

Abstract
Short Abstract
Many therapeutic applications require safe and efficient transport of drug carriers and their cargoes across cellular barriers in the body. This article describes an adaptation of established methods to evaluate the rate and mechanism of transport of drug nanocarriers (NCs) across cellular barriers, such as the gastrointestinal (GI) epithelium.
Long Abstract
Sub-micrometer carriers (nanocarriers; NCs) enhance efficacy of drugs by improving solubility, stability, circulation time, targeting, and release. Additionally, traversing cellular barriers in the body is crucial for both oral delivery of therapeutic NCs into the circulation or transport from the blood into tissues, where intervention is needed. NC transport across cellular barriers is achieved by: (i) the paracellular route, via transient disruption of the junctions that interlock adjacent cells, or (ii) the transcellular route, where materials are internalized by endocytosis, transported across the cell body, and secreted at the opposite cell surface (transyctosis). Delivery across cellular barriers can be facilitated by coupling therapeutics or their carriers with targeting agents that bind specifically to cell-surface markers involved in transport. Here, we provide methods to measure the extent and mechanism of NC transport across a model cell barrier, which consists of a monolayer of gastrointestinal (GI) epithelial cells grown on a porous membrane located in a transwell insert. Formation of a permeability barrier is confirmed by measuring transepithelial electrical resistance (TEER), transepithelial transport of a control substance, and immunostaining of tight junctions. As an example, ~200-nm polymer NCs are used, which carry a therapeutic cargo and are coated with an antibody that targets a cell-surface determinant. The antibody or therapeutic cargo is labeled with 125I for radioisotope tracing and labeled NCs are added to the upper chamber over the cell monolayer for varying periods of time. NCs associated to the cells and/or transported to the underlying chamber can be detected. Measurement of free 125I allows subtraction of the degraded fraction. The paracellular route is assessed by determining potential changes caused by NC transport to the barrier parameters described above. Transcellular transport is determined by addressing the effect of modulating endocytosis and transcytosis pathways.
Keywords: Drug delivery systems, targeted nanocarriers, transcellular transport, epithelial cells, tight junctions, transepithelial electrical resistance, endocytosis, transcytosis, radioisotope tracing, immunostaining
Introduction
Cellular barriers in the body act as a gateway between the external environment and internal compartments. This is the case for the epithelial lining separating the externally-exposed surface of the gastrointestinal (GI) tract and the bloodstream1–3. Cellular barriers also represent the interface between the bloodstream and the parenchyma and cellular components of tissues and organs. This is the case for the inner endothelial lining in blood vessels, such as the blood-lung barrier, the blood-brain barrier, etc.1. The ability to traverse these cellular barriers in the body is crucial for efficient delivery of therapeutic and diagnostic agents into the circulation and tissues/organs where intervention is needed.
To improve delivery of therapeutic or diagnostic agents, these compounds can be loaded into sub-micrometer nanocarriers (NCs). These drug delivery vehicles can be formulated with a variety of chemistries and structures to optimize drug solubility, protection, pharmacokinetics, release, and metabolism4,5. NCs can also be functionalized with affinity or targeting moieties (e.g., antibodies, peptides sugars, aptamers, etc.) to facilitate adhesion to areas of the body where the therapeutic action is required2,6. Targeting of NCs to determinants expressed on the surface of cellular barriers can further facilitate transport into and/or across these linings2,6.
The role of selectively transporting substances between two environments requires certain unique features among cell layers. One such feature is cell polarity, whereby the apical membrane facing the lumen of cavities varies from the basolateral membrane oriented towards the tissue interstitium, with respect to membrane morphology and composition of lipids, transporters, and receptors2. Another feature involves intercellular junctions connecting adjacent cells. Regulation of the proteins that form tight junctions, particularly junctional adhesion molecules (JAMs), occludins, and claudins, modulate the barrier function to selectively allow or not transport of substances between cells, known as paracellular transport, allowing passage of materials from the lumen to the basolateral space3. Binding of many natural and synthetic elements (leukocytes, molecules, particles, and drug delivery systems) to cellular barriers in the body can induce cell-junction opening, which may be transient and relatively innocuous or more prolonged and, therefore, unsafe with access of undesired substances across the barrier2,5,7–9. Consequently, this pathway can be assessed by measuring the transepithelial electrical resistance (TEER) and passive paracellular diffusion of molecules (herein called paracellular leakage), whereby decreased resistance to an electrical current or increased paracellular leakage of an inert compound into the basolateral space indicate opening of cell junctions, respectively5,10,11. To complement these methods, any of the tight junction proteins listed above can be stained to assess their integrity, where staining should appear concentrated at the cell-cell borders all around the cell periphery5,10,12.
Alternatively, drug delivery systems that target specific cell surface determinants, such as those associated to clathrin-coated pits or flask-shaped membrane invaginations called caveolae, may trigger vesicular uptake into cells by endocytosis, providing an avenue for drug delivery to intracellular compartments5,13. In addition, endocytosis may lead to trafficking of vesicles across the cell body for release at the basolateral side, a phenomenon known as transyctosis, or transcellular transport14. Therefore, knowledge of the kinetics and mechanism of endocytosis may be used to exploit intracellular and transcellular drug delivery, which offers a relatively safe and controlled mode of delivery compared to the paracellular route. The mechanism of endocytosis may be evaluated with modulators of classical pathways (clathrin- and caveolin-mediated endocytosis, and macropinocytosis) or non-classical routes (such as the case of cell adhesion molecule (CAM)-mediated endocytosis)5,13,15.
