

Abstract
The purpose of this study was to determine the system-wide consequences of deficiencies in two essential micronutrients, vitamin C and E, on the proteome using zebrafish (Danio rerio) as one of the few vertebrate models that similar to humans cannot synthesize vitamin C. We describe a label-free proteomics workflow to detect changes in protein abundance estimates dependent on vitamin regimes. We used ion mobility-enhanced data-independent tandem mass spectrometry to determine differential regulation of proteins in response to low dietary levels of vitamin C with or without vitamin E. The detection limit of the method was as low as 20 amol and the dynamic range was 5 orders of magnitude for the protein level estimates. Based on the quantitative changes obtained, we built a network of protein interactions that reflects the whole organism’s response to vitamin C deficiency. The proteomics-driven study revealed that in vitamin E deficient fish, vitamin C deficiency is associated with induction of stress response, astrogliosis, and a shift from glycolysis to glutaminolysis as an alternative mechanism to satisfy cellular energy requirements.
Keywords: Vitamin C, ascorbic acid, ion mobility, MSE, label free quantification, zebrafish, glycolysis, glutaminolysis
INTRODUCTION
The purpose of this study was to determine the consequences of deficiencies in dietary antioxidants, vitamin C and E, on the zebrafish proteome composition. The combination of deficiencies was used as this dietary manipulation is known to cause severe abnormalities1,2. Protein abundance estimates were obtained by label-free accurate quantification based on data-independent (MSE) acquisition. Functional interpretation of the systems-wide proteome responses revealed the biomolecular networks affected by vitamins C and E deficiency.
In zebrafish, the lack of vitamin E potentiates vitamin C deficiency and causes degenerative myopathy3. The classical roles of Vitamin C (ascorbic acid, AA) are in maintaining redox homeostasis4–6 and facilitating the recycling of vitamin E7,8. More recently, vitamin C has been identified as essential cofactor for multiple enzymes involved in the hydroxylation of procollagen9,10 and several dioxygenases that are involved in maintaining endothelial function with relevance to atherosclerosis11 and sepsis12,13. Inadequate vitamin C levels, potentially leading to hypovitaminosis, remain widespread health concerns in humans affecting particularly underprivileged populations. Recent research indicates that smoking14 and Western diets rich in fat and cholesterol15 exacerbate vitamin C deficiency.
In most animals, vitamin C is synthesized in the liver or kidneys and transferred to tissues via circulation16,17. Humans are not capable of synthesizing ascorbic acid due to the lack of the functional gene for L-gulonolactone oxidase18,19. For studying the effect of vitamin C deficiency, the most widely used animal model is the guinea pig that similarly to humans cannot synthesize vitamin C20. The gulonolactone oxidase knock-out (Gulo−/−) mouse is an established transgenic animal model for deciphering the physiological roles of vitamin C21,22. Similarly to humans, zebrafish also cannot synthesize vitamin C23–28. A first report using MS-based metabolomics to study the effect of vitamin C deficiency in zebrafish revealed that vitamin C deficiency affects the purine metabolism29. A classical antioxidant function of vitamin C entails the recycling of the tocopheroxyl radical that is generated when vitamin E (α-tocopherol, α-T) functions as a peroxyl radical, chain-breaking antioxidant30,31. In order to evaluate the interrelating roles of vitamin C and E we used a 2 × 2 design in the present study resulting in four dietary groups, namely vitamin E and C adequate (E+C+), vitamin E and C inadequate (E−C−), vitamin C adequate but inadequate in vitamin E (E−C+) and vitamin C inadequate but adequate in vitamin E (E+C−). We subsequently applied a label-free comparative proteomics approach for examining the systems-wide consequences of the different vitamin regimes in adult zebrafish.
In a typical “bottom up” shotgun experiment, a protein mixture is proteolytically digested with a protease, such as trypsin, to create peptides for further analysis. To both qualitatively and quantitatively analyze a complex mixture of peptides, liquid chromatography in combination with tandem mass spectrometry (LC-MS/MS) has become the predominantly used technique 32–34. The peptides are chromatographically separated using reversed-phase (RP) chromatography and subsequently subjected to collision-induced dissociation (CID) in a tandem mass spectrometer. The product ion spectra are searched against protein sequence databases to identify the peptides. Unlike comparative isotope-coded labeling methods (e.g. ICAT or iTRAQ), label-free mass spectrometry-based quantitative proteomic approaches are based on the fact that in electrospray ionization (ESI) the peak signal intensity is linearly proportional to the concentration of a peptide (over an approximate range of 5–5000 fmol)35,36. Currently two acquisition techniques have been technically realized and applied for detecting and quantifying peptides, namely data-dependent acquisitions (DDA) and data independent acquisitions (DIA).
Data-dependent acquisitions are based on tracking of precursor ion intensities on-the-go37. The peptide precursor ions that fulfill pre-selected features are sequentially subjected to MS/MS during the time course of the chromatography. Proteolytic digests in proteomics-type workflow are often complex and even when using ultra-performance liquid chromatography (UPLC) at every moment multiple peptides coelute. In DDA techniques, survey scan and MS/MS scans are used sequentially and compete for duty cycle time. Therefore, both the number of the most intense peaks subjected to MS/MS analysis and the timespan of a survey scan need to be optimized in order to acquire accurate mass information for the peptide precursor ions and to assure information-rich fragment ion spectra38,39. Albeit somewhat dependent on the mass spectrometry platform used, a possible caveat of DDAs is that a substantial portion of the acquisition cycle is spent acquiring fragment ion spectra; therefore, the intensities of the peptide ions are measured inaccurately and, consequently, are less suited for estimating peptide abundances39.
