. A few pathways regarding signal transduction and gene expression were also identified (Supporting Information, Table S4), possibly due to the many nuclear receptor and tyrosine kinase targets retrieved from the BioAssay database.
Tretinoin (or all trans retinoic acid, ATRA) can be used for treatment of acne91 and acute myeloid leukemia92, 93. Although tretinoin can activate retinoid acid nuclear receptors (RARs) 94, the underlying mechanisms responsible for its clinical effects are not quite clear95. The crystal structures of the primary target retinoid acid nuclear receptor gamma (RARG, PDB ID: 2LBD) and cellular retinoic acid binding protein type II (CRABP2, PDB ID: 1CBS) in complex with ARTA were retrieved. Based on the structures and the additional thirty eight proteins and eighty nine genes obtained by referring to BioAssay and GEO DataSets databases, the pathway of Retinoic Acid Receptors-mediated Signaling (BSID: 138039, shared by RARA/G, MAPK1, and VDR) was retrieved, supporting RARs as the primary targets of tretinoin. In addition, the pathway of Acute Myeloid Leukemia (BSID: 83117, shared by RARA, EIF4EBP1, MAPK1, MYC, and PPARD) was identified, consistent with tretinoin’s treatment for this disease. Extra cancer pathways were also retrieved (Table 2), such as Transcriptional Misregulation in Cancer (BSID: 523016), Pathways in Cancer (BSID: 83105), the Integrated Pancreatic Cancer Pathway (BSID: 711360), Small Cell Lung Cancer (BSID: 83118), and Non-small Cell Lung Cancer (BSID: 83119), indicating tretinoin may have potential therapeutic effects on these cancers. The cancer pathways agreed with previous discussion of potential roles of tretinoin for cancer treatment96–98. The relevant pathways to pancreatic and lung cancers retrieved in this analysis were supported by previous in vitro or in vivo tests, demonstrating that ATRA inhibited growth of human pancreatic cancer cells in vitro99, 100 and decreased metastasis of lung tumor in cancer-bearing mice101. The related pathways are shown in Figure 1(D), suggesting the primary targets RARs and their signaling pathways might be responsible for the clinical uses of tretinoin for the treatment of cancers.
Case B: antineoplastic drugs with identified pathways responsible for their clinical uses
Besides gefitinib and tamoxifen, a few drugs are antineoplastic drugs that kill cancer cells by targeting enzymes and receptors (such as flurouracil and bortezomib), or directly interacting with DNA or tubulin (such as doxorubicin, actinomycin, and vincristine, Table 1). All these drugs except bortezomib found biological pathways regarding particular cancers they were used for, supporting their clinical functions. The drugs targeting DNA and tubulin witnessed pathways regarding apoptosis and cell mitosis, which represented biological functions responsible for their drug effects. These relevant pathways are shown in Table 2.
Doxorubicin and actinomycin are anti-cancer agents by directly targeting DNA102–104. Both of them identified pathways responsible for apoptosis and variety of cancers (Table 2). The participation of the target genes and proteins into various cancer-related pathways supported the broad-spectrum usage of doxorubicin in cancer therapy105; the relevant pathways about apoptosis accounted for the biological and clinical functions of both drugs. Vincritine has its antineoplastic effects by binding to tubulin106. Pathways regarding cell mitosis were identified, contributing to the biological functions behind its drug effects.
Fluorouracil and bortezomib kill cancer cells by targeting thymidylate synthase (TYMS)107 and proteasome108, 109, respectively. The crystal structure of uridine phosphorylase 1 (UPP1) bound with fluorouracil (PDB ID: 3NBQ) was obtained. Pathways regarding the MOA of fluorouracil were retrieved, with those of Pyrimidine Metabolism (BSIDs: 82946 and 106281) shared by TYMS, UPP1 and other targets (Supporting Information, Table S4). The identified Integrated Pancreatic Cancer Pathway (BSID: 711360) supported the usage of fluorouracil for pancreatic cancer110. Bortezomib witnessed pathways underlying its MOA, such as the pathways of the Protein Processing in Endoplasmic Reticulum (BSID: 167325) and the Parkin-Ubiquitin Proteasomal System (BSID: 700638, Table 2).
Case C: drugs targeting nuclear receptors and other proteins
Some drugs have their clinical effects by targeting nuclear receptors, such as mifepristone at PGR (progesterone receptor)111, 112, rosiglitazone at PPAR (peroxisome proliferator-activated receptor)113, propofol at GABR (γ-aminobutyric acid (GABA) A receptors)114, 115, and triiodothyronine at THRs (thyroid hormone receptors)116. Some of them found pathways particular to their current drug effects. For example, rosiglitazone identified the Insulin (BSID: 83090) and PPAR (BSID: 83042) signaling pathways, which indicated the underlying MOAs accounting for its clinical function as an antidiabetic drug113.
Adenosine identified the pathway of Vascular Smooth Muscle Contraction (BSID: 96530) shared by its primary target ADOR (adenosine receptor) and secondary targets, supporting its role as an antiarrhythmic agent for treatment of heart disorders117.
DISCUSSION
Pathways enrichment analysis: a novel approach for drug repositioning
Pathways common to genes with significant expression change upon drug treatment were previously used to elucidate drug MOAs and repositioning by using gene expression profiles across multiple cell types and datasets4, 5, 21, 22. However, pathway enrichment analysis based on protein targets retrieved from large-scale public databases is still lacking. In this study, biological pathways shared by target proteins and genes retrieved via MMDB, PubChem BioAssay and GEO were identified and analyzed in an attempt to examine and predict drug MOAs and functions. Eleven investigated drugs in this study have identified enriched pathways among their drug targets that are relevant to their clinical uses and MOAs, indicating finding biological pathways may provide useful information of drug uses and MOAs, especially for the drugs with unknown underlying mechanisms.
