

Abstract
The class I terpenoid cyclase epi-isozizaene synthase (EIZS) utilizes the universal achiral isoprenoid substrate, farnesyl diphosphate, to generate epi-isozizaene as the predominant sesquiterpene cyclization product plus at least 5 minor sesquiterpene products, making EIZS an ideal platform for the exploration of fidelity and promiscuity in a terpenoid cyclization reaction. The hydrophobic active site contour of EIZS serves as a template that enforces a single substrate conformation and chaperones subsequently formed carbocation intermediates through a well-defined mechanistic sequence. Here, we have used the crystal structure of EIZS as a guide to systematically remold the hydrophobic active site contour in a library of 25 site-specific mutants. Remolded cyclization templates reprogram the reaction cascade not only by reproportioning products generated by the wild-type enzyme, but also by generating completely new products of diverse structure. Specifically, we have tripled the overall number of characterized products generated by EIZS. Moreover, we have converted EIZS into six different sesquiterpene synthases: F96A EIZS is an (E)-β-farnesene synthase, F96W EIZS is a zizaene synthase, F95H EIZS is a β-curcumene synthase, F95M EIZS is a β-acoradiene synthase, F198L EIZS is a β-cedrene synthase, and F96V EIZS and W203F EIZS are (Z)-γ-bisabolene synthases. Active site aromatic residues appear to be hot spots for reprogramming the cyclization cascade by manipulating the stability and conformation of critical carbocation intermediates. A majority of mutant enzymes exhibit only relatively modest 2–100-fold losses of catalytic activity, suggesting that residues responsible for triggering substrate ionization readily tolerate mutations deeper in the active site cavity.
INTRODUCTION
The family of terpenoid natural products, which includes steroids and carotenoids, contains more than 70,000 unique compounds identified to date (Dictionary of Natural Products, http://dnp.chemnetbase.com). This vast chemodiversity belies relatively simple roots in just two fundamental isoprenoid building blocks, the C5 compounds dimethylallyl diphosphate and isopentenyl diphosphate.1,2 These simple universal precursors can be coupled in head-to-tail fashion to generate longer acyclic isoprenoids such as geranyl diphosphate (C10), farnesyl diphosphate (C15), and geranylgeranyl diphosphate (C20),3,4 which in turn are substrates for terpenoid cyclases. While the specific mechanism of isoprenoid diphosphate cyclization varies from one cyclase to another, common mechanistic features include initial ionization of the allylic diphosphate substrate followed by a complex reaction sequence that proceeds through electrophilic attack on the internal double bonds involving multiple carbocation intermediates.5–9
Product chemodiversity for an individual terpenoid cyclase is a measure of its function as a template and chaperone. A terpenoid cyclase such as aristolochene synthase from Aspergillus terreus can be a strict chaperone and generate a single product with high fidelity.10 Alternatively, a terpenoid cyclase can serve as a more permissive chaperone and allow for multiple cyclization cascades leading to complex product mixtures. For example, γ-humulene synthase from Abies grandis generates 52 different sesquiterpene products from the substrate farnesyl diphosphate.11 Thus, the catalytic activity of a terpenoid cyclase is described not only by its reaction kinetics, but also by the number and structural diversity of the reaction products it generates.
The structural basis of fidelity and promiscuity in terpenoid cyclase reactions, particularly with regard to the code that relates the three-dimensional contour of an active site template with its cyclization product array, is a topic of intense interest in the field of enzyme structure and function. Recently, we determined the X-ray crystal structure of the sesquiterpene cyclase epi-isozizaene synthase (EIZS) from Streptomyces coelicolor at 1.6 Å resolution.12–14 EIZS is classified as a class I terpenoid cyclase because it utilizes a trinuclear magnesium cluster liganded by conserved DDXXD and NDXXSXXXE motifs15 to trigger formation of the initial substrate carbocation and inorganic pyrophosphate (PPi). Since EIZS is moderately promiscuous, generating at least 5 minor sesquiterpene products in addition to 79% epi-isozizaene from the universal sesquiterpene substrate farnesyl diphosphate (FPP),13,14 it is an ideal platform for the exploration of fidelity and promiscuity in a terpenoid cyclization reaction. Minor products generated by an enzyme may signal potential toe-holds for the introduction of new activities in nature and in the laboratory,16 and this perspective applies to the terpenoid cyclases. For example, limited mutations in the active site contour of γ-humulene synthase can minimize the formation of several major products and maximize the formation of a variety of normally minor products.17 Importantly, residues beyond those that define the immediate active site contour can also significantly influence the product ratios of a terpenoid cyclase.18
Here, we describe the preparation, assay, and comparison of 25 single-site mutants of EIZS (3 of which have been previously described13) in which several key aromatic and aliphatic amino acids comprising the hydrophobic active site contour have been systematically varied (Figure 1). We reasoned that by leaving intact the residues responsible for triggering initial substrate ionization (those residues that coordinate to divalent metal ions and residues that hydrogen bond with the diphosphate group of FPP), each mutant enzyme would be able to generate an initial allylic cation from substrate FPP that might possibly be chaperoned through an alternative reaction sequence due to the remolded active site contour. We focused on three types of residues: (1) the aromatic triad F95, F96, and F198 – the aromatic ring faces of these residues encircle the active site cavity and engage in cation-π interactions with a quaternary ammonium analogue, the benzyltriethylammonium cation (BTAC);13 (2) other aromatic residues that define the active site contour, W203, W325, and F332; and (3) aliphatic residues that contribute to the active site contour (L72, V329), or lie just beneath the solvent-accessible surface of the contour (A236).
Figure 1.
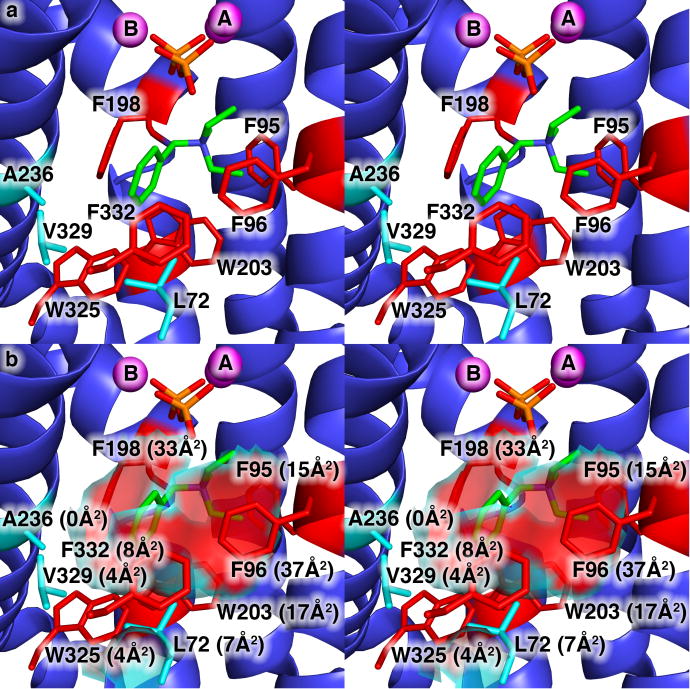
(a) View of the hydrophobic EIZS active site in the wild-type EIZS-Mg2+3-PPi-BTAC complex showing the locations of aromatic residues (red) and aliphatic residues (cyan) that largely define the active site contour shown in (b). Portions of the protein structure in the foreground, including the α-helix containing W325, V329, and F332, are cut away to enable the view of the active site. Mg2+ ions appear as magenta spheres; P and O atoms of the PPi anion are orange and red, respectively; C and N atoms of the BTAC cation are green and blue, respectively. The solvent-accessible surface area contributed by each residue is indicated in parentheses. Note that although A236 does not contribute to the solvent accessible surface area in the wild-type enzyme, it is sufficiently close to the surface that substitution of larger side chains will impact the surface contour.