Data-independent acquisition (DIA) techniques perform parallel fragmentation of precursor ions meaning that all peptide precursors are fragmented simultaneously regardless of their characteristics. For proteomic-type applications, currently most DIAs are realized by coupling nanoUPLC systems to high resolution quadrupole time-of-flight (Q-TOF) mass spectrometers. This combination of equipment takes advantage of the high peak and separation capacity of UPLCs and the measurement of precursor and product ions with accurate mass. In the DIA mode, the Q-TOF mass spectrometer conducts a low energy MS scan providing access to precursor ion mass and intensity data for quantitation and an elevated energy scan to obtain product ion information. A critical post acquisition step of the DIA technique is the alignment of the fragment ions with the respective precursor ion. DIA-based acquisitions enable quantification based on the peptide ion intensities. The estimation of protein abundance is based on the observation that the average signal intensity of the three most intense tryptic peptides relates to the protein abundance level regardless of protein size40. The estimation of protein levels (moles of each identified protein) is based on using an internal protein standard (spiked in at a defined amount).
Recently, a commercially available hybrid high resolution ion mobility quadrupole time-of-flight (IM-Q-TOF) mass spectrometry system that enables the acquisition of ion mobility-enhanced DIA (aka IM-MSE) data has become available41. In this instrument configuration, ions are separated according to their velocity with which they transverse the traveling wave ion mobility device. Parameters that affect the ion’s mobility are its charge, size and shape42. In ion mobility-enhanced MSE acquisitions, ions are separated according to their mobility and then dependent on the energy regime applied in the transfer region directly passed on to the TOF analyzer or collisionally dissociated and the resulting fragment ions are measured in the TOF analyzer. In the IM-MSE mode fragment ions are drift time-aligned with their precursors. An often touted advantage of ion mobility-enhanced MSE acquisitions is the orthogonal separation space gained additionally to the chromatographic separation of the peptides prior to collision induced dissociation leading to spectral decongestion, improved peptide detection and dynamic range, all leading cooperatively to more protein identifications43.
To investigate the consequences on protein networks of the combination of vitamin E and C deficiency in zebrafish, we profiled the changes in protein abundances. The observed proteome changes were then statistically evaluated and functionally annotated. The protein expression data were used for constructing a protein interaction network. Subnetworks were extracted that comprised proteins related to stress response, glycolysis and TCA cycles. The observed changes in protein abundances for zebrafish deficient in vitamins E and C suggest a metabolic switch from glycolysis to glutaminolysis as a means of alternative energy production.
MATERIALS AND METHODS
Chemicals
Dithiolthreitol (DTT) and iodoacetamide were purchased from Bio-Rad (Hercules, CA). MS-grade water, acetonitrile and formic acid (99%) were obtained from EMD Chemicals (Gibbstown, NJ). Sequencing-grade trypsin, resuspension buffer and protease MAX solution were purchased from Promega (Madison, WI). [Glu1]-fibrinopeptide B ([Glu1]-Fib) and Saccharomyces cerevisiae enolase digest were obtained from Waters (Milford, MA). PBS solution was purchased from Fisher (Fair Lawn, NJ).
Fish Husbandry
Housing of the wild-type tropical 5D strain zebrafish was carried out in the Sinnhuber Aquatic Research Laboratory at Oregon State University, Corvallis, Oregon. The study was performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC)44.
Feeding Experiment
The experiment was designed to allow analyzing changes to the proteome upon vitamin C supplementation of fish that were vitamin E adequate or deficient. To do that, fish were fed with predefined diets as previously described29. Briefly, at 42 day of age, 40 fish were divided in two groups and fed diets low in vitamin C (C−, 50 mg AA/kg diet, added as Stay-C®, DSM Nutritional Products, Parsippany, NJ) with vitamin E at sufficient levels (E+, 178 μmol RRR-α-tocopherol/kg diet) or deficient levels (E−, 22 μmol RRR-α-tocopherol/kg diet). Thus, two vitamin groups were created: E+C− and E−C− with 20 fish in each group. Induction of vitamin C deficiency continued for 56 days, after which half of the fish population in each group was harvested. The diet of remaining fish was altered to have a high amount of vitamin C (C+, 350 mg AA/kg diet, added as Stay-C®), thus creating another two vitamin groups: E+C+ and E−C+. After an additional 21 days, the fish were harvested. Thus, four groups with 10 fish in each were created, each having different vitamins levels that varied with supplementation: E+C+, E−C+, E+C− and E−C− (Supporting Information, Figure S1). Vitamin E and C concentrations have been published29.
Protein Extraction and Digestion
Each sample consisted of one whole flash-frozen fish. Whole flash-frozen fish was individually ground under liquid nitrogen using a mortar and pestle. The pipetting scheme used for preparing the protein extracts and digests for each sample is provided in the Supporting Information, Table S1. While still dry, the fish powder was transferred to a microcentrifuge tube and weighed (Supporting Information, Table S1, column 2). A PBS solution with ProteaseMAX (0.04%) was added to the micro tubes for the extraction of proteins; assuming the density as 1 mg/mL, its volume was adjusted to 6 times the fish weight, (Supporting Information, Table S1, column 5). The tubes containing the fish powder suspensions were submitted to three freeze-thaw cycles that included flash-freezing in liquid nitrogen for 2 minutes, thawing for 5 minutes, and sonicating for 15 minutes. After lysis, tubes were centrifuged at 4 °C for 10 minutes at 15,000 relative centrifugal force (rcf) and the supernatants were transferred to new microcentrifuge tubes (1.3 mL) while solid residue was discarded. The sample preparation workflow is shown in Supporting Information, Figure S2.
The concentration of proteins in each sample was determined by a photometric (Bradford) assay (Supporting Information, Table S1, column 6). Proteins in each sample were digested according to the manufacturer’s protocol (Promega Inc.). The pipetting scheme for preparing the digest of each sample is outlined in Supplementary Information, Table S1, columns 7,8, and 9. Briefly, a volume of solution containing 50 μg of proteins was taken to 93.5 μL with a freshly prepared 50 mM NH4HCO3. Next, 1 μL 0.5 M DTT (in Millipore water) was added to each vial and solutions were incubated at 56 °C for 20 min. Then, 2.7 μL 0.55 M iodoacetamide (in 50 mM NH4HCO3) was added and solutions were incubated at room temperature in the dark for 15 minutes. For protein digestion, 1 μL 1% ProteaseMAX Surfactant in (in 50 mM NH4HCO3) and 1.8 μL of trypsin (1 μg/μL in 50 mM acetic acid) were added. Solutions were incubated at 37 °C for 3 hours. The final volume of each solution of digested proteins was 100 μL. Solutions were centrifuged at 12,000 rcf for 10 seconds and trifluoroacetic acid (TFA) was added at a final concentration of 0.5%. After snap-freezing in liquid nitrogen, samples were stored at −20 °C until analyzed. At the end, we conducted LC-IM-MSE analyses of 8, 10, 10, and 9 samples of groups E+C+, E−C+, E+C− and E−C−, respectively (Supplementary information, Table S1).