Whereas intracellular trafficking is often studied in standard wells or coverslips, the absence of a basolateral compartment precludes cell polarization and the ability to study transport across cell layers. To overcome this obstacle, transport across cell monolayers has been long studied using transwell inserts10,11,16,17, which consist of an upper (apical) chamber, a porous permeable membrane where cells attach and form a tight monolayer, and a lower (basolateral) chamber (Fig. 1). In this configuration, transport can be measured in the apical-to-basolateral direction by administering a treatment into the upper chamber, following transport through the cell monolayer and the underlying porous membrane, and finally collecting the medium in the lower chamber for quantification of transported material. Transport in the basolateral-to-apical direction can also be measured by initial administration to the lower chamber and subsequent collection from the upper chamber5,10,12,16. Various techniques exist to verify the formation of permeability barrier on transwells, including TEER and paracellular transport assays, as described above. In addition, the permeable filter on which cells are cultured can be removed for imaging analysis (e.g., by fluorescence, confocal, electron microscopy), as further validation of the cell monolayer model as well as the mechanism of transport. Selection of the membrane type, which is available in different pore sizes, materials, and surface areas, depends on various factors such as the size of substances or objects to be transported, cell type, and imaging method16,18–20. Transwell inserts also facilitate controlled and accurate quantification of transport compared to complex mammalian systems, as volumes of the chambers and cell surface area are known constants. While many factors involved in in vivo delivery are eliminated, including the presence of intestinal mucus, shear stress, digestive enzymes, immune cells, etc., this small scale in vitro model provides useful preliminary information regarding transport.
Figure 1. Schematic of transepithelial transport of targeted nanocarriers across a gastrointestinal epithelial model.
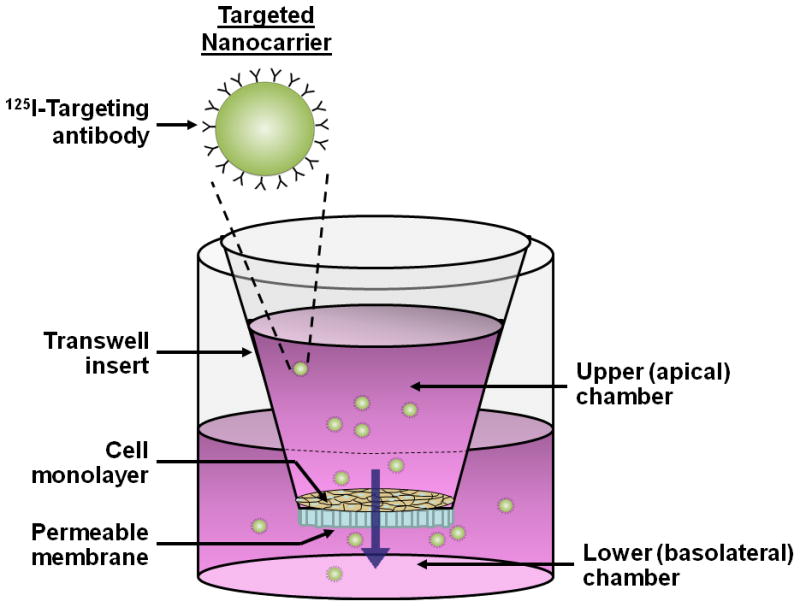
(A) As a platform to study transepithelial transport, Caco-2 cell monolayers are cultured on transwell inserts until a tight monolayer is formed (16–21 days post-seeding, TEER ≥ 350 Ω × cm2 over background). An example of a drug delivery vehicle, such as 125I-anti-ICAM NCs, is added to the upper (apical) chamber above the cell monolayer to study transport across cells and through the underlying porous membrane. The medium in the lower (basolateral) chamber is then collected for quantification of transported material. (B) Transport through the cell monolayer occurs via either a paracellular or transcellular/transcytosis route. Reproduced from Ghaffarian et al., J Controlled Rel, 2012, 163(1):25–33.
As an example to illustrate the adaptation of these methods to study NC transport across cellular barriers10,11,16,17, we describe here a case where the potential for NC transport across the GI epithelium was modeled by assessing passage of a model drug delivery system through a monolayer of human epithelial colorectal adenocarcinoma (Caco-2) cells. For this purpose, cells were cultured in transwell inserts, on a 0.8 μm-pore polyethylene terephthalate (PET) filter (6.4 mm-diameter), which is transparent and can be used for microscopy imaging. The status of the permeability barrier is validated by measuring TEER, apical-to-basolateral transport of a control substance, albumin, and fluorescence microscopy visualization of an element of the tight junctions, occludin protein. A model of targeted polymer NC is used, consisting of 100-nm, non-biodegradable polystyrene nanoparticles. NCs are coated by surface adsorption with a targeting antibody alone or a combination of a targeting antibody and a therapeutic cargo, where either component can be labeled with 125I for radioisotope tracing. In the selected example, the antibody recognizes intercellular adhesion molecule-1 (ICAM-1), a protein expressed on the surface of GI epithelial (and other) cells, which has been shown to facilitate intracellular and transcellular transport of drug carriers and their cargoes21. The cargo is alpha-Galactosidase (α-Gal), a therapeutic enzyme used for treatment of Fabry disease, a genetic lysosomal storage disorder22.
The coated NCs, of about 200-nm in size, are added to the apical chamber over the cell monolayer and incubated at 37°C for varying periods of time, after which 125I on NCs can be detected associated to the cell monolayer and/or transported to the basolateral chamber below the cells. Additional determination of free 125I allows subtraction of the degraded fraction and estimation of coated NC transport. The mechanism of transport is further assessed by examining changes in the permeability barrier pertaining to the paracellular route, through the parameters described above, while transcellular transport is determined by examining changes in transport when modulating pathways of endocytosis and transcytosis.
These methods provide valuable information regarding cellular barrier models, the extent and rate of transport of a drug delivery system, and the mechanism of such transport, altogether allowing evaluation of the potential for drug delivery across cellular barriers.
Protocol
1) Culturing a cell monolayer in transwell inserts
-
1.1
In a sterile, biosafety level 2 cell culture hood, place 0.8 μm-pore PET transwell inserts into a 24-well plate (4 wells per condition, for statistical significance) with forceps. All materials entering the hood should be sterilized with ethanol. Note: The pore size of the filter needs to be selected in accord to the mean size of the NC used, to allow transport across the membrane. Also, for statistically significant results, each experimental condition requires a minimum of four repeats (wells) within the same experiment, and a minimum of 3 independent experiments.
-
1.2
Prepare cell culture medium containing DMEM medium supplemented with 4.5 g/L glucose, 15% fetal bovine serum, and 1% Pen Strep, and heat to 37 °C in a water bath.
