

Abstract
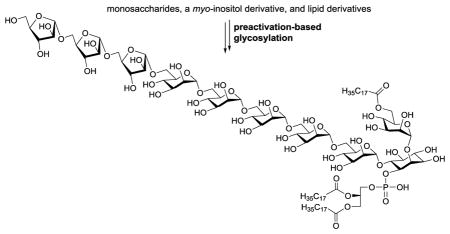
An analog of Mycobacterium tuberculosis lipoarabinomannan (LAM) has been synthesized containing the characteristic structures of all of its three major components; that is, a mannosylated phosphatidylinositol moiety, an oligomannan and an oligoarabinan. A highly convergent strategy was developed that is applicable to the synthesis of other LAM analogs. The synthetic miniature LAM should be useful for various biological studies.
Tuberculosis (TB) claims more than two million lives each year. Despite the enormous endeavor to combat this epidemic, there is still a trend of TB resurgence recently.1 Consequently, developing new TB therapies, in particular effective TB vaccines, has become an urgent topic.2,3 For this purpose, lipoarabinomannan (LAM), one of the major lipopolysaccharides in the cell envelope of Mycobacterium tuberculosis (Mtb), the causative pathogen of TB, and other mycobacteria as well, has attracted significant attention, as LAM is not only essential for mycobacterial growth and cell viability but also a major virulence factor that plays a critical role in bacterium interactions with the host immune system.4–8 Furthermore, LAMs have been revealed to be exposed on the bacterial cell surfaces,9 rendering them an ideal target for TB vaccine development.10
Although structurally diverse, LAMs share a conserved construct having a phospholipid, a lipidated mannose, and a complex arabinomannan polysaccharide attached to the myo-inositol 1-O-, 2-O- and 6-O-positions, respectively, as shown in Figure 1.4,11 In turn, arabinomannan consists of an inositol-attached mannan with an α-1,6-linked backbone and an arabinan with an α-1,5-linked backbone having 3-O-branches. There are additional mannose units randomly attached to the mannan 2-O-positions. Moreover, in some mycobacterial strains, there are short oligomannose caps at the arabinan non-reducing end to form mannosylated LAMs.12,13 It is interesting to observe that arabinomannans derived from some fast-growing Mycobacterium species were devoid of the mannose cap.12
Figure 1.

The structures of LAM and a LAM analog 1
Owing to its intriguing structure and bioactivity, LAM has become a popular subject for synthetic studies. As a result, a variety of LAM fragments or partial structures, such as arabinomannans, phosphatidylinositol mannosides (PIMs) and lipomannans (LMs), have been prepared and studied.14–37 However, to the best of our knowledge, there has been no reported synthesis of LAM analogs containing all its three major components, namely, mannan, arabinan and phosphatidylinositol. In view of the great potential of this type of LAM analog for studying the biological and immunological functions of LAMs, for developing LAM-based vaccines, and so on,5,8,10 we designed and prepared a miniature LAM 1 (Figure 1), which had a phospholipid, a lipidated mannose, and a short arabinomannan attached to the inositol 1-O-, 2-O-, and 6-O-positions, respectively. In LAMs, the linkage of arabinan to mannan has not been unequivocally proven, but it is most likely at the mannose 2-O-position.4 In our synthetic target, arabinan is attached to the alternative 6-O-position. Nevertheless, as discussed below, the two types of LAM analogs can be prepared by the same generally applicable synthetic strategy.