X-ray crystal structure determinations of selected mutants confirm the structure of the remolded active site contour and illuminate the binding mode of the analogue BTAC. Mutants retain varying levels of catalytic activity and exhibit altered product profiles; indeed, some mutants essentially lose the capacity to generate epi-isozizaene and instead generate completely new cyclization products. Specifically, we have tripled the overall product profile, represented by the number of characterized major and minor products generated from substrate FPP. Additionally, we have converted EIZS into six nominally different sesquiterpene synthases, each of which generates an alternative sesquiterpene as the predominant product.
MATERIALS AND METHODS
Mutagenesis and Kinetic Assays
A total of 22 new single-site mutants of EIZS were prepared by PCR mutagenesis. Mutations were introduced into the wild-type EIZS plasmid using forward and reverse primers for each mutant as listed in Table 1. Two optimal conditions for PCR amplification of insert were applied: (1) 100 ng of each forward and reverse primer, 3 μL of 10 mM dNTP mix, 100 ng of template plasmid, 5 μL of Pfu turbo polymerase buffer, and 1 unit of Pfu turbo polymerase diluted with water to a final volume of 50 μL; and (2) 150 ng of each forward and reverse primer, 3 μL of 10 mM dNTP mix, 150 ng of template plasmid, 5 μL of 10X PfuUltra High-Fidelity DNA polymerase buffer, and 1 unit of PfuUltra High-Fidelity DNA polymerase diluted with water to a final volume of 50 μL. Optimal PCR conditions required initial denaturation of the reaction mixture at 95 °C for 5 min, addition of polymerase followed by thirty cycles (1 min denaturation at 95 °C, 1 min annealing at Primer Tm – 5 or 10 °C, 7 min extension at 72 °C), and a final 10 min extension at 72 °C followed by a final hold at 4 °C. 1 μL of Dpn1 was added to the PCR mixture and incubated at 37 °C for 1.5 h to digest the template. PCR products were transformed into XL1-Blue cells for DNA isolation and sequencing. DNA was purified (Qiagen mini-prep kit) from cultures from single colonies, and DNA sequencing (DNA Sequencing Facility, University of Pennsylvania) confirmed incorporation of the mutations.
Table 1.
Mutagenic Primers Utilized for Generating EIZS Mutantsa
| Mutation | F/Rb | 5′–3′ Sequence |
|---|---|---|
| L72A | F R |
GTC TGT GCT ACA CGG ACg ctA TGG CGG GCT ACT ACC TG CAG GTA GTA GCC CGC CAT agc GTC CGT GTA GCA CAG AC |
| L72V | F R |
CTG TGC TAC ACG GAC gtg ATG GCG GGC TAC TAC GTA GTA GCC CGC CAT cac GTC CGT GTA GCA CAG |
| L72M | F R |
GTC TGT GCT ACA CGG ACa tgA TGG CGG GCT ACT ACC TG CAG GTA GTA GCC CGC CAT cat GTC CGT GTA GCA CAG AC |
| F95A | F R |
GAC TAC AGC GCG TGG gca TTC GTC TGG GAC GAC GTC GTC CCA GAC GAA tgc CCA CGC GCT GTA GTC |
| F95V | F R |
GAT AGC GGA CTA CAG CGC GTG Ggt tTT TGT CTG GGA CGA CCG TCA C GTG ACG GTC GTC CCA GAC AAA aac CCA CGC GCT GTA GTC CGC TAT C |
| F95M | F R |
GAT AGC GGA CTA CAG CGC GTG Gat gTT TGT CTG GGA CGA CCG TCA C GTG ACG GTC GTC CCA GAC AAA cat CCA CGC GCT GTA GTC CGC TAT C |
| F95H | F R |
GAT AGC GGA CTA CAG CGC GTG Gca tTT TGT CTG GGA CGA CCG TCA C GTG ACG GTC GTC CCA GAC AAA atg CCA CGC GCT GTA GTC CGC TAT C |
| F96V | F R |
GAT AGC GGA CTA CAG CGC GTG gtt CGT TGT CTG GGA CGA CCG TCA C GTG ACG GTC GTC CCA GAC AAC GAA cca CGC GCT GTA GTC CGC TAT C |
| F96L | F R |
GAC TAC AGC GCG TGG TTC tta GTC TGG GAC GAC CGT CAC GTG ACG GTC GTC CCA GAC taa GAA CCA CGC GCT GTA GTC |
| F96Y | F R |
GAT AGC GGA CTA CAG CGC GTG GTT Cta tGT CTG GGA CGA CCG TCA C GTG ACG GTC GTC CCA GAC ata GAA CCA CGC GCT GTA GTC CGC TAT C |
| F96W | F R |
GAC TAC AGC GCG TGG TTC tgg GTC TGG GAC GAC CGT CAC GTG ACG GTC GTC CCA GAC cca GAA CCA CGC GCT GTA GTC |
| F198V | F R |
GAA CTG CGC CGG CTC ACG gtt GCT CAC TGG ATC TGG AC GTC CAG ATC CAG TGA GCa acC GTG AGC CGG CGC AGT TC |
| F198L | F R |
CTC GAA CTG CGC CGG CTC ACG tta GCG CAC TGG ATC TGG ACC GAC GTC GGT CCA GAT CCA GTG CGC taa CGT GAG CCG GCG CAG TTC GAG |
| F198Y | F R |
GTA CCT CGA ACT GCG CCG GCT CAC Gta tGC ACA CTG GAT CTG GAC CGA C GTC GGT CCA GAT CCA GTG TGC ata CGT GAG CCG GCG CAG TTC GAG GTA C |
| A236G | F R |
GTC AGG AAT TCg gcG CCT GGT ACA AC GTT GTA CCA GGC gcc GAA TTC CTG AC |
| A236F | F R |
GCG TTG CTG AGT CAG GAA TTC ttt GCC TGG TAC AAC GAC CTC TGC GCA GAG GTC GTT GTA CCA GGC aaa GAA TTC CTG ACT CAG CAA CGC |
| A236M | F R |
GCG TTG CTG AGT CAG GAA TTC atg GCC TGG TAC AAC GAC CTC TGC GCA GAG GTC GTT GTA CCA GGC cat GAA TTC CTG ACT CAG CAA CGC |
| W325F | F R |
GGC AAT ATG CGG AAC TTC ttc AGT TCC GTC TAC TG CAG TAG ACG GAA CTG AAg aaG TTC CGC ATA TTG CC |
| V329A | F R |
CTG GTT CAG TTC Cgc gTA CTG GTT CCA CC GGT GGA ACC AGT Acg cGG AAC TGA ACC AG |
| V329G | F R |
CTG GTT CAG TTC Cgg tTA CTG GTT CCA CCA CGA GTC GAC TCG TGG TGG AAC CAG TAa ccG GAA CTG AAC CAG |
| V329I | F R |
CTG GTT CAG TTC Cat tTA CTG GTT CCA CCA CGA GTC GAC TCG TGG TGG AAC CAG TAa atG GAA CTG AAC CAG |
| V329M | F R |
CTG GTT CAG TTC Cat gTA CTG GTT CCA CCA CGA GTC GAC TCG TGG TGG AAC CAG TAc atG GAA CTG AAC CAG |
| F332A | F R |
CAG TTC CGT CTA CTG Ggc aCA CCA CGA GTC CGG C GCC GGA CTC GTG GTG tgc CCA GTA GAC GGA ACT G |
Lowercase letters represent the mutant codon introduced.