-
1.3
Dilute human epithelial colorectal adenocarcinoma (Caco-2) cells in cell medium and place 200–400 μl of the cell solution into the upper (apical) chamber of the transwell insert, at a density of 1.5 × 105 cells/cm2. Fill the lower (basolateral) chamber with 700–900 μl of cell medium.
-
1.4
Culture cells at 37 °C, 5% CO2, and 95% humidity for 16–21 days, replacing the medium in the upper and lower chamber every 3–4 days, using the volumes indicated in step 1.3. Replace medium in the following order to maintain the pressure above (rather than below) the cell monolayer: aspirate the medium from the lower chamber, aspirate the medium from the upper chamber, fill the upper chamber with fresh medium, and fill the lower chamber with fresh medium.
2) Validation of the GI epithelial permeability barrier using transepithelial electrical resistance (TEER) and immunostaining of tight junctions
-
2.1
for short-term storage (less than 2 weeks), submerge STX100 electrodes in an electrolyte solution (0.1–0.15 M KCl or NaCl). Connect the electrode cable to the electrode port on the EVOM volt-ohm meter so that the system is internally short-circuited and electrode symmetry is maintained. For long-term storage, rinse with dH2O and store in a dry, dark condition.
-
2.2
To sterilize the electrodes, immerse in ethanol for 15 min and allow to air dry for 15 s. Alternatively, the electrodes can be stored in a UV hood. Rinse the electrodes in a sterile electrolyte solution (phosphate buffer saline, PBS) or 0.1–0.15 M KCl or NaCl solution, before each resistance measurement.
-
2.3
With the volt-ohm meter set to the resistance setting, vertically place electrodes in a well containing the transwell insert, with the short electrode in the upper chamber and the long electrode in the lower chamber touching the bottom of the well. Once the EVOM reading stabilizes, record the resistance value (given in ohms; Ω) for each well.
-
2.4
Calculate resistivity (resistance normalized to area; Ω×cm2) of samples by subtracting background resistance (TEER of transwell inserts without cells) and multiplying by membrane surface area. Resistivity values are most often reported in the literature. - (See Discussion) -
-
2.5
Repeat measurements every 1–2 days until TEER values rise to a maximun and plateau, indicating formation of a permeability barrier, which typically takes 2–3 weeks from the moment of cell platting. Plateau TEER values and time necessary to achieve barrier integrity may vary according to cell passage number.
-
2.6
To verify the presence of tight junctions in cell monolayers with high TEER (equal to or above the threshold value for barrier formation), fix cells by incubation with cold 2% paraformaldehyde for 15 min. Then, wash cells with PBS and incubate them for 30 min at room temperature with 1 μg/ml anti-occludin. Wash cells again with PBS and incubate them for 30 min at room temperature with 7.5 μg/ml fluorescently-labeled secondary antibodies. Use wells with low TEER as a negative control for barrier formation. Note: As a substitute for anti-occludin, antibodies to alternative tight junction proteins may be used.
-
2.7
Carefully excise the filter membrane on which the fixed monolayer is attached, and mount onto slides for imaging using epifluorescence or confocal microscopy.
3) Evaluating transepithelial transport of targeted carriers
-
3.1
Label the targeting antibody (mouse monoclonal antibody against ICAM-1, anti-ICAM, in this example) with 125I, as previously described7. Use Bradford assay to estimate protein concentration and a gamma counter to determine 125I content on the antibody, then calculate the specific activity in counts per minute (CPM)/μg of labeled antibody. Note: To control for specificity, repeat the procedure by labeling mouse IgG, which will be used to prepare non-targeted coated NCs. To study transport of a therapeutic cargo, repeat the procedure labeling the cargo, e.g., alpha-Galactosidase (α-Gal) in our example. Alternatives may be used for the targeting agent, non-specific control, or therapeutic cargo. Alternative methods may also be used to label compounds and estimate the concentration of the labeled counterparts.
-
3.2
Couple the labeled targeting antibody (anti-ICAM in our example) to the surface of NCs. In this example, incubate polystyrene nanobeads (100-nm diameter) for 1 h at room temperature with 125I-anti-ICAM to allow surface adsorption, as described7. Note: For the non-specific control use 125I-IgG. To trace transport of a cargo, use a combination of targeting antibody and 125I-labeled cargo (α-Gal in our example).
-
3.3
Centrifuge at 12,000 rpm for 3 min and remove the non-coated counterparts in the supernatant by aspiration. Resuspend the pellet containing coated NCs using 1% bovine serum albumin (BSA) in PBS by pipetting, and sonicate at low power to disrupt potential particle aggregates (20–30 brief pulses).
-
3.4
for NC characterization, measure size, polydispersity, and ζ-potential of coated particles using dynamic light scattering (following vendor instructions), and quantify the number of 125I-labeled targeting antibody molecules (e.g., anti-ICAM) on the particle surface using a gamma counter. Note: These methods can be repeated for non-specific and therapeutic counterparts. The size of coated particles ranges around 200-nm.
-
3.5
Add 125I-antibody NCs, (e.g., 125I-anti-ICAM NCs; 56 nCi/ml) to the upper chamber above confluent Caco-2 monolayers with TEER ≥ 350 Ω × cm2 (see Discussion) over background (16–21 days post-seeding). Incubate at 37°C for one or more desired time intervals. Measure TEER (Section 2.3) before and after incubation to assess the effects of NCs on the permeability barrier. Note: This procedure can be repeated for non-specific and therapeutic counterparts.
-
3.6
Collect medium from the upper and lower chambers and wash them once with 0.5 ml DMEM at 37°C (upper chamber) or 1 ml dH2O (lower chamber). Collect the washes for measurement of total radioisotope content using a gamma counter.
-
3.7
for measurement of the cell fraction, excise the permeable filter, e.g., using a razor blade to cut around the edges, and incubate in a gamma counter tube with 1% Triton X-100 for 10 min (to release cell contents), before measuring cell-associated total radioactivity.
-
3.8
To measure 125I released from NCs during transport or due to potential degradation, first mix 300 μl of sample (from the upper, lower, or cell fractions) with 700 μl of 3% BSA in PBS and 200 μl trichloroacetic acid (TCA). Incubate at room temperature for 15 min. Meanwhile this time, measure the total radioactivity of this sample in a gamma counter.