Our synthetic plan for the target molecule 1 is depicted in Scheme 1. Since 1 contained acyl lipids in its structure, benzyl (Bn) ethers would be utilized as global protecting groups for the hydroxyls (2), which could potentially be readily deprotected without affecting the lipids. The lipids would be installed in the final stages, just before global deprotection, leading to a key intermediate 3 that had the 1-O-position of inositol and the 6-O-position of inositol 2-O-position-linked mannose orthorgonally protected with the p-methoxybenzyl (PMB) and tert-butyldimethylsilyl (TBS) groups, respectively. For 3, all of the 2-O-positions in the oligoarabinomannan moiety would be protected as acetates to ensure α-specific glycosylation reactions during the oligosaccharide assembly, owing to neighboring group participation. Disconnecting the glycosidic bond between the first and second mannose residues in 3 generated the heptasaccharide 5 and the pseudotrisaccharide 6. Notably, this synthetic design would entail attachment of an oligomannosyl donor to a relatively reactive primary alcohol by an α linkage that could be relatively easily and effectively realized. Orthogonally protected 6 was a rather versatile intermediate useful for the synthesis of various PIM, LM and LAM analogs and related structures, as demonstrated in our previous synthesis of a LM derivative.38 On the other hand, 5 could be assembled from 8–11, all of which were thioglycosides that would enable preactivation-based glycosylation reactions and one-pot synthesis. Moreover, it was anticipated that this synthetic strategy would be also applicable to LAM analogs having arabinans linked to the mannose 2-O-position, starting from a mannose derivative with an uniquely protected 2-O-position, instead of 10.
Scheme 1.

Retrosynthesis of the target molecule 1
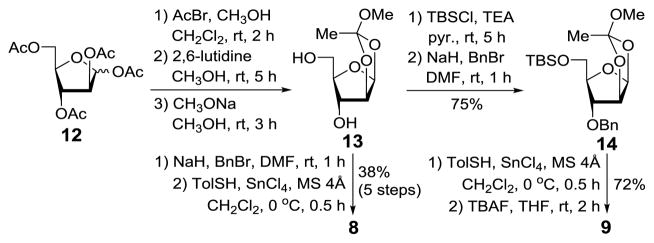
Monosaccharide building blocks 8 and 9 were prepared from peracetylated D-arabinofuranose 12 (Scheme 2).39 Bromination of 12, followed by intramolecular cyclization in the presence of 2,6-lutidine and deacetylation, afforded orthoester 13. Perbenzylation of 13 and SnCl4-promoted glycosylation with p-thiocresol (TolSH), via ring-opening of the orthoester, produced 8, which was obtained from 12 in five steps in a 38% overall yield. En route to 9, the two free hydroxyl groups in 13 were differentiated after regioselective silylation of the 5-OH group using TBSCl and benzylation of the 3-OH group to give 14. Finally, 14 was transformed into 9 after glycosylation with TolSH and SnCl4, as described above, and desilylation mediated by tetra-n-butylammonium fluoride (TBAF).
Scheme 2.
The synthesis of 6 and 7
Tetramannose 7 was convergently assembled by means of the preactivation-based glycosylation protocol (Scheme 3).40 After 1038 was activated at low temperature (−78 °C) with p-toluenesulfenyl triflate (TolSOTf) produced in situ from the reaction of p-toluenesulfenyl chloride (TolSCl) and silver triflate (AgOTf), 11 was added to achieve the glycosylation. This reaction was α-stereoselective, giving the disaccharide 15 in excellent yield (85%). Treatment of 15 with Et3N·3HF to remove the TBS group gave alcohol 16. Its glycosylation with 15 was achieved by the same preactivation protocol to give tetrasaccharide 17, of which all of the glycosidic linkages had α configuration, proved by the observed anomeric C-H coupling constants, which ranged from 169 to 175 Hz.41 Consecutively, the TBS protection was removed with Et3N·3HF to produce 7 as a glycosyl acceptor for further sugar chain elongation.
Scheme 3.