Considering the tremendous cost to develop new drugs, drug repositioning or repurposing has become a promising strategy to develop new therapeutic treatments for diseases10–14. Previous efforts from the bioinformatics point of view focused on the molecular modeling based on virtual screening of existing drug databases15–18, as well as computational biology that predicted new drug uses by establishing the relationship among drugs, genes, and diseases11–14, 19, 21. Most recently pathway enrichment analysis was applied for drug repositioning based on large-scale analysis of gene expression datasets21, 22. In this study, some drugs have identified relevant pathways responsible for diseases other than those currently treated by these drugs. Many of these pathways were supported by previous in vitro, in vivo test or clinical trials, indicating obtaining biological pathways shared by drug targets might be a novel approach for predicting new clinical uses of existing drugs. It’s noticed that the Integrated Pancreatic Cancer Pathway (BSID: 711360) was identified for many of the investigated drugs (especially those used for cancer therapy), and even indicated potential drug repositioning with drugs such as gefitinib, celecoxib, tamoxifen, and tretinoin. The involvement of this pathway with these drugs might be due to the integrated collection of multiple proteins based on different mechanistic path-ways relevant to pancreatic cancer (see description of this pathway in the BioSystems database: http://www.ncbi.nlm.nih.gov/biosystems/?term=711360), with many of the participating proteins also responsible for other caner pathways. The other point is, pathways pointing to diseases other than the current uses of a drug may not necessarily mean a novel therapeutic treatment. Some drugs were tested on diseases identified by such pathways but failed to become a treatment due to their pharmacokinetics properties or side effects. For example, although actinomycin identified a few pathways of cancers that are not currently used for, these relevant pathways do not indicate its new clinical uses due to its high cytotoxicity.
One thing to point out is, the pathways demonstrated in Table 2 were the examples of relevant pathways shared by the primary and secondary targets but might not indicate all the pathways that contributed to the pharmacological effects underlying the clinical uses of drugs. For example, doxorubicin is an antineoplastic drug with the function of inducing cell apoptosis by targeting DNA118. Besides the 20 relevant pathways demonstrated for in Table 2, several pathways of the participating molecules in the apoptosis signaling transduction were identified, such as those of TNF (Tumor necrosis factors), FAS (Fas cell surface death receptor) and caspases. Causing apoptosis is an important function of the immune system119. There were several retrieved pathways responsible for the immune system and the participating molecules, such as cytokines and interleukins. See Supporting Information, Table S4 for more pathways that could contribute to the pharmacological effects of the drugs.
Drug targets: direct or indirect interaction with drugs
The retrieved proteins and genes in this study can be divided into two categories: those physically bind with a drug and those without direct interaction with a drug but their activities or expression are affected as up- or down-stream proteins or by synergistic effects with primary proteins. Primary target proteins interact physically with a drug which causes its biological effects on this target. Many proteins retrieved from BioAssay database are also physically interacting with drugs, as suggested by their affinities with Kd, IC50, Ki, or EC50 values; for cell-based assays in BioAssay, an identified drug target might not be the exact pharmacological target and the drug effects on this target might be attributed to an up-stream protein that is directly targeted by this drug. The genes retrieved as drug targets via GEO DataSets were mainly according to their gene expression data, indicating that they might be regulated by the primary target within the same signal cascade. The identified pathways could involve targets with both direct and indirect interaction with drugs. It is reasonable that in a retrieved pathway, targets with physical binding with a drug coexist with other proteins belonging to the same signal cascade without direct drug interaction. Some identified pathways even involve multiple proteins with physical interaction with a drug. For example, TYMS is the primary target of fluorouracil; Uridine Phosphorylase 1 (UPP1) is also an enzyme catalyzing the metabolism of fluorouracil and its physical binding with this drug is supported by the crystal structure of UPP1 bound with fluorouracil (PDB ID: 3NBQ) retrieved from MMDB120. Both targets participated in the retrieved pathway of Pyrimidine Metabolism (BSID: 82946) that is responsible for the anti-neoplastic effect of fluorouracil (Table 2 and Supporting Information, Table S4). The involvement of targets with both direct and indirect interaction with drugs indicates that both categories of targets are significant for identification of biological pathways relevant to drug effects.
Drugs that failed to identify responsible pathways for their clinical uses
Disulfiram, mifepristone, propofol, triiodothyronine and valproic acid failed to find any significantly enriched pathways that are relevant to their biological functions or clinical uses common to the drug targets. The reasons might be: 1) the retrieved gene or protein targets were responsible for variety of cellular functions that were not particular to the pathways accounting for its primary target protein; 2) the retrieved target proteins or genes were not the direct targets of the drug but interacted with the drug in an indirect way as up- or down- stream proteins or by cross-talking with the primary pathways.; 3) the retrieved drug targets were limited to obtain relevant pathways. See Supporting Information for the detail discussion of these drugs.
Public databases: a resource for biomedical research on small molecules, bioactivities, target proteins and genes, and responsible biological systems
The enormous biological information in the MMDB, BioAssay, GEO and BioSystems databases provides advantage to support this study. The cross-link to protein structures at the PubChem Compound database provides a convenient way to exploit the crystal structures of drug targets associated with each drug in the MMDB database. It’s noticed that only a few resolved structures were retrieved for the investigated drugs, and some drugs even failed to find any crystal structures with human proteins. The limited structures retrieved from MMDB may be attributed to the lack of experimental data deposited into PDB. BioAssay is one of the public databases with the biggest collection of drug target information. One advantage of BioAssay database is the straightforward list of the drug bioactivity and targets, which presents users with an outline of drug target information. As a public repository with gene expression data, GEO DataSets makes a great complement to the target list in this study when the protein targets retrieved from MMDB and BioAssay are limited to obtain biological pathways relevant to drug effects. It should be pointed out that the usage of GEO DataSets in this study might be limited in that it’s time-consuming to explore the specific gene expression data when checking the literature associated with GEO DataSets records retrieved with each drug, partly due to the lack of cross-linking between GEO and PubChem.