F = forward primer, R = reverse primer.
Mutant proteins were expressed and purified using procedures identical to those previously reported.13 Steady-state kinetic parameters for the generation of hydrocarbon products by EIZS mutants were measured as previously described.13 Briefly, enzymes were solubilized in 50 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.5), 20% glycerol, 100 mM NaCl, 10 mM MgCl2, and 5 mM β-mercaptoethanol (BME). Each series of assays was performed 2–3 times using 0.025–50 μM [1-3H]FPP (100 mCi/mmol). The optimal concentration of each EIZS mutant was determined to be that at which the dependence of product formation on enzyme concentration was linear and substrate turnover was less than 10%. A 1-mL reaction mixture in a test tube was overlaid with 1 mL of hexane immediately after addition of substrate, covered with aluminum foil, and incubated for 12–15 min at 30 °C. The reaction was quenched by the addition of 75 μL of 500 mM EDTA (pH 8.0) and the mixture was vortexed for 20 s. The hexane extract was passed through a silica gel column (1 cm of silica gel in a Pasteur pipette) directly into a scintillation vial containing 5 mL of scintillation fluid. The aqueous phase was extracted with an additional 2 × 1 mL of hexane and passed through the same silica gel column. Finally, the column was washed with an additional 1 mL of diethyl ether. A Beckman scintillation counter was used to measure total product formation, and the substrate concentration versus rate of product formation data was fit by nonlinear regression using the program Prism to determine steady-state kinetic parameters, kcat and KM, based on the known total enzyme concentration.
Analysis of Sesquiterpene Product Arrays
80 μM FPP was incubated with 0.8 μM EIZS wild type or each mutant in 6 mL of buffer (50 mM PIPES (pH 6.5), 15 mM MgCl2, 100 mM NaCl, 20% glycerol, and 5 mM BME) and overlaid with 3 mL of HPLC grade n-pentane in a glass test tube at 30 °C for 18 h. The reaction mixture was extracted three times with n-pentane; pentane extracts were dried with anhydrous MgSO4, filtered and concentrated on an ice/water mixture under reduced pressure until the volume reached 100 μL. Extracts were analyzed using an Agilent 5890 Series II mass spectrometer with a 30 m × 0.25 mm HP5MS capillary column in EI positive mode using a temperature program with an initial hold at 60 °C for 2 min, a 20 °C/min temperature gradient from 60–280 °C, a final hold at 280 °C for 2 min, and a solvent delay of 3 min. Analysis of the organic extracts resulting from the incubation of FPP with the mutant cyclases by gas chromatography and mass spectrometry (GC-MS) revealed the formation of mixtures of sesquiterpene hydrocarbons with m/z 204. Compounds were identified by comparison of their individual mass spectra and chromatographic retention indices with those of authentic compounds using the MassFinder 4.0 Program and Database.19
X-ray Crystal Structure Determinations
The L72V, A236G, W325F and V329A EIZS mutants were crystallized by the hanging drop vapor diffusion method using the same conditions employed for crystallization of the wild-type enzyme.13 Briefly, a 4 μL drop of protein solution [8 mg/mL EIZS mutant, 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 1 mM TCEP, 2 mM sodium pyrophosphate, 2 mM BTAC] was added to a 4 μL drop of precipitant solution [100 mM Bis-Tris (pH 5.5), 25–28% polyethylene glycol 3350, 0.2 M (NH4)2SO4] and equilibrated against a 1 mL reservoir of precipitant solution at room temperature. Crystals of each mutant were isomorphous with those of the wild-type enzyme (space group P21, a = 53.2 Å, b = 47.4 Å, c = 75.4 Å, β = 95.5°)13 and diffracted to 1.76–2.30 Å resolution at the National Synchrotron Light Source (NSLS), beamline X-29, or the Advanced Photon Source (APS), NE-CAT beamline 24-ID-C. Data collection statistics are recorded in Table 2.
Table 2.