The synthesis of tetrasaccharide 7
Heptasaccharide 5 was constructed from monosaccharides 8 and 9 and tetrasaccharide 7 (Scheme 4) according to the preactivation-based iterative one-pot glycosylation protocol.40 Although this protocol has been quite broadly utilized in the synthesis of pyranosidic oligosaccharides, there are relatively few reports related to its application to furanosyl oligosaccharides.26,28 For each glycosylation, the thioglycosyl donor was first preactivated at −78 °C for 10 min with in situ generated TolSOTf as the promoter, and the coupling reaction was carried out at room temperature for ca. 20 min after the addition of a glycosyl acceptor in conjunction with 2,4,6-tri-tert-butylpyrimidine (TTBP), a sterically hindered base that was employed as a scavenger for trifluoromethanesulfonic acid formed from the reaction. Stoichiometric amount (1.0 equiv.) of TolSOTf and 0.9 equiv. of an acceptor (relative to the donor) were applied to each glycosylation to guarantee complete consumption of the acceptor, so as to minimize potential interference with the following reactions. Eventually, 5 was isolated in a 41% overall yield, suggesting an average of 75% yield for each glycosylation reaction. As a result of neighboring group participation, the glycosylation reactions were α-selective, since the anomeric carbon signals of all three newly formed arabinosyl linkages appeared at over 106.0 ppm, proving α-configuration.42,43 Compared to traditional methods utilized to prepare oligosaccharides, the use of preactivation glycosylation saved several steps in the glycosyl donor manipulation, e.g., anomeric deprotection and activation, and enabled one-pot synthesis to decrease the number of laborious column purification operations. Both helped to improve the overall synthetic efficiency. Our previous experience suggested that when complex oligosaccharides were used as glycosyl donors Schmidt glycosylation gave better results than thioglycosides.38 We therefore converted 5 into the trichloroacetimidate 18 following N-iodosuccinimide (NIS)-promoted hydrolysis of the thioglycoside and reaction of resulting hemiacetal with trichloroacetonitrile and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). Finally, 638 was glycosylated with 18 and trimethylsilyl triflate (TMSOTf), affording glycosylated inositol 3 stereoselectively in very good yield (79%). This result was especially impressive, considering the complex glycosyl donor and acceptor involved in the reaction. As planned, participation of the neighboring acetyl group in 18 assisted the glycosylation reaction with the relatively reactive primary alcohol of glycosyl acceptor 6. The structure of 3 was confirmed by MS and NMR spectra. For example, the 13C NMR (150 MHz, in CDCl3) spectrum of 3 displayed eight distinctive anomeric carbon signals at δ 106.2 (α-Ara), 106.0 (α-Ara), 106.0 (α-Ara), 98.6 (α-Man), 98.3 (α-Man), 98.26 (α-Man), 98.21 (α-Man), 98.1 (α-Man) and 97.9 (α-Man), with anomeric 1JC,H values for the mannosyl units ranging from 172 to 176 Hz.
Scheme 4.

The synthesis of glycosylated inositol 3
The endgame of this synthesis involved swapping the acetyl groups in 3 for benzyl protecting groups to allow regioselective installation of the lipids and complete global deprotection to give the target molecule (Scheme 5). First, 3 was deacetylated with sodium methoxide (NaOMe) in methanol (MeOH). This was followed by O-benzylation with BnBr and tetrabutylammonium iodide (TBAI). Next, the TBS group in the resultant 19 was selectively removed with Et3N·3HF, which paved the way for acylation of the 6-O-position with stearic acid using N,N′-dicyclohexyl-carbodiimide (DCC) as the condensation reagent to afford 21. Then, the inositol 1-O-position was phosphoglycero-lipidated after selective removal of the PMB group in 21. In view of the fact that the furanosyl arabinosides are rather acid labile,18 we treated 21 with very diluted (2%) trifluoroacetic acid (TFA) solution in dichloromethane (DCM) for a short period (6 h) for PMB group removal, which gave a 54% yield of 22 together with recovery of a significant amount of the starting material 21 (ca. 29%). Phosphoglycerolipidation of 22 by the two-step one-pot phosphoramidite method was smooth, upon reaction with freshly prepared 4 in the presence of 1H-tetrazole and in situ oxidation using meta-chloro-peroxybenzoic acid (m-CPBA), generating 2 (70%) as a diastereomeric mixture (1:1) due to the stereogenic phosphorus atom. Finally, global debenzylation of 2 was achieved under a hydrogen atmosphere using 10% Pd/C as the catalyst in a mixture of chloroform, methanol, and water (3:3:1) to obtain the synthetic target 1, which was confirmed by 1H and 31P NMR spectrometry and MALDI-TOF MS.
Scheme 5.