Liquid Chromatography - Mass Spectrometry
The analysis of all samples was performed using a Synapt G2 hybrid quadrupole time-of-flight mass spectrometer (Waters, Milford, MA) controlled by MassLynx 4.2 software (Waters). The sample peptide solution (9 μL) was mixed with 1 μL internal standard (Saccharomyces cerevisiae enolase digest, 1 pmol/μL). With 1 μL injection volume the amount of the internal standard per injection on column was 100 fmol. Peptides were separated with a nanoAcquity Ultra Performance LC system (Waters, Milford, MA) equipped with 100 μm × 100 mm BEH130 C18 column with a particle size of 1.7 μm (Waters, Milford, MA). The mobile phase A consisted of 0.1% formic acid in water, and B consisted of 0.1% formic acid in acetonitrile. Each sample was first retained on a trapping column and then washed using 99.5% A for 3 min at a flow rate 5 μL/min. Peptides were separated using a 120-min gradient (3–40% B for 90 min, 40–90% for 2 min, 90% B for 1 min, 90-3% B for 2 min and 3% B for 25 min) and then electrosprayed into the mass spectrometer, fitted with a nanoSpray source, at a flow rate of 400 nL/min. External calibration of the TOF analyzer was performed using NaI solution over the range of m/z 50 to 2000. The instrument operated in positive V-mode over the calibration range. Mass spectra were acquired in the MSE mode alternating between a low energy scan (6 eV) to acquire peptide precursor data and a high energy scan (ramping from 27 to 50 eV) to acquire fragmentation data. The capillary voltage was 2.5 kV and the source temperature was 40 °C. Scan time was 1.25 s. The instrument settings are listed in Table S2 (Supporting Information). An auxiliary pump delivered a [Glu1]-Fib solution as an external calibrant (lock-mass) with a concentration of 500 fmol/μL at a rate of 0.2 μL/min. For lock mass acquisition a low energy scan was acquired for 0.75 s every 60 s throughout a run.
Peak Detection
Elevated energy mass spectra were extracted, charge state deconvoluted, deisotoped and lock-mass corrected with [Glu1]-Fib (MH+ m/z 785.8426). All ion mobility-MSE samples were analyzed using IdentityE by ProteinLynx Global Server (PLGS) version 2.5 (Waters Corporation, Milford, MA). The following processing parameters and their respective settings were used: chromatographic peak width and MS TOF resolution, automatic; low energy threshold, 100 counts; elevated energy threshold, 10 counts; intensity threshold, 750 counts.
Protein Database Search
Waters’ IdentityE was configured to search Danio rerio protein database (26812 entries) with the digestion enzyme trypsin. The database was modified by adding the yeast enolase sequence (ENO1_YEAST, accession number P00924, Uniprot) that was spiked in as internal standard to facilitate label-free quantification. IdentityE searched the protein database with a fragment ion mass tolerance of 0.025 Da and a parent ion tolerance of 0.0100 Da. The iodoacetamide derivative of cysteine and the oxidation of methionine were specified as variable modifications of amino acids in IdentityE.
Protein Identification, Quantification and Validation
Scaffold (version 3.3.1, Proteome Software Inc., Portland, OR) was used to validate MSE-based peptide and protein identifications. Peptide probabilities were assigned by the Peptide Prophet algorithm45. Protein probabilities were assigned by the Protein Prophet algorithm 46. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. The peptide and protein probabilities were adjusted to allow the best quantification of proteins and keeping the false identification rates at low levels. For each sample, the numbers of peptides assigned with high confidence are compiled in Table S3 (Supplementary Information). In addition, Supplementary Information Table S5 - Excel file - “Motorykin SI Protein_sequence_coverage” compiles proteins identified, unique peptides assigned per protein and protein sequence coverage. This information was directly obtained from Scaffold.
The average of intensity of the three most intense peptides of each protein was normalized to that of the internal standard with an assigned amount of 100 fmol. Proteins could not be quantified if they ambiguously shared peptides that cannot be uniquely attributed to one protein. Ultimately the information about group name, sample name, protein ID and quantitative values for proteins was acquired from Scaffold and exported to spreadsheets. The data were then imported into Perseus (http://www.perseus-framework.org/) and statistically analyzed. Perseus was developed by the Max Planck Institute of Biochemistry, Martinsried, Germany, and designed to perform all downstream bioinformatics and statistics on proteomics output tables.
Western Blot
Three samples were randomly selected from each group. Samples were prepared for Western Blot analyses by diluting with sample buffer (5% mercaptoethanol in Laemmli buffer, Bio-Rad, Hercules, CA) to a concentration of 1 mg/mL of protein. For SDS-PAGE analysis 20 μg (20μL) of proteins were loaded into a pre-casted 10% gel (Bio-Rad). After electrotransfer to a nitrocellulose (NC) membrane (Bio-Rad), the latter was blocked overnight in 5% non-fat milk in TBS-T (10 mM Tris, pH 8, 150 mM NaCl, 0.05% Tween). The NC paper was blotted for 1 h with antibodies (Thermo, Rockford, IL) against pyruvate kinase M2 (Pkm2), glutamate dehydrogenase (Glud1) and glial fibrillary acidic protein (Gfap). A horseradish peroxidase-labeled anti-goat and anti-rabbit IgG (Bio-Rad) and an enhanced chemoluminescence kit (SuperSignal West Pico Chemiluminescent Substrate, Thermo) were used to detect signals on a film (Kodak, Rochester, NY). The molecular mass of a protein was estimated using a protein molecular standard (Bio-Rad).