-
3.9
Centrifuge TCA samples at 3,700 rpm for 5 min to separate intact protein (pellet) from the degraded protein or 125I fraction (supernatant). Quantify the radioactivity of the free125I fraction and subtract this value from total radioactivity measured before centrifugation. This will provide the amount of labeled-protein that is not degraded.
4) Mechanism of transepithelial transport of targeted nanocarriers
-
4.1
Label albumin with 125I as described in Section 3.1 and previously reported7. Note: Albumin is a 66.5 kDa protein and, hence, a relatively large substance. Although a valid control for passive transport of larger objects (e.g., 200-nm NCs used here), it should be substituted with smaller inert tracer molecules when studying transport of smaller drug carriers (see Discussion).
-
4.2
To assess paracellular transport using albumin paracellular leakage, culture Caco-2 monolayers on transwell inserts as described above.
-
4.3
To the upper chamber medium above the cell monolayer, add either 125I-albumin alone (negative control showing the basal level of leakage), or 125I-albumin and non-radiolabeled antibody-targeted NCs (as described in Section 3.5). Incubate at 37°C for the selected time interval(s), which should match those examined when testing NC transport. Measure TEER (Section 2.3) before, during, and after incubation, then collect all fractions for total 125I and free 125I measurements as indicated in Section 3. Note: Concomitant addition of 125I-albumin and non-radiolabeled control IgG NCs may be used as a control.
-
4.4
As a positive control for opening intercellular junctions, add cell media containing 5 mM H2O2 to the upper and lower chambers of the transwell insert, and incubate at 37°C for 30 min. Then, measure TEER (Section 2.3) and add 125I-albumin to the upper chamber for the selected time intervals. Measure TEER at various time points throughout the incubation to identify TEER value decay caused by H2O2-induced opening of the cell junctions.
-
4.5
In parallel experiments, evaluate transcellular transport, also called transcytosis, of 125I-targeted NCs by incubating confluent Caco-2 monolayers with either 50 μM monodansylcadaverine (MDC; inhibitor of clathrin endocytosis), 1 μg/ml filipin (inhibitor of caveolar endocytosis), 0.5 μM wortmannin (inhibitor of phosphatidylinositol 3 kinase (PI3K), involved in macropinocytosis), or 20 μM [5-(N-ethyl-N-isopropyl) amiloride] (EIPA, inhibitor of macropinocytosis and CAM-mediated endocytosis15).
Note: 125I-IgG NCs and 125I-albumin may be used as negative controls for the effect of these inhibitors, and other methods (e.g., siRNA techniques) may provide more selective inhibition.
-
4.6
Measure TEER (Section 2.3) before, during, and after incubation of cells with inhibitors and radiolabeled materials, as an additional control for the effect of the inhibitors on the monolayer permeability.
Representative Results
As validation of our cell model to study transepithelial transport of targeted NCs, Fig. 2 shows that Caco-2 cell monolayers plated at a density of 1.5×105 cells/cm2 reached confluence ~Day 12 and maintained monolayer integrity up to Day 18, indicated by TEER (Fig. 2A). This was validated by the presence of occludin-positive tight junctions (Fig. 2B) in monolayers with high TEER (390 Ω×cm2, Day 14), compared to poor tight junction labeling at low TEER (17 Ω×cm2, Day 5).
Figure 2. Caco-2 monolayers as a model for transepithelial transport.
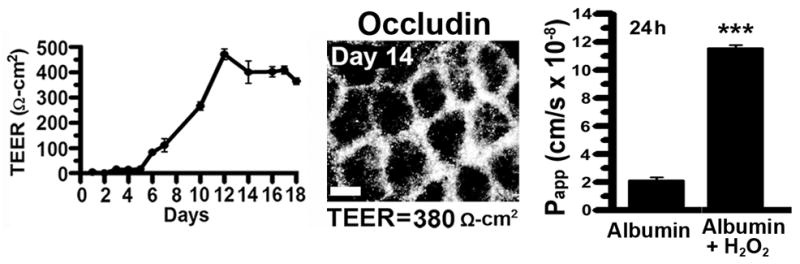
Caco-2 cells were grown on transwell inserts at 1.5×105 cells/cm2. (A) Transepithelial electrical resistance (TEER) was measured to assess monolayer integrity. Data are shown as means ± S.E.M; n=3. (B) Fluorescence microscopy of tight junctions immunolabeled with anti-occludin and Texas Red-labeled secondary antibody, on Days 5 or 14 (low versus high TEER values, respectively). Reproduced from Ghaffarian et al., J Controlled Rel, 2012, 163(1):25–33.
Tracing of the radioactive label on NC elements determines the total amount of these components (NC targeting antibody or cargo) initially added to the apical chamber, their cell associated fraction, and their fraction transported to the basolateral side. These values can be used to calculate various parameters that describe NC transport in a more relevant manner, particularly after subtraction of free 125I content of each fraction, which represents degraded counterparts. For instance, the number of antibody molecules, cargo molecules, or associated NCs that are transverse the cell monolayer can be calculated to estimate absolute transport. Also, the percentage of said molecules or NCs that is transported to the basolateral side with respect to the total number of molecules or NCs associated to the cell monolayer, estimates the efficiency of said transport. Finally, the apparent permeability coefficient (Papp), a standard parameter for the rate of transport, can be estimated. These parameters are calculated as it follows:
where CPM are the 125I counts-per-minute added to the upper chamber (CPMadded), the cell monolayer fraction (CPMcell fraction), or the lower chamber (CPMbasolateral), and NCadded or Moleculesadded are the number of NCs or molecules initially added to the upper chamber, A is the surface area of the filter membrane (cm2), Vol. is volume of medium in the upper chamber (ml), and t is time of incubation (s).