Further application of pathway analysis in drug repositioning
The current study involves identification of drug repositioning by investigating prioritized pathways based on available drug targets. The application of pathway analysis in drug repositioning can be from multiple perspectives. Novel therapeutic treatments can also be discovered by retrospectively identifying repurposed drug targets in terms of pathways analysis results. Therefore, drugs with a similar target profile common to a same pathway might be lead candidates for a disease without previous therapeutic indication.
The other point is, only pathways common to both primary and secondary targets are discussed in this study. Nevertheless, without the involvement of primary target, statistically enriched pathways shared by secondary targets may also indicate interesting and valuable pharmacological and clinical significance that is helpful for future drug design. The aforementioned applications of pathway analysis are underway in our group.
CONCLUSION
In conclusion, the pathway analysis based on drug target information outlined in this study might present a new approach to investigate the MOAs and additional clinical uses of drugs. Eleven investigated drugs have identified relevant pathways based on gene and protein targets retrieved from MMDB, BioAssay, and GEO DataSets; the remaining five drugs failed to identify enriched relevant pathways possibly due to the limited data available in the public databases. Interestingly, drugs gefitinib, celecoxib, tamoxifen, and tretinoin have retrieved biological pathways responsible for diseases other than their current uses, indicating that identifying biological pathways could be a new approach to exploit drug repositioning or repurposing. Overall, the data mining and pathway analysis applied in this study provides a new approach for identifying drug MOAs and repurposing, therefore possibly benefiting the development of new therapeutic treatments for diseases.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Library of Medicine (NLM).
ABBREVIATIONS
- ABAT
4-aminobutyrate aminotransferase
- ADOR
adenosine receptor
- ALDH
aldehyde dehydrogenase
- AR
androgen receptor
- CA2
carbonic anhydrase II
- COX2
cytochrome c oxidase subunit II
- CRABP2
cellular retinoic acid binding protein 2
- CYP2J2
cytochrome P450, family 2, subfamily J, polypeptide 2
- DUT
deoxyuridine triphosphatase
- EGFR
epidermal growth factor receptor
- EIF4EBP1
eukaryotic translation initiation factor 4E binding protein 1
- ERBB2/3/4
v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2/3/4
- ESR1/2
estrogen receptor 1/2
- FAS
Fas cell surface death receptor
- GABR
gamma-aminobutyric acid (GABA) A receptor
- GGT1/2
gamma-glutamyltransferase 1/2
- GLS
glutaminase
- GNAI1
guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1
- GNG11
guanine nucleotide binding protein (G protein), gamma 11
- LCK
lymphocyte-specific protein tyrosine kinase
- MAPK1
mitogen-activated protein kinase 1
- MKNK1
MAP kinase interacting serine/threonine kinase 1
- MYC
v-myc avian myelocytomatosis viral oncogene homolog
- PGR
progesterone receptor
- PPAR (D)
peroxisome proliferator-activated receptor (delta)
- PTGES
prostaglandin E synthase
- PTGS2
prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase)
- RARA/G
retinoic acid receptor, alpha/gamma
- RRM2(B)
ribonucleotide reductase M2 (B)
- SLK
STE20-like kinase
- SRC
v-src avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog
- THR
thyroid hormone receptor
- TK1
thymidine kinase 1, soluble
- TP53
tumor protein p53
- TNF
tumor necrosis factors
- TYMS
thymidylate synthetase
- UPP1
uridine phosphorylase 1
- VDR
vitamin D (1,25- dihydroxyvitamin D3) receptor
Footnotes
Supporting Information Available. 1) word file: discussion of relevant pathways identified for each drug; example of the data-mining and analysis regarding celecoxib. 2) excel file: Table S1. List of retrieved target proteins and genes by checking MMDB, PubChem Bioassay and GEO DataSets. Table S2. Drug CID numbers and links to search protein targets via PubChem Compound (short cut to MMDB) and BioAssay databases. Table S3. Genes and PDB IDs of resolved structures in complex with investigated drugs from MMDB. Table S4. List of biological pathways identified for each drug (each pathway shared by the primary targets and at least one secondary targets). Table S5, Official full names of all drug targets retrieved according to the Gene database. 3) others: in-house Ruby scripts and input and output files for pathway retrieval and data processing. This material is free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Chen B, Wild D, Guha R. PubChem as a source of polypharmacology. J Chem Inf Model. 2009;49(9):2044–2055. doi: 10.1021/ci9001876. [DOI] [PubMed] [Google Scholar]