Network Construction
To construct the vitamin C deficiency network, protein IDs were uploaded to STRING, Search Tool for the Retrieval of Interacting Genes (www.string-db.org) 47. The data found on protein interactions and the scores information was extracted. The protein network was constructed based on protein interactions using Cytoscape 2.8.2 (The Cytoscape Consortium). The latter is an open source platform for complex network analysis and visualization48.
RESULTS AND DISCUSSION
Overall Design and Strategy Applied for the Label-free Quantitative Proteomic Study
The goal of this study was to investigate the consequences of vitamin E and C deficiency on the zebrafish proteome. The analysis and validation of quantitative bottom-up shotgun proteomics datasets depends critically on the optimal combination of data processing software tools, because each commercially available software package can perform only a few steps of the data analysis pipeline (Figure 1). Here we implemented MassLynx and PLGS for peak extraction, alignment and database search; Scaffold for spectra validation and protein quantitation; Perseus for statistical analysis; STRING and Cytoscape for protein interactions analysis and network construction, respectively.
Figure 1.

Data analysis workflow for label-free quantitative proteomics using LC-IMS-MSE acquisitions. We implemented MassLynx and PLGS for peak extraction, alignment and database search, Scaffold for spectra validation and protein quantitation, Perseus for statistical analysis, and STRING and Cytoscape for protein interactions analysis and network construction.
Effects on Body Weight and Growth Rate
The experimental design of the feeding study afforded that zebrafish of the E+C− and E−C− groups were harvested after 98 days and the E−C+ and E+C+ groups were sacrificed after 119 days. We therefore expected that the different groups will result in different median fish body weights. Indeed, as shown in the Supporting Information, Figure S3A, the median fish weights were lower for E+C− and E−C− groups compared with E−C+ and E+C+ groups. The ANOVA test between all 4 groups did not show significant results, but the median body weights were significantly different between the E+C+ and E−C− groups (p-value=0.006, t-test). Also, not totally unexpected, the growth rate (median body mass/age) of fish adequate in both vitamins (E+C+ group) was approximately 3-times higher compared to the group deficient in both vitamins (E−C− group). Vitamin C deficiency seemed to have a more drastic effect on the growth rate compared to inadequate vitamin E levels (Supporting Information, Figure S3B).
Optimization of Sample Preparation and Analytical Workflow for the Comparative “Bottom up” Proteomics Study
To account for differences in fish body weights we normalized the total protein concentration, which resulted in solutions with equal total concentration of proteins independent of fish weights, age, and vitamin supplementation. The total protein concentration in each sample was verified to confirm the validity of the sample preparation technique and for further use. The median total concentration of proteins for all samples was 2.382 mg/mL with first quartile 2.258 mg/mL and third quartile 2.478 mg/mL. As was expected from the sample preparation that accounted for the differences in fish weights, the total concentration of proteins were distributed in a narrow range (Supporting Information, Figure S4). After lysis, insoluble pellets that remained in the microcentrifuge tubes were discarded. The supernatant was used for protein digestion.
Due to the complexity of the whole fish protein extracts we optimized the LC conditions to increase the number of peptides detected 49. For optimization, a protein extract was run using different LC gradients and the number of eluted peptides was compared. By changing the mobile phase composition over time, the gradient was optimized after each run to make the elution of peptides even across a chromatogram and to reduce the coelution of peptides. The gradients were named as “Gradient 1”, “Gradient 2”, “Gradient 3” and “Gradient 4”. The gradient profiles and respective chromatograms are presented in the Supporting Information, Figure S5. Using Venn diagrams, a comparison of the number of eluted peptides for each gradient is presented in the Supporting Information, Figure S6. “Gradient 4” resulted in the highest number of peptides detected and was used for all following LC-IM-MSE analyses.
Data Analysis Strategy Used for Obtaining Protein Abundance Estimates
Label-free approaches are liable to technical variability, such as LC retention time drift, nanospray instability and sample matrix effects. Therefore, to overcome these caveats label-free approaches depend on the use of an internal standard and the application of proper mathematical tools for data processing. Log transformation of data is commonly used to make the distribution of protein abundances more Gaussian, with subsequent application of parametric statistical tests, which have higher statistical power after data transformation50.
In this study, a digest of enolase from Saccharomyces cerevisiae was used as an internal standard. The same volume of the internal standard was added to each sample, making its concentration equal across samples. Data was acquired, deisotoped, peak aligned and exported to Scaffold for quantitative analysis. The quantification of proteins was done by normalization of the protein abundances to the internal standard. For the total of 2956 quantitative values obtained, a graph of protein amounts of the identified proteins from all samples against protein ranks was created (Figure 2). The value for the detection limit was extracted as the lowest amount of a protein that was calculated by Scaffold. The detection limit of this method was as low as 20 amol (Heat shock protein 8, sample E+C+#5), while the highest detected concentration was 2.3 pmol (Myosin light chain, sample E−C+#8). The dynamic range of estimates of protein amounts was calculated to be ca. 5 orders of magnitude.
Figure 2.
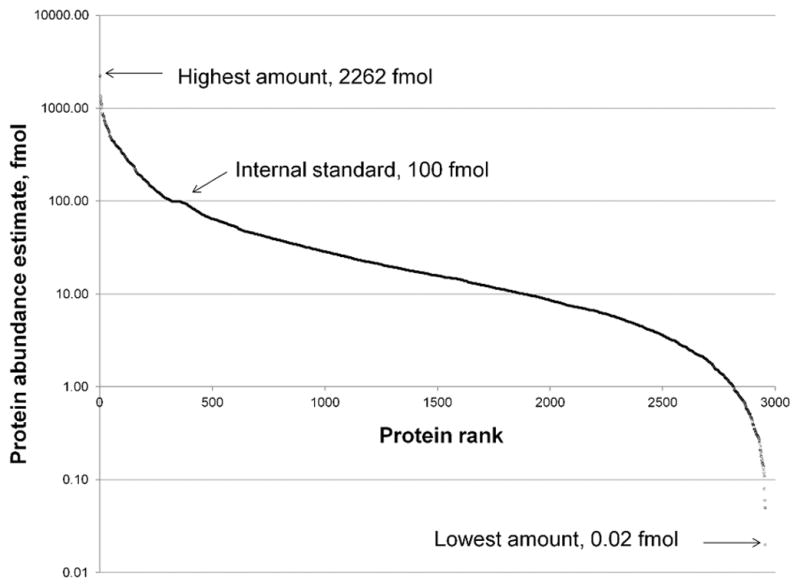
Dynamic range of the method based on protein abundance estimates. The plot has the predicted S-shape and the quantitative dynamic range covers 5 orders of magnitude based on 2956 data points derived from LC-IM-MSE data of all 37 samples. The value for the detection limit was identified as the lowest amount of a protein calculated by Scaffold software. The detection limit of this method was as low as 20 amol (Heat shock protein 8, sample E+C+#5), while the highest detected amount was 2.3 pmol (Myosin light chain, sample E−C+#8).