X-ray Crystal Structure Data Collection and Refinement Statistics
| EIZS structure | Unliganded wild-type | L72V Mg2+3-PPi-BTAC complex | F95M Mg2+3-PPi-BTAC complex | F95H Mg2+3-PPi-BTAC complex | A236G Mg2+3-PPi-BTAC complex | W325F Mg2+3-PPi complex | V329A Mg2+3-PPi-BTAC complex |
|---|---|---|---|---|---|---|---|
| Data Collection | |||||||
|
| |||||||
| Wavelength (Å) | 1.075 | 1.008 | 1.542 | 1.075 | 0.9795 | 0.9795 | 1.008 |
| Resolution (Å) | 50 – 2.40 | 50 – 2.10 | 50 – 2.45 | 50 – 2.10 | 50 – 1.76 | 50 – 2.46 | 50 – 1.95 |
| Unique reflections | 13,825 | 22,011 | 13,884 | 23,067 | 37,050 | 13,755 | 26,839 |
| Completeness (%)a | 95.8 (94.5) | 99.9 (99.2) | 99.9 (100.0) | 100.0 (100.0) | 99.2 (98.2) | 99.9 (99.9) | 97.3 (97.4) |
| Redundancy | 3.1 (2.9) | 3.6 (3.5) | 4.4 (4.2) | 5.4 (5.4) | 3.3 (3.1) | 3.6 (3.4) | 4.2 (3.7) |
| I/σa | 11.9 (2.4) | 12.2 (3.5) | 10.9 (3.5) | 11.9 (4.9) | 13.7 (2.7) | 7.6 (3.0) | 13.0 (3.0) |
| Rmergea,b | 0.091 (0.335) | 0.104 (0.301) | 0.119 (0.400) | 0.136 (0.323) | 0.071 (0.339) | 0.148 (0.427) | 0.116 (0.432) |
|
| |||||||
| Refinement | |||||||
|
| |||||||
| Rcryst / Rfreec | 0.191/0.250 | 0.173/0.206 | 0.185/0.224 | 0.160/0.204 | 0.164/0.198 | 0.182/0.231 | 0.152/0.201 |
| R.m.s. deviations | |||||||
| Bonds (Å) | 0.004 | 0.002 | 0.002 | 0.003 | 0.007 | 0.002 | 0.007 |
| Angles (°) | 0.8 | 0.7 | 0.7 | 0.7 | 1.1 | 0.6 | 1.0 |
| Dihedral angles (°) | 17 | 13 | 15 | 14 | 15 | 12 | 17 |
| Protein atoms | 2577 | 2789 | 2778 | 2795 | 2804 | 2770 | 2812 |
| Solvent atoms | 53 | 241 | 80 | 268 | 283 | 47 | 376 |
| Mg2+ cations | 3 | 3 | 3 | 3 | 3 | 3 | |
| PPi anion | 1 | 1 | 1 | 1 | 1 | 1 | |
| BTAC cation | 1 | 1 | 1 | 1 | 1 | ||
| glycerol | 1 | 4 | 1 | ||||
| SO42− anion | 2 | 1 | 1 | 1 | 1 | 2 | |
| HPO42− | 1 | ||||||
| Ramachandran plot (%)d | |||||||
| Most favored | 91.0 | 94.7 | 94.7 | 94.7 | 95.7 | 94.7 | 94.0 |
| Additional allowed | 8.6 | 5.3 | 5.3 | 5.3 | 4.3 | 5.3 | 6.0 |
| Generously allowed | 0.4 | ||||||
| PDB accession code | 4LTV | 4LXW | 4LTZ | 4LZ3 | 4LZ0 | 4LZC | 4LUU |
Values in parentheses refer to the highest shell of data.
Rmerge = Σ|Ih − 〈I〉h|/ΣIh, where 〈I〉h is the average intensity calculated from replicate reflections.
Rcryst = Σ| |Fo| − |Fc| |/Σ|Fo| for reflections contained in the test set held aside during refinement. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Calculated with PROCHECK version 3.4.4.
Crystals of unliganded wild-type EIZS were prepared by soaking crystals of the wild-type EIZS-Mg2+3-BTAC complex13 in a chelation buffer [5 mM 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid, 15 mM tetrasodium ethylenediaminetetraacetic acid, 100 mM Bis-Tris (pH 5.5), 30% polyethylene glycol 3350, 200 mM (NH4)2SO4, and 15% glycerol] for 7 days. Crystals diffracted X-rays to 2.4 Å resolution at NSLS beamline X-29 and were isomorphous with those of the liganded enzyme.13 Data collection statistics are recorded in Table 2.
F95M EIZS was crystallized by the sitting drop vapor diffusion method. A 0.6 μL drop of protein solution [15 mg/mL F95M EIZS, 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 2 mM TCEP, 2 mM sodium pyrophosphate, and 2 mM BTAC] was added to 0.6 μL of precipitant solution [100 mM Bis-Tris (pH 5.5), 200 mM (NH4)2SO4, and 25% polyethylene glycol 3350] and equilibrated against a 110 μL well reservoir of precipitant solution. Crystals appeared within one week. X-ray diffraction data (2.45 Å resolution) were collected on a Rigaku IV++ image plate area detector mounted on a Rigaku RU200HB rotating anode X-ray generator (8.048 keV/1.5406 Å); crystals were isomorphous with those of the wild-type enzyme.13 Data collection statistics are recorded in Table 2.
F95H EIZS was crystallized by the sitting drop vapor diffusion method. A 0.6 μL drop of protein solution [10 mg/mL F95H EIZS, 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 10 mM MgCl2, 10% glycerol, 2 mM TCEP, 2 mM sodium pyrophosphate, and 2 mM BTAC] was added to 0.6 μL of precipitant solution [40 mM KH2PO4, 16% polyethylene glycol 8000, and 20% glycerol] and equilibrated against a 110 μL well reservoir of precipitant solution. Crystals appeared in one week and diffracted X-rays to 2.1 Å resolution at NSLS beamline X-29. Crystals belonged to space group P212121 with unit cell parameters a = 46.7 Å, b = 75.6 Å, c = 107.6 Å (one monomer in the asymmetric unit).
Crystal structures were solved by molecular replacement using the program PHASER20,21; the atomic coordinates of wild-type EIZS less ligand and solvent molecules were used as a search probe for rotation and translation function calculations. Initial electron density maps of each mutant clearly revealed the substituted amino acid side chain, which was modeled into density and refined. The initial electron density map of unliganded EIZS revealed an empty active site. Iterative cycles of refinement and manual model building were performed with PHENIX and COOT, respectively.22,23 Ligands and water molecules were included in later cycles of refinement. Individual atomic B-factors were utilized in refinement. Data collection and refinement statistics for each structure are listed in Table 2. The r.m.s. deviations of Cα atoms between wild-type and mutant EIZS complexes were calculated with the graphics program COOT.
RESULTS
Catalytic Activity of EIZS Mutants
Steady-state kinetic parameters for all mutants reflect the generation of 3H-labeled hexane-extractable products and are reported in Table 3; data for the F96A, F198A, and W203F mutants, which were previously reported by Aaron and colleagues,13 are included for comparison. In general, KM values for mutants of aromatic and aliphatic active site residues are within an order of magnitude of those measured for wild-type EIZS. The most significant difference in KM is observed for V329M EIZS, which exhibits a 20-fold higher KM value compared with that of the wild-type enzyme. More significant changes are observed for the kcat values of various mutants; overall, changes in kcat tend to be greater for replacement of aromatic residues compared with substitution of aliphatic residues. In particular, substitution of F95, F96, and F198 by smaller aliphatic side chains results in 11–730-fold reductions in kcat. This aromatic triad encircles the active site (Figure 1) and is proposed to stabilize carbocationic transition states and intermediates through cation-π interactions.13
Table 3.