- 2.Cheng T, Li Q, Wang Y, Bryant SH. Identifying compound-target associations by combining bioactivity profile similarity search and public databases mining. J Chem Inf Model. 2011;51(9):2440–2448. doi: 10.1021/ci200192v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng T, Wang Y, Bryant SH. Investigating the correlations among the chemical structures, bioactivity profiles and molecular targets of small molecules. Bioinformatics. 2010;26(22):2881–2888. doi: 10.1093/bioinformatics/btq550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Covell DG. Integrating constitutive gene expression and chemoactivity: mining the NCI60 anticancer screen. PLoS One. 2012;7(10):e44631. doi: 10.1371/journal.pone.0044631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang R, Wallqvist A, Thanki N, Covell DG. Linking pathway gene expressions to the growth inhibition response from the National Cancer Institute's anticancer screen and drug mechanism of action. Pharmacogenomics J. 2005;5(6):381–399. doi: 10.1038/sj.tpj.6500331. [DOI] [PubMed] [Google Scholar]
- 6.Williams-DeVane CR, Wolf MA, Richard AM. DSSTox chemical-index files for exposure-related experiments in ArrayExpress and Gene Expression Omnibus: enabling toxico-chemogenomics data linkages. Bioinformatics. 2009;25(5):692–694. doi: 10.1093/bioinformatics/btp042. [DOI] [PubMed] [Google Scholar]
- 7.Williams-Devane CR, Wolf MA, Richard AM. Toward a public toxicogenomics capability for supporting predictive toxicology: survey of current resources and chemical indexing of experiments in GEO and ArrayExpress. Toxicol Sci. 2009;109(2):358–371. doi: 10.1093/toxsci/kfp061. [DOI] [PubMed] [Google Scholar]
- 8.Huang R, Wallqvist A, Covell DG. Anticancer metal compounds in NCI's tumor-screening database: putative mode of action. Biochem Pharmacol. 2005;69(7):1009–1039. doi: 10.1016/j.bcp.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Huang R, Wallqvist A, Covell DG. Comprehensive analysis of pathway or functionally related gene expression in the National Cancer Institute's anticancer screen. Genomics. 2006;87(3):315–328. doi: 10.1016/j.ygeno.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 10.Nair P. Second act: Drug repurposing gets a boost as academic researchers join the search for novel uses of existing drugs. Proc Natl Acad Sci U S A. 2013;110(7):2430–2432. doi: 10.1073/pnas.201300188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudley JT, Deshpande T, Butte AJ. Exploiting drug-disease relationships for computational drug repositioning. Brief Bioinform. 2011;12(4):303–311. doi: 10.1093/bib/bbr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med. 2011;3(96) doi: 10.1126/scitranslmed.3002648. 96ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 14.Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, Sage J, Butte AJ. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3(96) doi: 10.1126/scitranslmed.3001318. 96ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekins S, Williams AJ, Krasowski MD, Freundlich JS. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov Today. 2011;16(7–8):298–310. doi: 10.1016/j.drudis.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Lynch C, Pan Y, Li L, Ferguson SS, Xia M, Swaan PW, Wang H. Identification of novel activators of constitutive androstane receptor from FDA-approved drugs by integrated computational and biological approaches. Pharm Res. 2013;30(2):489–501. doi: 10.1007/s11095-012-0895-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pan Y, Chothe PP, Swaan PW. Identification of novel breast cancer resistance protein (BCRP) inhibitors by virtual screening. Mol Pharm. 2013;10(4):1236–1248. doi: 10.1021/mp300547h. [DOI] [PubMed] [Google Scholar]
- 18.Pan Y, Li L, Kim G, Ekins S, Wang H, Swaan PW. Identification and validation of novel human pregnane X receptor activators among prescribed drugs via ligand-based virtual screening. Drug Metab Dispos. 2011;39(2):337–344. doi: 10.1124/dmd.110.035808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Agarwal P. A pathway-based view of human diseases and disease relationships. PLoS One. 2009;4(2):e4346. doi: 10.1371/journal.pone.0004346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, Zmoos AF, Vaka D, Tran KQ, Zhou M, Krasinska K, Riess JW, Neal JW, Khatri P, Park KS, Butte AJ, Sage J. A Drug Repositioning Approach Identifies Tricyclic Antidepressants as Inhibitors of Small Cell Lung Cancer and Other Neuroendocrine Tumors. Cancer Discov. 2013 doi: 10.1158/2159-8290.CD-13-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith SB, Dampier W, Tozeren A, Brown JR, Magid-Slav M. Identification of common biological pathways and drug targets across multiple respiratory viruses based on human host gene expression analysis. PLoS One. 2012;7(3):e33174. doi: 10.1371/journal.pone.0033174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madej T, Addess KJ, Fong JH, Geer LY, Geer RC, Lanczycki CJ, Liu C, Lu S, Marchler- Bauer A, Panchenko AR, Chen J, Thiessen PA, Wang Y, Zhang D, Bryant SH. MMDB: 3D structures and macromolecular interactions. Nucleic Acids Res. 2012;40(Database issue):D461–D464. doi: 10.1093/nar/gkr1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35(Database issue):D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q, Cheng T, Wang Y, Bryant SH. PubChem as a public resource for drug discovery. Drug Discov Today. 2010;15(23–24):1052–1057. doi: 10.1016/j.drudis.