Next, data were exported to Perseus and log2 transformed. To estimate the biological variation between fish samples, the coefficient of determination, R2, was calculated based on the protein estimates between biological replicates. As an example, Supporting Information, Figure S7 shows the correlation plot between the biological replicates E−C+ #9 and E−C+ #8 resulting in a R2 value of 0.654. Additional correlation plots for the E−C+ group are shown in Supporting Information, Figure S8. As expected, variation between samples was observed. The correlation of protein amounts between different fish (within one feeding group) was found to be stronger in samples containing higher numbers of proteins.
Proteome Changes Linked to Vitamin C Deficiency
To investigate the proteome changes caused by vitamin C deficiency in vitamin E-adequate fish we evaluated the fold changes of the protein estimates obtained for the E+C+ versus the E+C− group. For quantitative analysis, peptide identifications were accepted if they could be established at a greater than 95% probability. Protein identifications were accepted if they could be established at greater than 99.0% probability and contained at least 2 identified peptides. The quantitative values calculated for the comparison of groups C+ vs. C− (both sufficient in vitamin E) were based on 8134 spectra with peptide FDR 0.1% and 203 protein with protein FDR 2.0%. Next, internal standard and contaminants were removed and quantitation filter applied. All proteins that were not detected at least 2 times across a group were filtered out. This conservative approach resulted in 119 proteins. Among them, 61% were up-regulated in vitamin C-deficient fish (72 of 119). A change in concentration of 2-fold or more was observed for 28% of the proteins in this group.
The same quantitative analysis was done to investigate the proteome changes caused by vitamin C deficiency in vitamin E deficient fish. For quantitative analysis peptide identifications were accepted if they could be established at a greater than 50% probability. Protein identifications were accepted if they could be established at greater than 99.9% probability and contained at least 2 identified peptides. The quantitative values calculated for the comparison of the C+ vs C− group (both deficient in vitamin E) were based on 11308 spectra with peptide FDR 0.2% and 221 protein with protein FDR 2.3%. Again, internal standard and contaminants were removed and quantitation filter applied. This resulted in a list of 112 proteins (Supplementary Table S3). Among them, 52% were up-regulated in vitamin C-deficient fish (58 of 112). A change in concentration of 2-fold or more was observed for 29% of the proteins.
Figure 3A shows a volcano plot for the distribution of p-values versus fold-changes calculated for the proteins that were assigned for the comparison of the E+C+ vs. E+C− groups (E+C+/E+C− transition). A negative logarithm of the p-values and the logarithm of the ratio between amount of proteins from E+C+ and E+C− groups are displayed on the y- and x-axes, respectively. Dashed lines show a cut-off of 2-fold change and a p-value threshold of 0.05 to define differential regulation of proteins between groups. Four proteins, namely type II cytokeratin, alpha tropomyosin, myosin heavy polypeptide 6 and fatty acid binding protein 6, showed a significant change upon vitamin C deficiency in vitamin E adequate fish and all were upregulated. Regulation of alpha tropomyosin by α-tocopherol aligns well with previously published data 51.
Figure 3.

Volcano plot representation of protein abundance changes caused by vitamin C deficiency.
Side-by-side comparison of the volcano plots of protein abundance changes reveals the permissive role of vitamin E in vitamin C deficient fish. In the group low in vitamin E the lack of vitamin C causes changes in proteins associated with pathways of energy metabolism (pyruvate kinase and glutamate dehydrogenase) and other protein with roles in stress response.
In the Figure 3B the volcano plot is shown for evaluating differential regulation of proteins in group E−C+ vs. group E−C− (E−C+/E−C− transition). We found that three proteins were down-regulated (pyruvate kinase M2b, fatty acid binding protein 11a and peptidyl-prolyl cis-trans isomerase) and six up-regulated (glial fibrillary acidic protein, ventricular myosin heavy chain, glutamate dehydrogenase 1, heat shock protein 90kDa alpha, LOC100002040 protein and novel myosin family protein CH211-158M24.10-001) as a consequence of vitamin C deficiency in vitamin E deficient fish.
A side-by-side comparison of the two volcano plots reveals the permissive role of vitamin E in the effect of vitamin C deficiency on the fish. In the vitamin E deficient fish the additional lack of vitamin C causes changes in expression levels of proteins responsible for energy metabolism, namely pyruvate kinase and glutamate dehydrogenase, along with other marker proteins. In order to obtain functional insight into proteins and pathways that are affected by vitamin C deficiency we functionally annotated the combined proteomic data sets of the E−C− and E−C+ groups using the Gene Ontology (GO) classification (Supporting Information, Table S4).
The Responsive Protein Interaction Network of Vitamin C Deficiency
A protein interaction network was constructed to provide a comprehensive visualization of the proteins identified, their abundance estimates as well as differential regulation caused by vitamin C deficiency in fish low on vitamin E (Supporting Information, Figure S9). In the network each node represents a protein found in either group E−C+ or E−C−. The change in protein amounts between vitamin C adequate and deficient fish is color coded: green nodes represent proteins that increased their amounts, red represent those that decreased, and grey nodes show proteins that were not quantified. The size of a node represents an average of the protein abundance estimates in the E−C− group. The width of a line connecting proteins represents the strength of proteins interaction, as extracted from STRING. Figure 4 provides a closer look at sub-networks that encompassed proteins that were differentially regulated in response to vitamin E and C deficiency. Upregulated proteins were members of the subnetworks representing glutaminolysis, TCA cycle, and stress response.