Kinetic Parameters for Wild-Type and Mutant EIZS Enzymes
| Enzyme | kcat (ms−1) | KM (nM) | kcat/KM (M−1s−1) |
|---|---|---|---|
| WT | 84 ± 1 | 330 ± 20 | 260000 ± 10000 |
|
| |||
| Aromatic | Mutants | ||
|
| |||
| F95A | 1.8 ± 0.2 | 672 ± 2 | 2700 ± 300 |
| F95V | 7.4 ± 0.4 | 1700 ± 300 | 4400 ± 600 |
| F95M | 3.16 ± 0.09 | 275 ± 4 | 11500 ± 500 |
| F95H | 1.72 ± 0.02 | 650 ± 30 | 2600 ± 100 |
|
| |||
| F96Aa | 0.24 ± 0.02 | 800 ± 100 | 310 ± 60 |
| F96V | 1.15 ± 0.02 | 580 ± 30 | 199 ± 7 |
| F96L | 2.57 ± 0.02 | 820 ± 60 | 3100 ± 200 |
| F96Y | 0.19 ± 0.01 | 1800 ± 100 | 107 ± 2 |
| F96W | 0.35 ± 0.02 | 310 ± 70 | 1200 ± 400 |
|
| |||
| F198Aa | 0.30 ± 0.02 | 1200 ± 200 | 250 ± 50 |
| F198V | 1.66 ± 0.09 | 2800 ± 400 | 600 ± 60 |
| F198L | 4.75 ± 0.05 | 4200 ± 400 | 1100 ± 100 |
| F198Y | 15.04 ± 0.06 | 4100 ± 100 | 3600 ± 100 |
|
| |||
| W203Fa | 0.34 ± 0.03 | 1500 ± 200 | 230 ± 40 |
|
| |||
| W325F | 22 ± 2 | 160 ± 60 | 14000 ± 5000 |
|
| |||
| F332A | 0.105 ± 0.008 | 360 ± 90 | 290 ± 80 |
|
| |||
| Aliphatic | Mutants | ||
|
| |||
| L72A | 8.81 ± 0.09 | 3000 ± 100 | 2900 ± 100 |
| L72V | 44 ± 3 | 600 ± 100 | 70000 ± 20000 |
| L72M | 11 ± 2 | 3200 ± 900 | 3400 ± 400 |
|
| |||
| A236G | 66 ± 2 | 450 ± 50 | 150000 ± 20000 |
| A236M | — | — | — |
| A236F | 0.0424 ± 0.0007 | 230 ± 50 | 190 ± 40 |
|
| |||
| V329A | 126 ± 7 | 470 ± 80 | 270000 ± 50000 |
| V329G | 19.8 ± 0.1 | 1590 ± 60 | 12500 ± 500 |
| V329I | 22.8 ± 0.3 | 770 ± 40 | 29000 ± 1000 |
| V329M | 0.640 ± 0.004 | 6500 ± 300 | 98 ± 3 |
mutants previously reported (ref. 13).
Two tryptophan residues, W203 and W325, although only marginally exposed to the central active site cavity compared to F95 and F96, were substituted by phenylalanine in order to probe the importance of their aromatic indole side chains. Whereas W203F EIZS exhibits a 250-fold reduction in kcat compared to wild-type EIZS, W325F EIZS shows only a modest 4-fold reduction in kcat. Thus, W203 has more of an influence on catalysis, probably because W203 contributes more surface area to the active site contour than W325 (Figure 1). The F332A mutant exhibits an 800-fold reduction in kcat, consistent with the fact that the edge of this aromatic residue also defines part of the active site contour.
Substitution of the aliphatic residues L72, V329, and A236 with either larger or smaller aliphatic side chains appears to be better tolerated, with kcat values comparable to or within an order of magnitude of that measured for the wild-type enzyme (kcat = 0.084 s−1), except for V329M EIZS, for which kcat is reduced more than 100-fold to 6.4 × 10−4 s−1. Interestingly, A236M EIZS yields no detectable products, while A236F EIZS exhibits nearly a 2000-fold reduction in kcat, suggesting that a small side-chain at residue 236 is mandatory for efficient catalysis. Accordingly, A236G EIZS exhibits nearly the same steady-state kinetic parameters as wild-type EIZS.
Sesquiterpene Product Arrays Generated by EIZS Mutants
Sesquiterpene hydrocarbon mixtures generated by each of the EIZS mutants were subjected to analysis and quantification by gas chromatography-mass spectrometry (GC-MS) (Table 4). Sesquiterpene products identified in these mixtures are shown in Figure 2. In general, replacement of F95, F96, or F198 in the aromatic triad compromises the generation of epi-isozizaene (18) – generally more so for F96 and F198, which each contribute greater solvent-accessible surface area to the active site contour than does F95 (Figure 1). It appears that the fidelity of epi-isozizaene formation is most sensitive to substitution for F96, particularly by smaller aliphatic residues – the F96A, F96V, and F96L mutants generate less than 10% epi-isozizaene. Only F96Y EIZS, representing a nearly isosteric substitution of the aromatic ring of residue 96, retains the fidelity of epi-isozizaene generation, in spite of a substantial 2400-fold reduction in catalytic efficiency (kcat/KM). It is nonetheless notable that the product distribution can be significantly reprogrammed by only a single amino acid substitution for each member of the aromatic triad, often with retention of significant catalytic activity. For example, the F95M mutation converts EIZS into a predominant β-acoradiene synthase, generating a mixture consisting of 68% β-acoradiene (13) and only 32% epi-isozizaene (18), with only a 23-fold reduction in catalytic efficiency. F95M EIZS is thus directed to make an entirely different predominant cyclization product with relatively good catalytic efficiency. Analysis of the biosynthetic manifold (Figure 3) suggests that the observed change in product distribution is the consequence of diversion of the normal cyclization of the homobisabolyl cation intermediate.
Table 4.