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2013;41(Database issue):D8f–D20. doi: 10.1093/nar/gks1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Zhou Z, Han L, Karapetyan K, Dracheva S, Shoemaker BA, Bolton E, Gindulyte A, Bryant SH. PubChem's BioAssay Database. Nucleic Acids Res. 2012;40(Database issue):D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, Bryant SH. The NCBI BioSystems database. Nucleic Acids Res. 2010;38(Database issue):D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keseler IM, Bonavides-Martinez C, Collado-Vides J, Gama-Castro S, Gunsalus RP, Johnson DA, Krummenacker M, Nolan LM, Paley S, Paulsen IT, Peralta-Gil M, Santos-Zavaleta A, Shearer AG, Karp PD. EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 2009;37(Database issue):D464–D470. doi: 10.1093/nar/gkn751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, Kanapin A, Lewis S, Mahajan S, May B, Schmidt E, Vastrik I, Wu G, Birney E, Stein L, D'Eustachio P. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37(Database issue):D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the Pathway Interaction Database. Nucleic Acids Res. 2009;37(Database issue):D674–D679. doi: 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelder T, Pico AR, Hanspers K, van Iersel MP, Evelo C, Conklin BR. Mining biological pathways using WikiPathways web services. PLoS One. 2009;4(7):e6447. doi: 10.1371/journal.pone.0006447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pico AR, Kelder T, van Iersel MP, Hanspers K, Conklin BR, Evelo C. WikiPathways: pathway editing for the people. PLoS Biol. 2008;6(7):e184. doi: 10.1371/journal.pbio.0060184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(Database issue):D668–D672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.NCBI. NCBI help Mannual. Bethesda, MD: 2010. Entrez Programming Utilities Help. [Google Scholar]
- 40.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011;39(Database issue):D52–D57. doi: 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.Baselga J, Albanell J, Ruiz A, Lluch A, Gascon P, Guillem V, Gonzalez S, Sauleda S, Marimon I, Tabernero JM, Koehler MT, Rojo F. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J Clin Oncol. 2005;23(23):5323–5333. doi: 10.1200/JCO.2005.08.326. [DOI] [PubMed] [Google Scholar]
- 43.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305(5687):1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 45.Mohammed A, Janakiram NB, Li Q, Madka V, Ely M, Lightfoot S, Crawford H, Steele VE, Rao CV. The epidermal growth factor receptor inhibitor gefitinib prevents the progression of pancreatic lesions to carcinoma in a conditional LSL-KrasG12D/+ transgenic mouse model. Cancer Prev Res (Phila) 2010;3(11):1417–1426. doi: 10.1158/1940-6207.CAPR-10-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Kleeff J, Giese N, Buchler MW, Korc M, Friess H. Gefitinib ('Iressa', ZD1839), a selective epidermal growth factor receptor tyrosine kinase inhibitor, inhibits pancreatic cancer cell growth, invasion, and colony formation. Int J Oncol. 2004;25(1):203–210. [PubMed] [Google Scholar]
- 47.Parker JJ, Dionne KR, Massarwa R, Klaassen M, Foreman NK, Niswander L, Canoll P, Kleinschmidt-Demasters BK, Waziri A. Gefitinib selectively inhibits tumor cell migration in EGFR-amplified human glioblastoma. Neuro Oncol. 2013 doi: 10.1093/neuonc/not053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colquhoun AJ, McHugh LA, Tulchinsky E, Kriajevska M, Mellon JK. Combination treatment with ionising radiation and gefitinib ('Iressa', ZD1839), an epidermal growth factor receptor (EGFR) inhibitor, significantly inhibits bladder cancer cell growth in vitro and in vivo. J Radiat Res. 2007;48(5):351–360. doi: 10.1269/jrr.07014. [DOI] [PubMed] [Google Scholar]
- 49.Lubet RA, Lu Y, Bode AM, You M, Verney ZM, Steele VE, Townsend R, Juliana MM, Grubbs CJ. Efficacy of the EGFr inhibitor Iressa on development of chemically-induced urinary bladder cancers: dose dependency and modulation of biomarkers. Oncol Rep. 2011;25(5):1389–1397. doi: 10.3892/or.2011.1200. [DOI] [PubMed] [Google Scholar]
- 50.Lubet RA, Steele VE, Juliana MM, Grubbs CJ. Screening agents for preventive efficacy in a bladder cancer model: study design, end points, and gefitinib and naproxen efficacy. J Urol. 2010;183(4):1598–1603. doi: 10.1016/j.juro.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 51.Brand TM, Iida M, Li C, Wheeler DL. The nuclear epidermal growth factor receptor signaling network and its role in cancer. Discov Med. 2011;12(66):419–432. [PMC free article] [PubMed] [Google Scholar]
- 52.Van den Eynde M, Baurain JF, Mazzeo F, Machiels JP. Epidermal growth factor receptor targeted therapies for solid tumours. Acta Clin Belg. 2011;66(1):10–17. doi: 10.2143/ACB.66.1.2062508. [DOI] [PubMed] [Google Scholar]
- 53.Fratto ME, Santini D, Vincenzi B, Silvestris N, Azzariti A, Tommasi S, Zoccoli A, Galluzzo S, Maiello E, Colucci G, Tonini G. Targeting EGFR in bilio-pancreatic and liver carcinoma. Front Biosci (Schol Ed) 2011;3:16–22. doi: 10.2741/s128. [DOI] [PubMed] [Google Scholar]
- 54.Modjtahedi H, Essapen S. Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs. 2009;20(10):851–855. doi: 10.1097/CAD.0b013e3283330590. [DOI] [PubMed] [Google Scholar]
- 55.Chang YS, Chung JH, Shin DH, Chung KY, Kim YS, Chang J, Kim SK. Retinoic acid receptor-beta expression in stage I non-small cell lung cancer and adjacent normal appearing bronchial epithelium. Yonsei Med J. 2004;45(3):435–442. doi: 10.3349/ymj.2004.45.3.435. [DOI] [PubMed] [Google Scholar]