Figure 4.

Protein-protein interaction network visualization of sub-networks that contain proteins that showed changes in expression levels upon vitamin C deficiency in the vitamin E deficient state.
In the network each node represents a protein found in either group E−C+ or E−C−. The change in protein amounts between vitamin C adequate and deficient fish is color coded: green nodes represent proteins that increased their amounts, red represent those that decreased, grey nodes show proteins that were not quantified. The size of a node represents an average of the protein abundance estimates in the E+C− group. The width of a line connecting proteins represents the strength of proteins interaction, as extracted from STRING software. Subnetworks of proteins involved in glycolysis and the TCA cycle are highlighted as well as proteins associated with stress response.
Biological Inferences of Altered Protein Expressions in Vitamin E and C Deficient Zebrafish
The interaction network highlights the modulation of several proteins involved in two interconnected metabolic pathways, the glycolysis and TCA cycle (Figure 5). Proteins are shown as upregulated (green arrow up), downregulated (red arrow down), unquantified (gray arrow), and not detected (no arrow) with their respective numerical fold changes (Supporting Information, Table S4). Proteins that showed a statistically significant fold change are marked with an asterisk.
Figure 5.

Schematic of the metabolic pathways that were associated with proteins that showed changes in abundance estimates as a consequence of vitamin C deficiency.
To analyze the consequences of vitamin C deficiency, quantitative data was combined with metabolic pathways linked to energy production, namely glycolysis, TCA cycle and glutaminolysis. Proteins that were upregulated are indicated by a green arrow up, downregulated proteins are marked with a red arrow down, detected but not quantified proteins are marked with a double arrow in gray, and not detected proteins are unmarked. Proteins are labeled with their respective numerical fold-changes observed for vitamin C deficiency (in the vitamin E deficient group). Proteins that showed a statistically significant fold change are marked with an asterisk.
Upon vitamin C deficiency in groups low on vitamin E, glycolytic enzymes were downregulated. The last step in the glycolysis pathway involves a transfer of the phosphate group from phosphoenolpyruvate to ADP, producing a molecule of pyruvate and a molecule of ATP. This step is catalyzed by pyruvate kinase (PK), which was significantly downregulated in vitamin E and C-deficient fish (p-value=0.009, t-test for log transformed data). In humans and other mammals, four isoforms of pyruvate kinase are expressed: the L and R isoforms are found in liver and red blood cells, the M1 isoform is expressed in most adult tissues, and the M2 isoform, a splice variant of M1 and which is the predominant form found in proliferating cells and tumor cells 52,53. In the current study only pyruvate kinase M2 was detected, but not M1 nor L/R. Western Blot analysis confirmed the presence and change of pyruvate kinase M2 (data not shown). Under the conditions of suppressed glycolysis there will be less production of energy (ATP) and reducing power (NADH), the main products of the cycle. We may therefore speculate that in vitamin E and C-deficient fish compensatory mechanisms for the production of energy are activated. Kirkwood et al. studied vitamin C deficient zebrafish at the metabolome level and reported activation of the purine nucleotide cycle as possible compensatory mechanism to satisfy intracellular ATP requirements29.
After conversion of glutamine to glutamate, glutamate dehydrogenase catalyzes the production of α-ketoglutarate, which subsequently enters the TCA cycle. The conversion of glutamine to lactate is commonly referred as glutaminolysis 54,55. Our quantitative proteomic study indicates that several proteins that play key roles in glutaminolysis are elevated in vitamin E and C-deficient fish (E−C− group). Our data sets show a significant increase in levels of glutamate dehydrogenase (Glud1b) under vitamin E and C deficiency compared with E deficiency alone (p-value=0.046, t-test for log transformed data). The change in this protein was confirmed with Western Blot (data not shown). Malate dehydrogenase and lactate dehydrogenase, enzymes involved in glutaminolysis, were increased as well. The elevation of several key enzymes involved in glutaminolysis supports the notion that vitamins E and C deficiency promotes a metabolic phenotype in which glutaminolytic pathways are activated for alternative energy production (ATP) and reducing power (NADPH) in cells under conditions of suppressed glycolysis.
In addition, our quantitative proteomics screens revealed that Glial fibrillary acidic protein (Gfap) was significantly elevated (27 times) in fish deficient in both vitamins (p-value=0.002, t-test for log transformed data), while there was no significant difference in Gfap expression between E+C+ and E−C+ groups consistent with the notion that vitamin E seems to exhibit a permissive role in governing the proteome biology of adult zebrafish. The adverse effect of deficiency of both vitamins has been reported before 2. The identification of Gfap was based on a conservative probability for protein identification of at least 85% and at least 2 unique peptides with peptide identification probability of at least 50%. The similarity between zebrafish and human Gfap (GFAP_HUMAN, entry P14136 in Uniprot) is 65% as calculated by Blast search. Despite the good homology of the human and zebrafish protein the human antibody used in the Western blot analyses showed non-specific binding and Western blots were not conclusive. Gfap is a recognized marker of neurologic injury and trauma and is released during astrogliosis in higher vertebrates 56–58.
CONCLUSIONS
We applied a label-free comparative proteomics approach for examining the systems-wide consequences of insufficient levels of two essential micronutrients, vitamins E and C, in adult zebrafish. The experimental design adopted allowed the study of vitamin C deficiency at the whole organism level. Using a label-free quantitative proteomic workflow it was possible to assess for the first time changes in the vertebrate proteome upon vitamin C deficiency. The label-free quantitative bottom-up strategy with LC-Ion Mobility-MSE is a powerful approach to determine protein abundance estimates in zebrafish. We report sensitivity of the method as low as 20 amol and a dynamic range of 5 orders of magnitude for protein level estimates. Our results indicate the modulation of expression of proteins involved in stress response and metabolic pathways associated with energy production. Our findings suggest that severe vitamin C deficiency potentiated by vitamin E deficiency causes the suppression of glycolysis and the activation of glutaminolysis as an alternative way to fulfill cellular energy requirements. Glial fibrillary acidic protein (Gfap) showed a significant overexpression in fish low in both vitamins C and E, proving that severe vitamin C deficiency in combination with low vitamin E status causes injury of the central nervous system.