The final assembly of the LAM analog 1
In summary, we have achieved the first LAM mimic 1 that contained all of the three main components of LAMs by a highly convergent synthetic strategy. As a miniature LAM having homogeneous and defined structure, 1 should be useful for various biological and immunological studies of LAMs and for the development of LAM-based TB vaccines. We are currently working on the synthesis of a series of LAM analogs by the strategy described here and are using them to probe the structure-immunological activity relationships of LAMs.
Supplementary Material
Acknowledgments
This work was supported in part by National Major Scientific and Technological Special Project for New Drugs Development (2012ZX09502001) and National High Technology Research and Development (863) Program of China (2012AA021504) and the National Institutes of Health (R01 GM090270).
Footnotes
Supporting Information Available. Experimental procedures, 1H, 13C and 31P NMR spectra of the synthetic intermediates and the final products were available free of charge from the website at http://pubs.acs.org.
References
- 1.Raviglione MC, Smith IM. N Engl J Med. 2007;356:656. doi: 10.1056/NEJMp068273. [DOI] [PubMed] [Google Scholar]
- 2.Russell DG. Nat Rev Mol Cell Biol. 2001;2:569. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 3.Sacchettini JC, Rubin EJ, Freundlich JS. Nat Rev Microbiol. 2008;6:41. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- 4.Brennan PJ, Nikaido H. Annu Rev Biochem. 1995;64:29. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 5.Vercellone A, Nigou J, Puzo G. Front Biosci. 1998;3:E149. doi: 10.2741/a372. [DOI] [PubMed] [Google Scholar]
- 6.Koul A, Herget T, Klebl B, Ullrich A. Nat Rev Microbiol. 2004;2:189. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 7.Russell DG. Nat Rev Microbiol. 2007;5:39. doi: 10.1038/nrmicro1538. [DOI] [PubMed] [Google Scholar]
- 8.Briken V, Porcelli SA, Besra GS, Kremer L. Mol Microbiol. 2004;53:391. doi: 10.1111/j.1365-2958.2004.04183.x. [DOI] [PubMed] [Google Scholar]
- 9.Pitarque S, Larrouy-Maumus G, Payre B, Jackson M, Puzo G, Nigou J. Tuberculosis. 2008;88:560. doi: 10.1016/j.tube.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flynn JL. Tuberculosis. 2004;84:93. doi: 10.1016/j.tube.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Nigou J, Gilleron M, Puzo G. Biochimie. 2003;85:153. doi: 10.1016/s0300-9084(03)00048-8. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee D, Lowell K, Rivoire B, McNeil MR, Brennan PJ. J Biol Chem. 1992;267:6234. [PubMed] [Google Scholar]
- 13.Khoo KH, Tang JB, Chatterjee D. J Biol Chem. 2001;276:3863. doi: 10.1074/jbc.M004010200. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Stocker BL, Seeberger PH. J Am Chem Soc. 2006;128:3638. doi: 10.1021/ja0565368. [DOI] [PubMed] [Google Scholar]
- 15.Boonyarattanakalin S, Liu X, Michieletti M, Lepenies B, Seeberger PH. J Am Chem Soc. 2008;130:16791. doi: 10.1021/ja806283e. [DOI] [PubMed] [Google Scholar]
- 16.Jayaprakash KN, Lu J, Fraser-Reid B. Angew Chem, Int Ed. 2005;44:5894. doi: 10.1002/anie.200500505. [DOI] [PubMed] [Google Scholar]