- 56.Pearson MA, Fabbro D. Targeting protein kinases in cancer therapy: a success? Expert Rev Anticancer Ther. 2004;4(6):1113–1124. doi: 10.1586/14737140.4.6.1113. [DOI] [PubMed] [Google Scholar]
- 57.Ventura JJ, Nebreda AR. Protein kinases and phosphatases as therapeutic targets in cancer. Clin Transl Oncol. 2006;8(3):153–160. doi: 10.1007/s12094-006-0005-0. [DOI] [PubMed] [Google Scholar]
- 58.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352(11):1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 59.Yelland MJ, Nikles CJ, McNairn N, Del Mar CB, Schluter PJ, Brown RM. Celecoxib compared with sustained-release paracetamol for osteoarthritis: a series of n-of-1 trials. Rheumatology (Oxford) 2007;46(1):135–140. doi: 10.1093/rheumatology/kel195. [DOI] [PubMed] [Google Scholar]
- 60.Brandao RD, Veeck J, Van de Vijver KK, Lindsey P, de Vries B, van Elssen CH, Blok MJ, Keymeulen K, Ayoubi T, Smeets HJ, Tjan-Heijnen VC, Hupperets PS. A randomised controlled phase II trial of pre-operative celecoxib treatment reveals anti-tumour transcriptional response in primary breast cancer. Breast Cancer Res. 2013;15(2):R29. doi: 10.1186/bcr3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chow LW, Tung SY, Ng TY, Im SA, Lee MH, Yip AY, Toi M, Gluck S. Concurrent celecoxib with 5-fluorouracil/epirubicin/cyclophosphamide followed by docetaxel for stages II - III invasive breast cancer: the OOTR-N001 study. Expert Opin Investig Drugs. 2013;22(3):299–307. doi: 10.1517/13543784.2013.766715. [DOI] [PubMed] [Google Scholar]
- 62.Morisaki T, Umebayashi M, Kiyota A, Koya N, Tanaka H, Onishi H, Katano M. Combining celecoxib with sorafenib synergistically inhibits hepatocellular carcinoma cells in vitro. Anticancer Res. 2013;33(4):1387–1395. [PubMed] [Google Scholar]
- 63.Patel AR, Chougule MB, I T, Patlolla R, Wang G, Singh M. Efficacy of aerosolized celecoxib encapsulated nanostructured lipid carrier in non-small cell lung cancer in combination with docetaxel. Pharm Res. 2013;30(5):1435–1446. doi: 10.1007/s11095-013-0984-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perroud HA, Rico MJ, Alasino CM, Queralt F, Mainetti LE, Pezzotto SM, Rozados VR, Scharovsky OG. Safety and therapeutic effect of metronomic chemotherapy with cyclophosphamide and celecoxib in advanced breast cancer patients. Future Oncol. 2013;9(3):451–462. doi: 10.2217/fon.12.196. [DOI] [PubMed] [Google Scholar]
- 65.Wang YJ, Niu XP, Yang L, Han Z, Ma YJ. Effects of celecoxib on cycle kinetics of gastric cancer cells and protein expression of cytochrome C and caspase-9. Asian Pac J Cancer Prev. 2013;14(4):2343–2347. doi: 10.7314/apjcp.2013.14.4.2343. [DOI] [PubMed] [Google Scholar]
- 66.Zhang B, Ma X, Li Z, Gao X, Wang F, Liu L, Shen G, Sang Y, Li M, Li Y, Zhao J, Wei Y. Celecoxib enhances the efficacy of 15-hydroxyprostaglandin dehydrogenase gene therapy in treating murine breast cancer. J Cancer Res Clin Oncol. 2013;139(5):797–807. doi: 10.1007/s00432-013-1381-9. [DOI] [PubMed] [Google Scholar]
- 67.Zhang DQ, Guo Q, Zhu JH, Chen WC. Increase of cyclooxygenase-2 inhibition with celecoxib combined with 5-FU enhances tumor cell apoptosis and antitumor efficacy in a subcutaneous implantation tumor model of human colon cancer. World J Surg Oncol. 2013;11:16. doi: 10.1186/1477-7819-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Al-Wadei HA, Al-Wadei MH, Ullah MF, Schuller HM. Celecoxib and GABA cooperatively prevent the progression of pancreatic cancer in vitro and in xenograft models of stress-free and stress-exposed mice. PLoS One. 2012;7(8):e43376. doi: 10.1371/journal.pone.0043376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosendahl AH, Gundewar C, Said K, Karnevi E, Andersson R. Celecoxib synergizes human pancreatic ductal adenocarcinoma cells to sorafenib-induced growth inhibition. Pancreatology. 2012;12(3):219–226. doi: 10.1016/j.pan.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 70.Ding X, Zhu C, Qiang H, Zhou X, Zhou G. Enhancing antitumor effects in pancreatic cancer cells by combined use of COX-2 and 5-LOX inhibitors. Biomed Pharmacother. 2011;65(7):486–490. doi: 10.1016/j.biopha.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 71.Arjona-Sanchez A, Ruiz-Rabelo J, Perea MD, Vazquez R, Cruz A, Munoz Mdel C, Tunez I, Muntane J, Padillo FJ. Effects of capecitabine and celecoxib in experimental pancreatic cancer. Pancreatology. 2010;10(5):641–647. doi: 10.1159/000288708. [DOI] [PubMed] [Google Scholar]
- 72.Padillo FJ, Ruiz-Rabelo JF, Cruz A, Perea MD, Tasset I, Montilla P, Tunez I, Muntane J. Melatonin and celecoxib improve the outcomes in hamsters with experimental pancreatic cancer. J Pineal Res. 2010;49(3):264–270. doi: 10.1111/j.1600-079X.2010.00791.x. [DOI] [PubMed] [Google Scholar]
- 73.Koch A, Bergman B, Holmberg E, Sederholm C, Ek L, Kosieradzki J, Lamberg K, Thaning L, Ydreborg SO, Sorenson S. Effect of celecoxib on survival in patients with advanced non-small cell lung cancer: a double blind randomised clinical phase III trial (CYCLUS study) by the Swedish Lung Cancer Study Group. Eur J Cancer. 2011;47(10):1546–1555. doi: 10.1016/j.ejca.2011.03.035. [DOI] [PubMed] [Google Scholar]