Supplementary Material
Acknowledgments
Funding Sources
This study was made possible in part by grants from the National Institutes of Health, P30ES000210, S10RR025628 and R01HD062109.
The authors thank Dr. Cristobal Miranda for assistance in sample preparation.
Footnotes
Author Contributions
The experiments were done by IVM. The manuscript was written by IVM and CSM. The experimental design was developed by MGT, RLT and CSM. The fish were grown by RLT. The manuscript was reviewed by all members before final submission.
Notes
The authors declare no competing financial interest.
Table S1, Pipetting scheme used for sample lysis and preparation of digests; Table S2, Synapt G2 HDMS instrument settings used for Ion-Mobility-MSE acquisitions; Table S3, The number of peptides and proteins assigned in each of the biological samples subjected to LC-IM-MSE analysis; Table S4, Proteins identified in the E−C+ and the E−C− groups, GO annotations and fold-changes observed in the vitamin C deficient fish (E−C−/E−C+ transition). Table S5 (Excel file), Protein Table for Study (A) E+C+ vs E+C− and (B) E−C+ vs E−C−: Compilation of proteins found in each samples, unique peptides identified per protein and respective sequence coverage; Figure S1, Design of the feeding experiment; Figure S2, Workflow used for preparing the samples for the label-free “bottom up” quantitative proteomics study; Figure S3, Fish characteristics dependent on the vitamin regime fed; Figure S4, A boxplot of the distribution of the total protein concentration after extraction and lysis; Figure S5, Gradient profiles with respective base peak chromatograms; Figure S6, Venn’s diagrams comparing the number of peptides eluting from a column and identified with LC-IM-MSE; Figure S7, Correlation plot between E−C+ sample 9 and E−C+ sample 8; Figure S8, Correlation plot matrix of the E−C+ group. Figure S9, The responsive protein network for the vitamin transition C+/C− and both groups deficient in vitamin E status (E−C+ vs E−C+). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Burk RF, Christensen JM, Maguire MJ, Austin LM, Whetsell WO, May JM, Hill KE, Ebner FF. J Nutr. 2006;136:1576–1581. doi: 10.1093/jn/136.6.1576. [DOI] [PubMed] [Google Scholar]
- 2.Hill KE, Montine TJ, Motley AK, Li X, May JM, Burk RF. Am J Clin Nutr. 2003;77:1484–8. doi: 10.1093/ajcn/77.6.1484. [DOI] [PubMed] [Google Scholar]
- 3.Lebold KM, Löhr CV, Barton CL, Miller GW, Labut EM, Tanguay RL, Traber MG. Comp Biochem Physiol C Toxicol Pharmacol. 2013;157:382–9. doi: 10.1016/j.cbpc.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buettner GR, Schafer FQ. Teratology. 2000;62:234. doi: 10.1002/1096-9926(200010)62:4<234::AID-TERA10>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 5.Halliwell B. Free Radic Res. 1999:261–72. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- 6.Kalyanaraman B, Darley-Usmar VM, Wood J, Joseph J, Parthasarathy S. J Biol Chem. 1992;267:6789–95. [PubMed] [Google Scholar]
- 7.Bruno RS, Leonard SW, Atkinson J, Montine TJ, Ramakrishnan R, Bray TM, Traber MG. Free Radic Biol Med. 2006;40:689–97. doi: 10.1016/j.freeradbiomed.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 8.Buettner G. Arch Biochem Biophys. 1993:535–43. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 9.Peterkovsky B, Undenfriend S. Proc Natl Acad Sci U S A. 1965;53:335–42. doi: 10.1073/pnas.53.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myllylä R, Kuutti-Savolainen ER, Kivirikko KI. Biochem Biophys Res Commun. 1978;83:441–448. doi: 10.1016/0006-291x(78)91010-0. [DOI] [PubMed] [Google Scholar]
- 11.Diaz MN, Frei B, Vita JA, Keaney JF. N Engl J Med. 1997;337:408–16. doi: 10.1056/NEJM199708073370607. [DOI] [PubMed] [Google Scholar]
- 12.Wilson JX. Biofactors. 2009;35:5–13. doi: 10.1002/biof.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armour J, Tyml K, Lidington D, Wilson JX. J Appl Physiol. 2001;90:795–803. doi: 10.1152/jappl.2001.90.3.795. [DOI] [PubMed] [Google Scholar]
- 14.Piyathilake CJ, Macaluso M, Hine RJ, Vinter DW, Richards EW, Krumdieck CL. Cancer Epidemiol Biomarkers Prev. 1995;4:751–8. [PubMed] [Google Scholar]
- 15.Frikke-Schmidt H, Tveden-Nyborg P, Birck MM, Lykkesfeldt J. Br J Nutr. 2011;105:54–61. doi: 10.1017/S0007114510003077. [DOI] [PubMed] [Google Scholar]
- 16.Bánhegyi G, Braun L, Csala M, Puskás F, Mandl J. Free Radic Biol Med. 1997;23:793–803. doi: 10.1016/s0891-5849(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 17.Chatterjee IB, Majumder AK, Nandi BK, SN Ann N Y Acad Sci. 1975:24–47. doi: 10.1111/j.1749-6632.1975.tb29266.x. [DOI] [PubMed] [Google Scholar]
- 18.Delanghe JR, Langlois MR, De Buyzere ML, Na N, Ouyang J, Speeckaert MM, Torck Ma. Genes Nutr. 2011;6:341–6. doi: 10.1007/s12263-011-0237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. J Biol Chem. 1994;269:13685–8. [PubMed] [Google Scholar]
- 20.Grollman AP, Lehninger AL. Arch Biochem Biophys. 1957;69:458–467. doi: 10.1016/0003-9861(57)90510-6. [DOI] [PubMed] [Google Scholar]