Sesquiterpene Product Arrays Generated by EIZS Mutants
| Enzyme | Relative average percentage of sesquiterpene products (product numbering shown in Figure 1)
|
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | N/Aa | |
| WTb | 5 | 2 | 1 | 2 | 9 | 79 | |||||||||||||
| WTc | 1 | 1 | 5 | 93 | |||||||||||||||
| WTd | 99 | 1 | |||||||||||||||||
|
| |||||||||||||||||||
| L72A | 6 | 6 | 13 | 67 | 8 (2) | ||||||||||||||
| L72V | 3 | 2 | 14 | 43 | 38 | ||||||||||||||
| L72M | 7 | 2 | 10 | 80 | |||||||||||||||
|
| |||||||||||||||||||
| F95A | 4 | 3 | 35 | 1 | 56 (3) | ||||||||||||||
| F95V | 26 | 2 | 66 | 6 (2) | |||||||||||||||
| F95M | 68 | 32 | |||||||||||||||||
| F95H | 50 | 2 | 44 | 4 (2) | |||||||||||||||
|
| |||||||||||||||||||
| F96Ab | 70 | 7 | 9 | 8 | 7 (1) | ||||||||||||||
| F96V | 44 | 14 | 5 | 6 | 31 (1) | ||||||||||||||
| F96L | 2 | 4 | 10 | 8 | 3 | 16 | 2 | 30 | 14 | 9 | 2 (1) | ||||||||
| F96Y | 3 | 1 | 18 | 78 | |||||||||||||||
| F96W | 9 | 65 | 3 | 23 (3) | |||||||||||||||
|
| |||||||||||||||||||
| F198Ab | 5 | 24 | 6 | 20 | 13 | 12 | 20 (3) | ||||||||||||
| F198V | 3 | 3 | 2 | 48 | 6 | 3 | 4 | 13 | 10 | 8 (3) | |||||||||
| F198L | 1 | 61 | 14 | 24 | |||||||||||||||
| F198Y | 89 | 5 (2) | |||||||||||||||||
|
| |||||||||||||||||||
| W203Fb | 6 | 47 | 7 | 7 | 14 | 21 (4) | |||||||||||||
|
| |||||||||||||||||||
| A236G | 2 | 70 | 28 (4) | ||||||||||||||||
| A236M | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| A236F | 8 | 15 | 77 | ||||||||||||||||
|
| |||||||||||||||||||
| W325F | 45 | 50 | 5 (3) | ||||||||||||||||
|
| |||||||||||||||||||
| V329A | 4 | 3 | 85 | 8 (1) | |||||||||||||||
| V329G | 2 | 8 | 6 | 2 | 7 | 75 | |||||||||||||
| V329I | 2 | 98 | |||||||||||||||||
| V329M | 33 | 62 | 5 (1) | ||||||||||||||||
|
| |||||||||||||||||||
| F332A | 1 | 37 | 56 | 6 (3) | |||||||||||||||
N/A, not assigned; value in parentheses indicates number of unknown products detected by GC-MS.
Product array reported in ref. 13.
Assay run at 20° C.
Assay run at 4° C.
Figure 2.
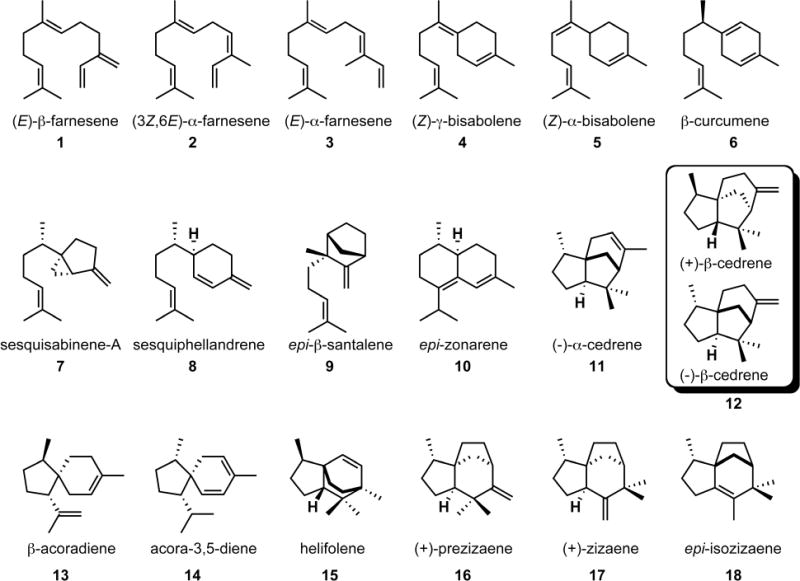
Sesquiterpene products generated by EIZS mutants as detected by GC-MS analysis and identified by comparison of their individual mass spectra and chromatographic retention indices with those of authentic compounds in the MassFinder 4.0 Database.
Figure 3.
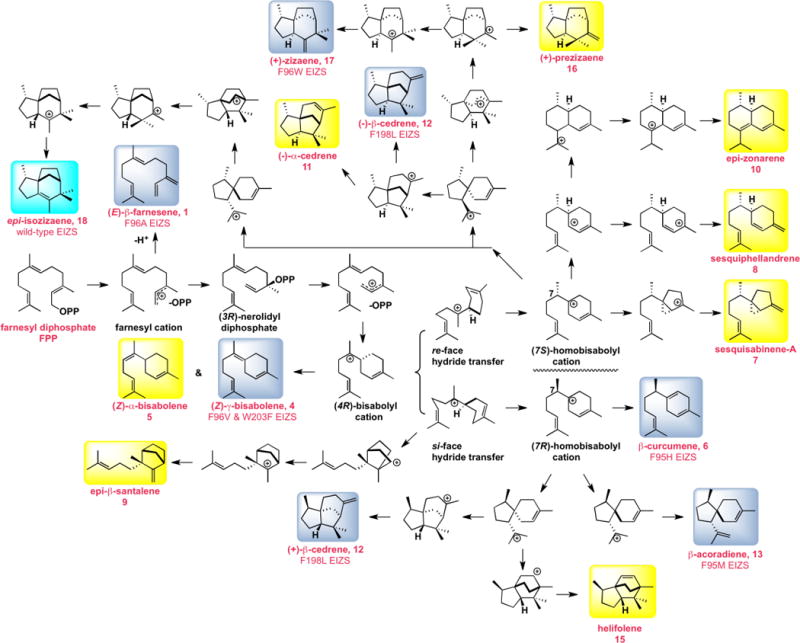
Biosynthetic manifold illustrating the proposed mechanism of FPP cyclization reactions catalyzed by EIZS and its site-specific mutants. Product numbering is identical to that in Figure 2. The predominant products of wild-type EIZS and certain EIZS mutants are highlighted in cyan and blue boxes, respectively, and side products are highlighted in yellow boxes; product percentages are listed in Table 4. For clarity, farnesene isomers 2 and 3 generated at low levels (1%) by wild-type EIZS are not shown, nor is acora-3,5-diene (14) generated at low levels (2%) by F95V EIZS. Regarding product stereochemistry, it should be noted that the determination of the absolute configuration of the (+)-epi-isozizaene produced by EIZS has been previously reported.12 Although the absolute configurations of (+)-prezizaene (16) and (+)-zizaene (17) produced by EIZS mutants have not been directly determined,14 they can be provisionally assigned from the natural cyclization mechanism, since they represent simple late-stage diversions of the pathway. By contrast, generation of both enantiomers of β-cedrene (12) has been directly determined for F198L EIZS by direct chiral GC-MS comparison with authentic standards, as described in the text. The formation of both (−) and (+)-12 requires that EIZS or its mutants be capable of generating both the (7S)- and (7R)-homobisabolyl cations, as shown. The absolute configurations of the remaining aberrant sesquiterpene cyclization products have not been determined and are arbitrarily illustrated for (Z)-α-bisabolene (5), β-curcumene (6), sesquisabinene A (7), sesquiphellandrene (8), epi-β-santalene (9), epi-zonarene (10), (−)-α-cedrene (11), β-acoradiene (13), and helifolene (15). Formation of the enantiomers of any of these compounds can readily be accommodated by simple variations of the mechanistic scheme.