- 74.Gore E, Bae K, Langer C, Extermann M, Movsas B, Okunieff P, Videtic G, Choy H. Phase I/II trial of a COX-2 inhibitor with limited field radiation for intermediate prognosis patients who have locally advanced non-small-cell lung cancer: radiation therapy oncology group 0213. Clin Lung Cancer. 2011;12(2):125–130. doi: 10.1016/j.cllc.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 75.Klenke FM, Abdollahi A, Bischof M, Gebhard MM, Ewerbeck V, Huber PE, Sckell A. Celecoxib enhances radiation response of secondary bone tumors of a human non-small cell lung cancer via antiangiogenesis in vivo. Strahlenther Onkol. 2011;187(1):45–51. doi: 10.1007/s00066-010-2116-3. [DOI] [PubMed] [Google Scholar]
- 76.Komaki R, Wei X, Allen PK, Liao Z, Milas L, Cox JD, O'Reilly MS, Chang JY, McAleer MF, Jeter M, Blumenschein GR, Jr, Kies MS. Phase I study of celecoxib with concurrent irinotecan, Cisplatin, and radiation therapy for patients with unresectable locally advanced non-small cell lung cancer. Front Oncol. 2011;1:52. doi: 10.3389/fonc.2011.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Aruajo AM, Mendez JC, Coelho AL, Sousa B, Barata F, Figueiredo A, Amaro T, Azevedo I, Soares M. Phase II study of celecoxib with cisplatin plus etoposide in extensive-stage small cell lung cancer. Cancer Invest. 2009;27(4):391–396. doi: 10.1080/07357900802232756. [DOI] [PubMed] [Google Scholar]
- 78.Natale RB. Irinotecan, cisplatin/carboplatin, and COX-2 inhibition in small-cell lung cancer. Oncology (Williston Park) 2003;17(7 Suppl 7):22–26. [PubMed] [Google Scholar]
- 79.Jendrossek V. Targeting apoptosis pathways by Celecoxib in cancer. Cancer Lett. 2013;332(2):313–324. doi: 10.1016/j.canlet.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 80.Dempke W, Rie C, Grothey A, Schmoll HJ. Cyclooxygenase-2: a novel target for cancer chemotherapy? J Cancer Res Clin Oncol. 2001;127(7):411–417. doi: 10.1007/s004320000225. [DOI] [PubMed] [Google Scholar]
- 81.Shao N, Feng N, Wang Y, Mi Y, Li T, Hua L. Systematic review and meta-analysis of COX-2 expression and polymorphisms in prostate cancer. Mol Biol Rep. 2012;39(12):10997–11004. doi: 10.1007/s11033-012-2001-5. [DOI] [PubMed] [Google Scholar]
- 82.Wang R, Guo L, Wang P, Yang W, Lu Y, Huang Z, Tang C. Chemoprevention of cancers in gastrointestinal tract with cyclooxygenase 2 inhibitors. Curr Pharm Des. 2013;19(1):115–125. doi: 10.2174/13816128130116. [DOI] [PubMed] [Google Scholar]
- 83.Kraus S, Naumov I, Arber N. COX-2 active agents in the chemoprevention of colorectal cancer. Recent Results Cancer Res. 2013;191:95–103. doi: 10.1007/978-3-642-30331-9_5. [DOI] [PubMed] [Google Scholar]
- 84.Garcia Rodriguez LA, Cea-Soriano L, Tacconelli S, Patrignani P. Coxibs: pharmacology, toxicity and efficacy in cancer clinical trials. Recent Results Cancer Res. 2013;191:67–93. doi: 10.1007/978-3-642-30331-9_4. [DOI] [PubMed] [Google Scholar]
- 85.Jordan VC. Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. Br J Pharmacol. 2006;147(Suppl 1):S269–S276. doi: 10.1038/sj.bjp.0706399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tomao S, Romiti A, Massidda B, Ionta MT, Farris A, Zullo A, Brescia A, Santuari L, Frati L. A phase II study of gemcitabine and tamoxifen in advanced pancreatic cancer. Anticancer Res. 2002;22(4):2361–2364. [PubMed] [Google Scholar]
- 87.Lee I, Lee YH, Mikulski SM, Shogen K. Effect of ONCONASE +/− tamoxifen on ASPC-1 human pancreatic tumors in nude mice. Adv Exp Med Biol. 2003;530:187–196. doi: 10.1007/978-1-4615-0075-9_18. [DOI] [PubMed] [Google Scholar]
- 88.Lamy R, Conroy T, Brunaud L, Bresler L. [Tamoxifen for metastatic pancreatic adenocarcinoma: a complete response] Gastroenterol Clin Biol. 2001;25(10):912–913. [PubMed] [Google Scholar]
- 89.Wenger FA, Kilian M, Mautsch I, Jacobi CA, Schimke I, Saul GJ, Guski H, Muller JM. Influence of octreotide and tamoxifen on tumor growth and liver metastasis in N-nitrosobis(2-oxopropyl)amineinduced pancreatic cancer in Syrian hamsters. Horm Res. 2000;54(2):74–77. doi: 10.1159/000053235. [DOI] [PubMed] [Google Scholar]
- 90.Eckert H, Bajorath J. Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches. Drug Discov Today. 2007;12(5–6):225–233. doi: 10.1016/j.drudis.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 91.Stefanaki C, Stratigos A, Katsambas A. Topical retinoids in the treatment of photoaging. J Cosmet Dermatol. 2005;4(2):130–134. doi: 10.1111/j.1473-2165.2005.40215.x. [DOI] [PubMed] [Google Scholar]
- 92.Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, Degos L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76(9):1704–1709. [PubMed] [Google Scholar]
- 93.Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, Gu LJ, Wang ZY. Use of alltrans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72(2):567–572. [PubMed] [Google Scholar]