- 17.Fraser-Reid B, Chaudhuri SR, Jayaprakash KN, Lu J, Ramarnurty CVS. J Org Chem. 2008;73:9732. doi: 10.1021/jo802000p. [DOI] [PubMed] [Google Scholar]
- 18.Fraser-Reid B, Lu J, Jayaprakash KN, Lopez JC. Tetrahedron: Asymmetry. 2006;17:2449. [Google Scholar]
- 19.Ainge GD, Compton BJ, Hayman CM, Martin WJ, Toms SM, Larsen DS, Harper JL, Painter GF. J Org Chem. 2011;76:4941. doi: 10.1021/jo200588u. [DOI] [PubMed] [Google Scholar]
- 20.Ainge GD, Parlane NA, Denis M, Hayman CM, Larsen DS, Painter GF. Bioorg Med Chem. 2006;14:7615. doi: 10.1016/j.bmc.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Ainge GD, Hudson J, Larsen DS, Painter GF, Gill GS, Harper JL. Bioorg Med Chem. 2006;14:5632. doi: 10.1016/j.bmc.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 22.Joe M, Bai Y, Nacario RC, Lowary TL. J Am Chem Soc. 2007;129:9885. doi: 10.1021/ja072892+. [DOI] [PubMed] [Google Scholar]
- 23.Stadelmaier A, Biskup MB, Schmidt RR. Eur J Org Chem. 2004:3292. [Google Scholar]
- 24.Ainge GD, Parlane NA, Denis M, Dyer BS, Harer A, Hayman CM, Larsen DS, Painter GF. J Org Chem. 2007;72:5291. doi: 10.1021/jo070639m. [DOI] [PubMed] [Google Scholar]
- 25.Dyer BS, Jones JD, Ainge GD, Denis M, Larsen DS, Painter GF. J Org Chem. 2007;72:3282. doi: 10.1021/jo0625599. [DOI] [PubMed] [Google Scholar]
- 26.Wang HR, Ning J. J Org Chem. 2003;68:2521. doi: 10.1021/jo026325a. [DOI] [PubMed] [Google Scholar]
- 27.Hölemann A, Stocker BL, Seeberger PH. J Org Chem. 2006;71:8071. doi: 10.1021/jo061233x. [DOI] [PubMed] [Google Scholar]
- 28.Deng LM, Liu X, Liang XY, Yang JS. J Org Chem. 2012;77:3025. doi: 10.1021/jo300084g. [DOI] [PubMed] [Google Scholar]
- 29.D’Souza FW, Ayers JD, McCarren PR, Lowary TL. J Am Chem Soc. 2000;122:1251. [Google Scholar]
- 30.Lu J, Fraser-Reid B. Chem Commun. 2005:862. doi: 10.1039/b413694b. [DOI] [PubMed] [Google Scholar]
- 31.Watanabe Y, Yamamoto T, Ozaki S. J Org Chem. 1996;61:14. [Google Scholar]
- 32.Ishiwata A, Akao H, Ito Y. Org Lett. 2006;8:5525. doi: 10.1021/ol062198j. [DOI] [PubMed] [Google Scholar]
- 33.Mereyala HB, Hotha S, Gurjar MK. Chem Commun. 1998:685. [Google Scholar]
- 34.D’Souza FW, Lowary TL. Org Lett. 2000;2:1493. doi: 10.1021/ol005907g. [DOI] [PubMed] [Google Scholar]
- 35.Cao B, Williams SJ. Nat Prod Rep. 2010;27:919. doi: 10.1039/c000604a. [DOI] [PubMed] [Google Scholar]
- 36.Patil PS, Hung SC. Org Lett. 2010;12:2618. doi: 10.1021/ol1008137. [DOI] [PubMed] [Google Scholar]
- 37.Patil PS, Hung SC. Chem Eur J. 2009;15:1091. doi: 10.1002/chem.200802189. [DOI] [PubMed] [Google Scholar]
- 38.Jian G, Guo Z. J Org Chem. 2013;78:12717. doi: 10.1021/jo4021979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kam BL, Barascut JL, Imbach JL. Carbohydr Res. 1979;69:135. [Google Scholar]
- 40.Huang X, Huang L, Wang H, Ye XS. Angew Chem, Int Ed. 2004;43:5221. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 41.Podlasek CA, Wu J, Stripe WA, Bondo PB, Serianni AS. J Am Chem Soc. 1995;117:8635. [Google Scholar]
- 42.Mizutani K, Kasai R, Nakamura M, Tanaka O, Matsuura H. Carbohydr Res. 1989;185:27. [Google Scholar]
- 43.Yin H, D’Souza FW, Lowary TL. J Org Chem. 2002;67:892. doi: 10.1021/jo010910e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.