By contrast, replacement of the phenyl ring by an imidazole group in F95H EIZS results in the generation of a mixture of 50% β-curcumene (6) and 44% epi-isozizaene (18), accompanied by only 2% β-acoradiene (13), along with an additional 4% of 2 products of unassigned structure. Although the overall catalytic efficiency is compromised 100-fold in this mutant, the rigidity and perhaps the polarity of the substituted imidazole side chain apparently favor early termination of the cyclization cascade by direct deprotonation of the homobisabolyl cation. This suggests that F95 plays a key role in stabilizing and directing the chemistry of this monocyclic intermediate (Figure 3).
Similarly, substitution of F198 with the progressively smaller aliphatic residues leucine, valine, and alanine reduces the proportion of epi-isozizaene to 24%, 10%, and 0% of the total sesquiterpene products, respectively. Notably, F198L EIZS generates instead 61% β-cedrene (12), of which 80% is (−)-β-cedrene and 20% is (+)-β-cedrene based on GC-MS comparison with authentic standards; F198L EIZS also generates 24% epi-isozizaene (18), 14% zizaene (17), and 1% sesquisabinene-A (7). While the catalytic efficiency of this mutant is 236-fold lower than that of wild-type EIZS, it is notable that a single amino acid substitution is sufficient to convert EIZS predominantly into a β-cedrene synthase. Importantly, the generation of (+)-β-cedrene indicates that synthetic sequences proceeding through the alternative (7R)-homobisabolyl enantiomer, rather than the normal (7S)-enantiomer leading to (−)-β-cedrene and epi-isozizaene formation, are feasible. The absolute configurations of the remaining aberrant sesquiterpene cyclization products shown in Figure 3 have not been determined and are arbitrarily illustrated.
Interestingly, F96V EIZS and W203F EIZS generate 44% and 47% (Z)-γ-bisabolene (4), respectively, as part of more complex product mixtures containing significantly reduced proportions of epi-isozizaene (6% and 14%, respectively). Early termination of the cyclization sequence by proton elimination from the (4R)-bisabolyl cation intermediate to yield 4 suggests that the steric bulk of each of these residues, as well as presumed cation-π interactions with F96, play a key role in directing the biosynthetic fate of the (4R)-bisabolyl cation.
Finally, the F96W substitution generates a mixture of 65% zizaene (17), 9% (3Z,6E)-α-farnesene (2), and only 3% epi-isozizaene (18), along with 23% of 3 unidentified sesquiterpene hydrocarbon products. An increase in steric bulk at residue 96 thus redirects the cyclization of the (7S)-homobisabolyl cation to favor attack on the opposite face of the side-chain trisubstituted double bond. By contrast, as previously reported,13 F96A EIZS, bearing a much smaller, non-aromatic methyl side chain, generates a mixture containing 70% of the acyclic sesquiterpene (E)-β-farnesene (1) – replacement of this key aromatic side chain with a methyl group thus results in the quenching of the initially formed farnesyl cation. While catalytic efficiency is reduced more than 800-fold in this mutant relative to wild-type EIZS, the catalytic efficiency for enzymatic formation of (E)-β-farnesene by F96A EIZS (kcat/KM = 310 M−1s−1) is nonetheless only ~14-fold lower than that reported for the wild-type wormwood (E)-β-farnesene synthase (kcat/KM = 4500 M−1s−1).24
Substitutions for the aliphatic residues L72, A236, and V329 mostly result in less dramatic changes in sesquiterpene product spectrum compared to replacement of aromatic residues. All such mutants generate mixtures containing at least 60% epi-isozizaene, with the exception of L72V EIZS, which generates a mixture of 43% epi-isozizaene (18), 14% zizaene (17), 3% sesquisabinene-A (7), 2% β-cedrene (12), and 38% of an as yet unidentified sesquiterpene hydrocarbon product. The majority of these aliphatic mutants generate zizaene in addition to epi-isozizaene, representing the alternative cyclization stereochemistry for the common (7S)-homobisabolyl cation intermediate (Figure 3). Since the side chains of these aliphatic amino acids make smaller contributions to the active site contour of wild-type EIZS, the effects of their replacement may be limited. The overall catalytic activity as well as the cyclization fidelity both strongly depend on the presence of a small residue at position 236, which is normally buried just beneath the solvent-accessible surface of the active site contour. Thus, A236G EIZS generates a mixture of 70% epi-isozizaene (18), 2% zizaene (17), and 28% of a mixture of 4 unidentified sesquiterpenes with a near-normal catalytic rate, whereas the corresponding A236F EIZS generates a mixture of 77% epi-isozizaene (18), 15% zizaene (17), and 8% β-cedrene (12) with a nearly 2000-fold reduction in kcat. By contrast, A236M EIZS is completely devoid of activity.
Crystal Structures of Unliganded EIZS and Selected Mutants
X-ray crystal structures of unliganded EIZS and of selected mutants were determined at resolutions of 1.76–2.46 Å. The structure of unliganded EIZS, the first to be determined of the unliganded wild-type enzyme, reveals an open active site conformation that is essentially identical to that previously observed in the corresponding unliganded D99N mutant,13 with a root-mean-square (r.m.s.) deviation of 0.51 Å for 308 Cα atoms between the two structures. Interestingly, active site aromatic residue F96 adopts a different conformation in the unliganded enzymes compared with the structures of all other wild-type or mutant EIZS enzymes complexed with 3 Mg2+ ions and PPi, whether or not the carbocation mimic, benzyltriethyl ammonium cation (BTAC), is also bound.13 The change in conformation of F96 triggered by the binding of Mg2+3-PPi and consequent full active site closure corresponds to rotations of ~105° and ~10° about side chain torsion angles χ1 and χ2, respectively (Figure 4). Such induced fit conformational changes in the active site contour of a terpenoid cyclase are unusual, but not unprecedented. For example, F578 of bornyl diphosphate synthase undergoes a significant conformational change upon ligand binding.25 Comparisons of high-resolution unliganded and liganded contours of other terpenoid cyclases, however, generally reveal conformational changes that reflect rigid-body movements rather than significant modification of side chain conformations.
Figure 4.