In our example, when the radioactive isotope is labeling the targeting antibody, this analysis reveals the degree and efficiency of transport in terms of antibodies or NCs transported per cell and percentage of cell monolayer-associated antibodies or NCs that were transported (Fig. 3A and 3B), as well as the rate of transport in terms of Papp (Fig. 3D). These parameters, estimated in our example for 125I-anti-ICAM NCs, were also compared to those of control 125I-IgG NCs to demonstrate the transport efficiency relative to a non-targeted counterpart (Fig. 3C and 3D).
Figure 3. Transport of anti-ICAM nanocarriers across Caco-2 cell monolayers.

Confluent Caco-2 monolayers grown on transwell inserts were incubated with 125I-anti-ICAM NCs or 125I-IgG NCs added to the apical chamber. (A–C) 125I content in the basolateral chamber was measured at the indicated time points, to calculate the amount of NCs transported per cell (see Representative Results). (B) Percent of transported NCs was calculated as the ratio of carriers found in the basolateral fraction to that in the combined basolateral and cell fractions. (D) Apparent permeability coefficients (Papp) were calculated as described in Representative Results, reflecting rates of transport of 125I-anti-ICAM NCs or 125I-IgG NCs. Data are shown as means ± S.E.M; n=4. Reproduced from Ghaffarian et al., J Controlled Rel, 2012, 163(1):25–33.
When the radioactive label is incorporated on the NC cargo, the described parameters reflect transport of the cargo, which in our example was α-Gal enzyme, used for treatment of a genetic lysosomal storage disorder known as Fabry disease (Table 1). In addition to calculating NC transport by tracing said cargo, transepithelial delivery of this therapeutic enzyme can be estimated, e.g., by expressing it as molecules or mass of α-Gal transported per cell.
Table 1. Transepithelial transport of α-Gal by anti-ICAM nanocarriers.
Transcellular transport of anti-ICAM/125I-α-Gal NCs across confluent Caco-2 monolayers was assessed at 3 h and 24 h. Radioisotope content of apical, basolateral, and cell fractions were converted into various values relevant to transport, as described in Representative Results. Reproduced from Ghaffarian et al., J Controlled Rel, 2012, 163(1):25–33.
| Total NCs associated/cell | Intracellular NCs/cell | Transported NCs/cell | % Transported | Transported α-Gal molecules/cell × 105 | pg α-Gal transported/cell × 10−3 | Papp (×10−8 cm/s) | |
|---|---|---|---|---|---|---|---|
|
|
|||||||
| 3 h | 663.4 ± 116.0 | 434.0 ± 76.8 | 243.6 ± 15.4 | 44.4 ± 6.7 | 1.8 ± 0.2 | 9.7 ± 1.2 | 8.1 ± 1.1 |
| 24 h | 2024.8 ± 409.3 | 1218.9 ± 255.6 | 806.9 ± 164.1 | 40.6 ± 2.0 | 3.9 ± 0.5 | 21.3 ± 2.5 | 1.9 ± 0.4 |
NCs = nanocarriers; α-Gal = α-galactosidase; Total NCs associated/cell = Intracellular NCs/cell + NCs transported/cell; % Transported = (NCs transported/Total NCs associated) × 100; Papp = apparent permeability coefficient (cm/s). See Methods for further description of these parameters. Data are shown as means ± S.E.M. (n = 8 wells); (-TNF-α Caco-2 cells).
Finally, this method attributes the transport pathway to the paracellular route or the transcellular route. For instance, in our example, EIPA reduced transport of 125I-anti-ICAM NCs across Caco-2 cell monolayers (Fig. 4A), whereas MDC, filipin, and wortmannin did not affect transport levels with respect to the control condition (no inhibitor). This suggests that anti-ICAM NCs utilize CAM-mediated endocytosis for transcellular transport, but not clathrin-, caveolar-, or macropinocytosis-related transcytosis. Moreover, paracellular transport was ruled out using measurements of TEER (Fig. 4B) and an albumin passive transport (Fig. 4C), whereby a decrease in TEER and an increase in albumin paracellular leakage into the lower chamber during transport of anti-ICAM NCs would have indicated disruption of intercellular junctions. However, incubation with anti-ICAM NCs did not alter TEER or 125I-albumin paracellular leakage over a period of 48h, similar to control IgG NCs that are not transported. As a positive control for opening of the cell junctions and paracellular transport, H2O2 decreased TEER to basal levels and markedly enhanced 125I-albumin leakage to the basolateral chamber.
Figure 4. Mechanism of transport of anti-ICAM nanocarriers across Caco-2 monolayers.

(A) Transcellular transport of 125I-anti-ICAM NCs across confluent Caco-2 monolayers was assessed at 24 h in the absence or presence of 20 μM EIPA, 1 μg/ml filipin, 50 μM MDC, or 0.5 μM wortmannin. (B) TEER was measured during transport of 125I-anti-ICAM NCs across Caco-2 monolayers, to assess paracellular transport. TEER values in the absence of NCs are shown as controls (the dashed interval marks S.E.M of the mean value). Incubation with 5 mM H2O2 is a positive control for opening of intercellular junctions. (C) Paracellular protein leakage, measured as the apparent permeability coefficient (Papp) of 125I-Albumin crossing the cell monolayer in the absence or presence of 5 mM H2O2, IgG NCs, or anti-ICAM NCs, was measured and calculated as in Fig. 3. Data are shown as means ± S.E.M.; n≥4. Reproduced from Ghaffarian et al., J Controlled Rel, 2012, 163(1):25–33.
Discussion
Using the methods discussed above, a cell model for studying transport of targeted NCs across cellular barriers can be established, such as the example provided for Caco-2 epithelial cells, which is relevant to evaluating transport from the GI lumen into the blood in the case of oral drug delivery systems. Culturing of GI epithelial cell monolayers in transwell inserts enabled measurement of TEER and fluorescence immunostaining of tight junctions to confirm formation of a cell permeability barrier. Subsequently, radiolabeling of targeting and therapeutic agents with 125I provided rapid and sensitive quantification of targeted NCs into and across GI cell monolayers. Finally, mechanistic studies were applied using endocytic inhibitors, TEER, and an albumin paracellular leakage assay to assess whether transport occurs through a vesicular route versus a paracellular route.