- 94.Yu S, Levi L, Siegel R, Noy N. Retinoic acid induces neurogenesis by activating both retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) J Biol Chem. 2012;287(50):42195–42205. doi: 10.1074/jbc.M112.410381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arteaga MF, Mikesch JH, Qiu J, Christensen J, Helin K, Kogan SC, Dong S, So CW. The histone demethylase PHF8 governs retinoic acid response in acute promyelocytic leukemia. Cancer Cell. 2013;23(3):376–389. doi: 10.1016/j.ccr.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1(3):181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 97.Hu Y, Liu HX, He Y, Fang Y, Fang J, Wan YJ. Transcriptome profiling and genome-wide DNA binding define the differential role of fenretinide and all-trans RA in regulating the death and survival of human hepatocellular carcinoma Huh7 cells. Biochem Pharmacol. 2013;85(7):1007–1017. doi: 10.1016/j.bcp.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kelly WK, Osman I, Reuter VE, Curley T, Heston WD, Nanus DM, Scher HI. The development of biologic end points in patients treated with differentiation agents: an experience of retinoids in prostate cancer. Clin Cancer Res. 2000;6(3):838–846. [PubMed] [Google Scholar]
- 99.Li J, Orr B, White K, Belogortseva N, Niles R, Boskovic G, Nguyen H, Dykes A, Park M. Chmp 1A is a mediator of the anti-proliferative effects of all-trans retinoic acid in human pancreatic cancer cells. Mol Cancer. 2009;8:7. doi: 10.1186/1476-4598-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gupta S, Pramanik D, Mukherjee R, Campbell NR, Elumalai S, de Wilde RF, Hong SM, Goggins MG, De Jesus-Acosta A, Laheru D, Maitra A. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clin Cancer Res. 2012;18(1):280–289. doi: 10.1158/1078-0432.CCR-11-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Siddikuzzaman Grace VM. Inhibition of metastatic lung cancer in C57BL/6 mice by liposome encapsulated all trans retinoic acid (ATRA) Int Immunopharmacol. 2012;14(4):570–579. doi: 10.1016/j.intimp.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 102.Fornari FA, Randolph JK, Yalowich JC, Ritke MK, Gewirtz DA. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol Pharmacol. 1994;45(4):649–656. [PubMed] [Google Scholar]
- 103.Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976;36(8):2891–2895. [PubMed] [Google Scholar]
- 104.Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci U S A. 1985;82(16):5328–5331. doi: 10.1073/pnas.82.16.5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.CLINIC M. Doxorubicin (Intravenous Route) Micromedex. 2012 [Google Scholar]
- 106.Johnson IS, Armstrong JG, Gorman M, Burnett JP., Jr The Vinca Alkaloids: A New Class of Oncolytic Agents. Cancer Res. 1963;23:1390–1427. [PubMed] [Google Scholar]
- 107.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 108.Bonvini P, Zorzi E, Basso G, Rosolen A. Bortezomib-mediated 26S proteasome inhibition causes cellcycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphoma. Leukemia. 2007;21(4):838–842. doi: 10.1038/sj.leu.2404528. [DOI] [PubMed] [Google Scholar]
- 109.Gelman JS, Sironi J, Berezniuk I, Dasgupta S, Castro LM, Gozzo FC, Ferro ES, Fricker LD. Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PLoS One. 2013;8(1):e53263. doi: 10.1371/journal.pone.0053263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.CLINIC M. Fluorouracil (Intravenous Route, Injection Route) In. 2011 [Google Scholar]
- 111.Fiala C, Gemzel-Danielsson K. Review of medical abortion using mifepristone in combination with a prostaglandin analogue. Contraception. 2006;74(1):66–86. doi: 10.1016/j.contraception.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 112.Heikinheimo O, Kekkonen R, Lahteenmaki P. The pharmacokinetics of mifepristone in humans reveal insights into differential mechanisms of antiprogestin action. Contraception. 2003;68(6):421–426. doi: 10.1016/s0010-7824(03)00077-5. [DOI] [PubMed] [Google Scholar]
- 113.Mayerson AB, Hundal RS, Dufour S, Lebon V, Befroy D, Cline GW, Enocksson S, Inzucchi SE, Shulman GI, Petersen KF. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes. 2002;51(3):797–802. doi: 10.2337/diabetes.51.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kotani Y, Shimazawa M, Yoshimura S, Iwama T, Hara H. The experimental and clinical pharmacology of propofol, an anesthetic agent with neuroprotective properties. CNS Neurosci Ther. 2008;14(2):95–106. doi: 10.1111/j.1527-3458.2008.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trapani G, Altomare C, Liso G, Sanna E, Biggio G. Propofol in anesthesia. Mechanism of action, structure-activity relationships, and drug delivery. Curr Med Chem. 2000;7(2):249–271. doi: 10.2174/0929867003375335. [DOI] [PubMed] [Google Scholar]
- 116.Mosby's Medical Dictionary. Elsevier; 2009. Triiodothyronine. [Google Scholar]
- 117.Mitchell J, Lazarenko G. Wide QRS complex tachycardia. Diagnosis: Supraventricular tachycardia with aberrant conduction; intravenous (IV) adenosine. CJEM. 2008;10(6):572–573. 581. [PubMed] [Google Scholar]
- 118.Mizutani H, Tada-Oikawa S, Hiraku Y, Kojima M, Kawanishi S. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci. 2005;76(13):1439–1453. doi: 10.1016/j.lfs.2004.05.040. [DOI] [PubMed] [Google Scholar]
- 119.Ekert PG, Vaux DL. Apoptosis and the immune system. Br Med Bull. 1997;53(3):591–603. doi: 10.1093/oxfordjournals.bmb.a011632. [DOI] [PubMed] [Google Scholar]
- 120.Roosild TP, Castronovo S. Active site conformational dynamics in human uridine phosphorylase 1. PLoS One. 2010;5(9):e12741. doi: 10.1371/journal.pone.0012741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.