- 21.Harrison FE, Meredith ME, Dawes SM, Saskowski JL, May JM. Brain Res. 2010;1349:143–52. doi: 10.1016/j.brainres.2010.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Telang S, Clem AL, Eaton JW, Chesney J. Neoplasia. 2007;9:47–56. doi: 10.1593/neo.06664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dabrowski K. Biol Chem Hoppe Seyler. 1990;371:207–14. doi: 10.1515/bchm3.1990.371.1.207. [DOI] [PubMed] [Google Scholar]
- 24.Touhata K, Toyohara H, Mitani T, Kinoshita M, Satou M, Sakaguchi M. Fish Sci. 1995;61:729–730. [Google Scholar]
- 25.Toyohara H, Nakata T, Touhata K, Hashimoto H, Kinoshita M, Sakaguchi M, Nishikimi M, Yagi K, Wakamatsu Y, Ozato K. Biochem Biophys Res Commun. 1996;223:650–3. doi: 10.1006/bbrc.1996.0949. [DOI] [PubMed] [Google Scholar]
- 26.Phromkunthong W, Storch V, Braunbeckw T. J Appl Ichthyol. 1994;10:146–153. [Google Scholar]
- 27.Dabrowski K, Ciereszko A. Experientia. 1996;52:97–100. doi: 10.1007/BF01923351. [DOI] [PubMed] [Google Scholar]
- 28.Drouin G, Godin JR, Pagé B. Curr Genomics. 2011;12:371–8. doi: 10.2174/138920211796429736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirkwood JS, Lebold KM, Miranda CL, Wright CL, Miller GW, Tanguay RL, Barton CL, Traber MG, Stevens JF. J Biol Chem. 2012;287:3833–41. doi: 10.1074/jbc.M111.316018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brigelius-Flohe R, Traber MG. FASEB J. 1999;13:1145–1155. [PubMed] [Google Scholar]
- 31.Traber MG, Atkinson J. Free Radic Biol Med. 2007;43:4–15. doi: 10.1016/j.freeradbiomed.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCormack AL, Schieltz DM, Goode B, Yang S, Barnes G, Drubin D, YJ Anal Chem. 1997;69:767–76. doi: 10.1021/ac960799q. [DOI] [PubMed] [Google Scholar]
- 33.Peng J, Elias JE, Thoreen CC, Licklider LJ, GS J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 34.Washburn MP, Wolters D, Yates JR. Nat Biotechnol. 2001;19:242–7. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 35.Voyksner RD, Lee H. Rapid Commun Mass Spectrom. 1999;13:1427–37. doi: 10.1002/(SICI)1097-0231(19990730)13:14<1427::AID-RCM662>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 36.Purves RW, Gabryelski W, Li L. Rapid Commun Mass Spectrom. 1998;12:695–700. [Google Scholar]
- 37.Croker CG, Pearcy JO, Stahl DC, Moore RE, Keen DA, Lee TD. J Biomol Tech. 2000;11:135–141. [PMC free article] [PubMed] [Google Scholar]
- 38.Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-perez JM, Nicholson JK. Rapid Commun Mass Spectrom. 2006:1989–1994. doi: 10.1002/rcm.2550. [DOI] [PubMed] [Google Scholar]
- 39.Geromanos SJ, Vissers JPC, Silva JC, Dorschel Ca, Li G-Z, Gorenstein MV, Bateman RH, Langridge JI. Proteomics. 2009;9:1683–95. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 40.Silva JC, Denny R, Dorschel C, Gorenstein MV, Li GZ, Richardson K, Wall D, Geromanos SJ. Mol Cell Proteomics. 2006;5:589–607. doi: 10.1074/mcp.M500321-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Ritter S. Chem Eng news. 2007;85:61–65. [Google Scholar]
- 42.Salbo R, Bush MF, Naver H, Campuzano I, Robinson CV, Pettersson I, Jørgensen TJD, Haselmann KF. Rapid Commun Mass Spectrom. 2012;26:1181–93. doi: 10.1002/rcm.6211. [DOI] [PubMed] [Google Scholar]
- 43.Hoaglund-Hyzer CS, Li J, Clemmer DE. Anal Chem. 2000;72:2737–40. doi: 10.1021/ac0000170. [DOI] [PubMed] [Google Scholar]
- 44.Miller GW, Labut EM, Lebold KM, Floeter A, Tanguay RL, Traber MG. J Nutr Biochem. 2011 doi: 10.1016/j.jnutbio.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 46.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 47.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, Von Mering C. Nucleic Acids Res. 2011;39:D561–8. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Righetti PG, Castagna A, Herbert B, Reymond F, Rossier JS. Proteomics. 2003;3:1397–407. doi: 10.1002/pmic.200300472. [DOI] [PubMed] [Google Scholar]
- 50.Ramsey FL, Schafer DW. Cengage Learning. 2012. The Statistical Sleuth: A Course in Methods of Data Analysis; p. 760. [Google Scholar]
- 51.Azzi A, Breyer I, Feher M, Ricciarelli R, Stocker A, Zimmer S, Zingg J. J Nutr. 2001;131:378S–81S. doi: 10.1093/jn/131.2.378S. [DOI] [PubMed] [Google Scholar]
- 52.Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. Structure. 1998;6:195–210. doi: 10.1016/s0969-2126(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 53.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Semin Cancer Biol. 2005;15:300–8. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 54.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Proc Natl Acad Sci U S A. 2007;104:19345–50. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJC, Lorkiewicz PK, Higashi RM, Fan TWM, Dang CV. Cell Metab. 2012;15:110–21. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vos PE, Lamers KJB, Hendriks JCM, van Haaren M, Beems T, Zimmerman C, van Geel W, de Reus H, Biert J, Verbeek MM. Neurology. 2004;62:1303–10. doi: 10.1212/01.wnl.0000120550.00643.dc. [DOI] [PubMed] [Google Scholar]
- 57.Eng LF, Ghirnikar RS, Lee YL. Neurochem Res. 2000;25:1439–51. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 58.Schiff L, Hadker N, Weiser S, Rausch C. Mol Diagn Ther. 2012;16:79–92. doi: 10.2165/11631580-000000000-00000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.