An important parameter in establishing similar models is the selection of a cell type, which should reflect the physiological nature of the cellular barrier under study. Various epithelial and endothelial cells from human or animal origin are commercially available16,18–20. These can be grown alone as single cultures as described here, or as co-cultures of these cells with other cell types (e.g., mucus-secreting cells, immune cells, pericytes, etc.)16,18–20. In such co-culture form, transwell insert conditions can be modulated to adjust the respective growth rates and requirements for differentiation factors in the cell media16,18–20. For example, a blood-brain barrier model may involve co-cultivation of brain microvascular endothelial cells and astrocytes or pericytes on the opposite surface of the porous filter, and the respective media for each cell type should be placed in each chamber19. In addition, depending on the cell line, extracellular matrix components may be required for effective cell attachment to the porous membrane18–20. It is important to consider that each particular model will render different basal levels with regard to permeability barrier parameters and transport rates and mechanism, hence control values need to be obtained and these methods optimized for each situation.
Transwell inserts such as the ones described here, have been widely used to study transport of materials across cell monolayers, from the apical into a basolateral compartment or vice versa5,10–12,16,17. This cannot be achieved by culturing cells on classical wells or coverslips because such systems preclude this form of transport as they do not offer two independent compartments and hence may not give a realistic view of cell sorting. For example, materials naturally destined for release from the basolateral membrane may be shunted to other cell compartments in a coverslip model, making it difficult to resolve the mechanism of transcellular transport. Moreover, in contrast to coverslips, the transwell configuration gives rise to physiologically-relevant features associated with cell differentiation and membrane polarity10,11,16–19. For example, Caco-2 cells grown on transwells exhibit the characteristics of mature enterocytes, including the presence of microvilli and tight junctions, dome formation, and production of brush border enzymes10,11,17. Other parameters can induce a more physiologically relevant phenotype, such as shear stress in the case of GI epithelial cells (e.g. mucus flow and contractile movements) and vascular endothelial cells (e.g. blood flow), which can be applied in transwells using agitation or pumps10,16, and co-cultures to produce the necessary growth and differentiation factors for certain cell types16,18–20. Nevertheless, one must consider that cell lines often differ from cells in body tissues, e.g., they may express certain determinants at different levels and, hence, present varied functions and regulations. For instance, Caco-2 cells do not contain all transporter proteins and enzymes required for activation and transport of oral therapeutics in vivo, such as CYP3A4, a drug-metabolizing enzyme10,17,23. In these cases, cell line sub-clones, transfection, or addition of transcriptional agents can be used modulate the cell line as to better reflect physiological conditions17,23,24.
In selecting an optimal transwell model, the various features of the permeable filter must be taken into account. Permeable filters are available in different pore sizes, ranging from 0.4 to 8 μm, and must accommodate the size of transported cargo. For instance, smaller pore sizes (0.4 – 3 μm) are typically used for studies of transport of small chemical compounds, while pores 3 μm or larger are used for cell invasion, chemotaxis, and motility studies in which microbial, immune, and migrating cells are transported. Pore size and density also affects cell density and differentiation, as some cells may migrate through pores upon seeding, precluding monolayer formation. In addition, the material of the permeable support influences imaging clarity, for assessment of cell viability and monolayer formation, and cell attachment. Polyethylene terephthalate (PET) filters are transparent, allowing visibility under phase contrast microscopy, in contrast to translucent polycarbonate filters. For enhanced cell attachment, permeable supports pre-coated with collagen or fibronectin are commercially available. Moreover, the filter diameter, which is adjusted for different well plates, may influence permeability, whereby larger diameters (e.g. in 6-well plates) tend to increase paracellular leakiness of the cell monolayer.
Once a transwell model is established, including the appropriate selection of cell type, media composition, and permeable filter, the status of the cell monolayer can be assessed during the culturing and experimental stages using TEER. However, TEER values vary with 1) innate biological factors associated with cell type, source, and passage number; 2) culturing conditions, such as the aforementioned transwell characteristics, seeding density, days in culture, media composition and pH; 3) physical factors, such as temperature, media volume, sampling schedule, and degree of agitation/washing; and 4) pathological and/or chemical factors that modulate cell junction integrity (e.g., cytokines, immune cells, oxidative stress, pharmacological additives, etc.)10,16,17. Due to the various biological and environmental factors affecting TEER, a minimum baseline value indicating barrier formation must be established for each system by regularly monitoring TEER during the culture period and after experimental manipulations. Values that are deemed adequate for barrier formation, which can range between 150 – 1600 Ω × cm2 16, also depend on the size of the substance to be transported such that passive paracellular diffusion of that substance is restricted; while TEER ≥ 350 Ω × cm2 seems to preclude passive diffusion of relatively large NCs such as the ones shown here (200-nm), this may not be the case for smaller drug delivery systems, in which tighter monolayers (e.g., ≥ 700 Ω × cm2) are required. This baseline value can also be assessed using controls for disrupting the permeability barrier, including H2O2, penetrating peptides, calcium chelators (e.g., EDTA), protein kinases and phosphatases, and various other regulatory molecules25–27.
Complementary to TEER, many permeability assays exist to verify monolayer integrity and assess paracellular transport. Like albumin, other membrane-impermeable tracer molecules of various sizes, such as mannitol, lucifer yellow, inulin, and dextrans, which can be coupled with a UV, fluorescent, or radioactive label to measure and compare permeability rates (Papp) to known values for these solutes. When evaluating paracellular transport, it is important to use a tracer molecule that is smaller than the transported compound under study. For example, paracellular transport of 200-nm particles would open intercellular junctions enough to cause paracellular leakage of smaller, yet still relatively large molecules, such as albumin used in our example. In contrast, paracellular transport of smaller drug delivery vehicles such as dendrimers (~5-nm) should be evaluated using molecules < 5-nm, such as mannitol. Paracellular leakage studies therefore indicate the size of cell junction opening, yet due to long incubation times they cannot identify transient disruptions of cell junctions.
Another mode of evaluating barrier integrity is to image the tight junctions that connect adjacent cells. In this study, we immunostained occludin for fluorescence microscopy, yet various other proteins present in the tight junction can be used for this analysis, including transmembrane proteins of the junction adhesion molecule (JAM), claudin, and occludin families. Here, we stained the extracellular domain to bypass a permeabilization step, yet intracellular domains can also be stained. Supplementary to this is the use of controls for non-specific staining, using fluorescent secondary antibodies only, absence of tight junctions (here, we used cells with a monolayer showing low TEER), or disruption of tight junctions using the junction-modulating agents listed above.
In our example of transepithelial transport we use ICAM-1-targeted NCs, but the transwell system also accommodates transport of other drug vehicle formulations, therapeutic and diagnostic compounds, innate compounds found in the body, or a combination of the above, with valuable implications for clinical studies or understanding of fundamental pathways. The methods we describe to quantify transport across cell monolayers involve radioisotope labeling of the targeting agent or therapeutic cargo in the NC coat, yet this strategy can also be used to track NCs with diverse chemistries and structures, including linear or branched polymers, dendrimers, particles, micelles, liposomes, etc.4,5,28,29. Different isotopes (e.g., 125I, 3H, 32P, etc.) are available to quantify transport of a wide range of small and large molecules: not only drug carriers but also chemical and biological therapeutics, without affecting the functional or structural integrity of the compound, unlike bulky fluorescent labels. Radiolabeling provides greater quantitative sensitivity and speed compared to other strategies that measure transport, such as gel electrophoresis, PCR, mass spectrometry, HPLC, and fluorescence spectrometry. This label can also be used to determine the degree of degradation (e.g., protein targeting agent and cargo in our example) by assessing free iodine content, which is fast and accurate compared to alternative protein activity assays, including UV spectrophotometry and Western blot. However, since free iodine does not entirely reflect alterations in protein conformation or activity that may occur in the absence of degradation, the above alternative methods may be used to further evaluate the integrity of transported materials. Also, when administering compounds with free radioisotopes into the apical chamber, it is unclear whether the label found in the cell monolayer and transported fractions result from real protein degradation or diffusion of free iodine from the apical space. To circumvent this limitation, the apical NC concentration may be applied for a brief time interval to allow association to the cell monolayer, followed by removal of the apical medium in exchange for fresh medium, and a chase period for evaluating transport. Although an amount of free radioisotopes from the original pool may initially leak into the other fractions, this value can be determined from samples with no chase period and subtracted from conditions involving a chase period.
In terms of assessing the mechanism of transport, the same methods described for validating monolayer integrity (TEER, leakage assays, and tight junction staining) can be used to evaluate the paracellular route. For transcellular transport, pharmacological inhibitors of determinants involved in intracellular trafficking (e.g., transferrin associated to clathrin pathways, cholera toxin B associated to caveolar pathways, etc.) can be used, whereby effective concentrations to specifically inhibit those determinants need to be elucidated for each system. Alternatives to the inhibitors used in our study include chlorpromazine and potassium depletion, specific for clathrin-dependent endocytosis, and genistein and methyl-β-cyclodextrin (MβCD), specific for caveolae-mediated uptake30. Other strategies that may avoid potential non-specific effects of pharmacological inhibitors involve co-localization of transported cargo with these determinants using fluorescence or radioisotope labeling, utilization of specific ligands as controls (e.g., transferrin or cholera toxin B, as indicated above), and siRNA to knockdown the expression of regulatory elements of these pathways.
In this study, we demonstrate equations for converting radioactivity data to various parameters that provide key preliminary information regarding transepithelial transport. For example, the degree of transepithelial transport is represented by NCs or molecules transported per cell, the efficiency of transporting content that binds or enters cells is represented by percentage of cell-associated NCs or molecules transported, and the rate of transport, which allows for comparison of permeability between different solutes, is signified by Papp. In order to understand the potential of targeted NCs for large-scale delivery of therapeutics (e.g., the dose needed for in vivo administration), these equations may be modified to estimate transport values on a macroscopic scale. For instance, the amount of a therapeutic cargo transported per unit surface area of intestinal tissue (mg/cm2) as well as the potential delivery across the GI tract of a patient may be estimated from the following equations,
where the Specific Activity represents the measured CPM/mass unit of cargo (see Protocol, Step 3.1), and TA represents the total absorptive surface of the small intestine (2,000,000 cm2). Therefore, the data resulting from our methods gives rise to a wide range of possible quantitative analyses that describe transport in relevant terms.
Advancing from transwell inserts, models that better reflect the physiological complexity of transport include “Ussing” chambers and microfluidic devices, which provide dynamic fluid flow and a contained chamber that minimizes contact with air, as in blood vessels. These models also allow tissue explants to be cultured for ex vivo studies, prior to validation in vivo17,18.
In conclusion, the methods used in this study have provided valuable preliminary information regarding the transport of a model drug delivery system, in terms of efficiency, across cell monolayers. Using this cell culture model we demonstrated the potential of the ICAM-1-targeted nanocarrier platform in the context of oral drug delivery, paving the way for subsequent in vivo studies, yet may be modified for other systems requiring transport across cellular barriers found in the body.
Acknowledgments
This work was supported by a fellowship of the Howard Hughes Medical Institute and National Science Foundation to R.G, and funds awarded to S.M. by the National Institutes of Health (Grant R01-HL98416) and the American Heart Association (Grant 09BGIA2450014).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/50638.
Disclosures:
The authors declare that they have no competing financial interests.
Contributor Information
Rasa Ghaffarian, Email: rghaffar@umd.edu, 5115 Plant Sciences Bldg., Fischell Department of Bioengineering, University of Maryland, College Park, College Park, U.S.A.
Silvia Muro, Email: muro@umd.edu, 5115 Plant Sciences Bldg., Fischell Department of Bioengineering and Institute for Bioscience and Biotechnology Research, University of Maryland, College Park, College Park, U.S.A, Tel: (301) 405-4777, Fax: (301) 314-9075.
References
- 1.Deli MA. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim Biophys Acta. 2009;1788:892–910. doi: 10.1016/j.bbamem.2008.09.016. S0005-2736(08)00304-0 [pii] [DOI] [PubMed] [Google Scholar]
- 2.Mrsny RJ. Lessons from nature: “Pathogen-Mimetic” systems for mucosal nano-medicines. Adv Drug Deliv Rev. 2009;61:172–192. doi: 10.1016/j.addr.2008.09.009. S0169-409X(08)00266-4 [pii] [DOI] [PubMed] [Google Scholar]
- 3.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. nri2653 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur J Pharm Biopharm. 2009;71:431–444. doi: 10.1016/j.ejpb.2008.09.026. S0939-6411(08)00382-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sadekar S, Ghandehari H. Transepithelial transport and toxicity of PAMAM dendrimers: implications for oral drug delivery. Adv Drug Deliv Rev. 2012;64:571–588. doi: 10.1016/j.addr.2011.09.010. S0169-409X(11)00241-9 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muro S. Challenges in design and characterization of ligand-targeted drug delivery systems. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.05.052. S0168–3659(12)00511-1 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volkheimer G. Persorption of particles: physiology and pharmacology. Adv Pharmacol Chemother. 1977;14:163–187. doi: 10.1016/s1054-3589(08)60188-x. [DOI] [PubMed] [Google Scholar]
- 8.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 9.Jung T, et al. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50:147–160. doi: 10.1016/s0939-6411(00)00084-9. S0939-6411(00)00084-9 [pii] [DOI] [PubMed] [Google Scholar]
- 10.Hubatsch I, Ragnarsson EG, Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc. 2007;2:2111–2119. doi: 10.1038/nprot.2007.303. nprot.2007.303 [pii] [DOI] [PubMed] [Google Scholar]
- 11.Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (Caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96:736–749. S0016508589001009 [pii] [PubMed] [Google Scholar]
- 12.Tavelin S, Grasjo J, Taipalensuu J, Ocklind G, Artursson P. Applications of epithelial cell culture in studies of drug transport. Methods Mol Biol. 2002;188:233–272. doi: 10.1385/1-59259-185-X:233. 1-59259-185-X-233 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59:748–758. doi: 10.1016/j.addr.2007.06.008. S0169-409X(07)00096-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuma PL, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev. 2003;83:871–932. doi: 10.1152/physrev.00001.2003. 83/3/871 [pii] [DOI] [PubMed] [Google Scholar]
- 15.Muro S, et al. A novel endocytic pathway induced by clustering endothelial ICAM-1 or PECAM-1. J Cell Sci. 2003;116:1599–1609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 16.Shah P, Jogani V, Bagchi T, Misra A. Role of Caco-2 cell monolayers in prediction of intestinal drug absorption. Biotechnol Prog. 2006;22:186–198. doi: 10.1021/bp050208u. [DOI] [PubMed] [Google Scholar]
- 17.Delie F, Rubas W. A human colonic cell line sharing similarities with enterocytes as a model to examine oral absorption: advantages and limitations of the Caco-2 model. Crit Rev Ther Drug Carrier Syst. 1997;14:221–286. [PubMed] [Google Scholar]
- 18.Kuhnline Sloan CD, et al. Analytical and biological methods for probing the blood-brain barrier. Annu Rev Anal Chem (Palo Alto Calif) 2012;5:505–531. doi: 10.1146/annurev-anchem-062011-143002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatherell K, Couraud PO, Romero IA, Weksler B, Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood-brain barrier using mono-, co-, and tri-cultivation Transwell models. J Neurosci Methods. 199:223–229. doi: 10.1016/j.jneumeth.2011.05.012. S0165-0270(11)00270-6 [pii] [DOI] [PubMed] [Google Scholar]
- 20.Kasper J, et al. Flotillin-involved uptake of silica nanoparticles and responses of an alveolar-capillary barrier in vitro. Eur J Pharm Biopharm. 2012 doi: 10.1016/j.ejpb.2012.10.011. S0939-6411(12)00337-2 [pii] [DOI] [PubMed] [Google Scholar]
- 21.Ghaffarian R, Bhowmick T, Muro S. Transport of nanocarriers across gastrointestinal epithelial cells by a new transcellular route induced by targeting ICAM-1. J Control Release. 2012;163:25–33. doi: 10.1016/j.jconrel.2012.06.007. S0168-3659(12)00500-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu J, et al. Enhanced endothelial delivery and biochemical effects of alpha-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J Control Release. 2011;149:323–331. doi: 10.1016/j.jconrel.2010.10.031. S0168-3659(10)00845-X [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmiedlin-Ren P, et al. Mechanisms of enhanced oral availability of CYP3A4 substrates by grapefruit constituents. Decreased enterocyte CYP3A4 concentration and mechanism-based inactivation by furanocoumarins. Drug Metab Dispos. 1997;25:1228–1233. [PubMed] [Google Scholar]
- 24.Hughes J, Crowe A. Inhibition of P-glycoprotein-mediated efflux of digoxin and its metabolites by macrolide antibiotics. J Pharmacol Sci. 2010;113:315–324. doi: 10.1254/jphs.10109fp. JST.JSTAGE/jphs/10109FP [pii] [DOI] [PubMed] [Google Scholar]
- 25.Wielinga PR, de Waal E, Westerhoff HV, Lankelma J. In vitro transepithelial drug transport by on-line measurement: cellular control of paracellular and transcellular transport. J Pharm Sci. 1999;88:1340–1347. doi: 10.1021/js980497z. [pii] [DOI] [PubMed] [Google Scholar]
- 26.Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100:201–217. doi: 10.1042/BC20070116. BC20070116 [pii] [DOI] [PubMed] [Google Scholar]
- 27.Kapus A, Szaszi K. Coupling between apical and paracellular transport processes. Biochem Cell Biol. 2006;84:870–880. doi: 10.1139/o06-202. o06-202 [pii] [DOI] [PubMed] [Google Scholar]
- 28.Hood ED, et al. Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J Control Release. 2012;163:161–169. doi: 10.1016/j.jconrel.2012.08.031. S0168-3659(12)00653-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simone E, et al. Endothelial Targeting of Polymeric Nanoparticles Stably Labeled with Radioisotopes: from Synthesis to PET Imaging. Biomaterials. 2012 doi: 10.1016/j.biomaterials.2012.04.036. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vercauteren D, et al. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol Ther. 18:561–569. doi: 10.1038/mt.2009.281. mt2009281 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]