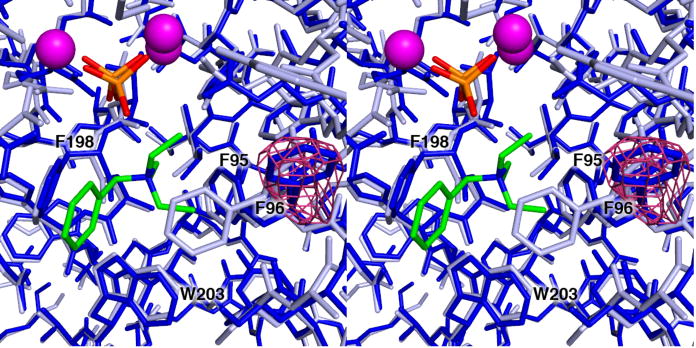
Omit electron density map contoured at 2.2σ showing the conformation of F96 in the unliganded enzyme (dark blue), compared with the conformation of this residue in EIZS-ligand complexes as represented by the wild-type EIZS-Mg2+3-PPi-BTAC complex (light blue). Mg2+ ions appear as magenta spheres; P and O atoms of the PPi anion are orange and red, respectively; C and N atoms of the BTAC cation are green and blue, respectively. Selected active site residues are labeled. The aromatic side chain of F96 undergoes a significant induced-fit conformational change as the terpenoid cyclase active site undergoes the transition from a fully open to a fully closed conformation.
X-ray crystal structures of Mg2+3-PPi-BTAC complexes were determined for the L72V, F95M, F95H, A236G, and V329A mutants of EIZS, while the structure of the Mg2+3-PPi complex was determined for the W325F mutant. Each of these mutants was selected because it exhibits significant residual catalytic activity (indeed, V329A exhibits enhanced catalytic activity; see Table 3), and/or interesting differences in the array of resulting sesquiterpene products compared to the wild-type enzyme (Table 4). All of these mutants, except for F95H EIZS, were readily crystallized under the same conditions as the wild-type protein. In general, each single amino acid substitution causes minimal structural perturbations, with r.m.s. deviations from the structure of the wild-type EIZS-Mg2+3-PPi complex as follows: L72V, 0.13 Å (340 Cα atoms); F95M, 0.17 Å (340 Cα atoms); F95H, 0.22 Å (340 Cα atoms); A236G 0.16 Å (340 Cα atoms); V329A, 0.08 Å (340 Cα atoms); W325F, 0.18 Å (338 Cα atoms). The binding mode of the Mg2+3-PPi cluster is generally unchanged in each mutant, except that the PPi anion rotates slightly in F95M EIZS such that the bridging oxygen atom moves ~ 0.6 Å, and the Mg2+C ion moves ~1.0–1.5 Å in the F95M and W325F mutants (Figure 5). Thus, site-specific mutations can be made in the hydrophobic region of the active site without significantly perturbing neighboring residues, including those that play an important role in triggering substrate ionization.
Figure 5.
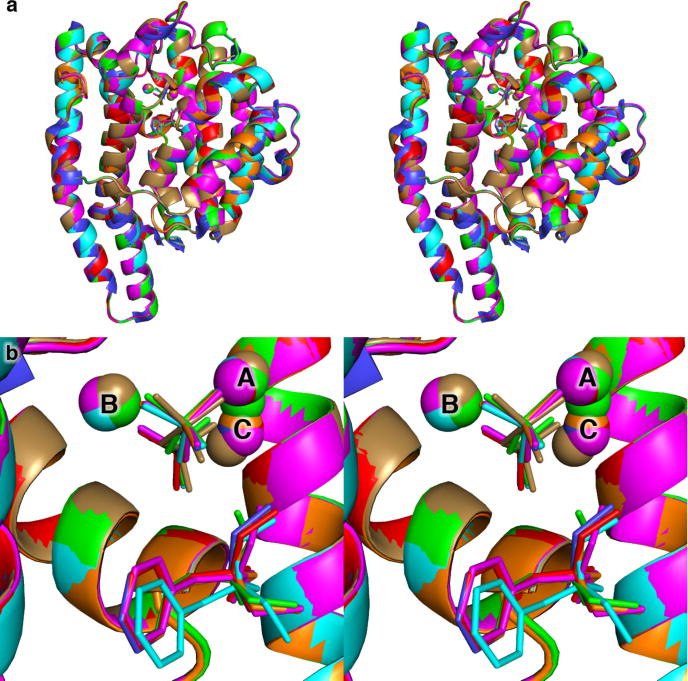
Superposition of wild-type (blue), L72V (cyan), F95M (green), F95H (magenta), A236G (orange), and V329A (red) EIZS mutants complexed with 3 Mg2+3 ions, PPi, and BTAC, and W325F EIZS complexed with 3 Mg2+3 ions and PPi (tan).
The position and orientation of BTAC is unchanged in A236G EIZS and V329A EIZS, which still generate epi-isozizaene as the major sesquiterpene cyclization product (70% and 85%, respectively; see Table 4). However, BTAC rotates by ~20° and moves ~0.5 Å deeper into the active site of L72V EIZS as a consequence of the smaller side chain at this position (Figure 5). The proportion of epi-isozizaene generated is reduced to 43% in this mutant. Interestingly, BTAC appears to be absent from the active site of W325F EIZS, although some uninterpretable electron density peaks are observed that could correspond to disordered BTAC; the formation of epi-isozizaene generated by this mutant is reduced to 50%. It thus appears that the movement or disorder of BTAC in crystalline complexes with EIZS mutants correlates with a higher potential for alternative cyclization sequences. Possibly, more authentic mimics of carbocation intermediates in the EIZS-catalyzed cyclization mechanism, such as (4R)-azabisabolene,26 which mimics the (4R)-bisabolyl cation intermediate, would provide an even more sensitive indicator of the potential for redirected cyclization cascades in EIZS mutants.
DISCUSSION
Active Site Mutations Dramatically Enhance Product Chemodiversity
We have demonstrated that the hydrophobic active site contour of EIZS can be remolded with single amino acid substitutions to adopt a predictable, alternative shape based on analysis of three-dimensional structures of wild-type and mutant enzymes. Furthermore, in a library of 25 site-specific mutants, we have tripled the overall number of major and minor characterized products, and we have converted EIZS into six different sesquiterpene synthases that each generate an alternative predominant cyclization product or, in one case, an elimination product. Thus, F96W EIZS is a zizaene synthase, F95H EIZS is a β-curcumene synthase, F95M EIZS is a β-acoradiene synthase, F198L EIZS is a β-cedrene synthase, and F96V EIZS and W203F EIZS are (Z)-γ-bisabolene synthases, while F96A EIZS is a simple (E)-β-farnesene synthase (Table 4). New cyclization products include simple abortive derailment products resulting from direct quenching of natural carbocationic intermediates, such as the quenching of the (R)-bisabolyl cation to yield (Z)-α-bisabolene and (Z)-α-bisabolene, while other new cyclization products are, in effect, shunt metabolites resulting from diversion of the natural cyclization cascade and subsequent quenching of the derived carbocations, such as the alternative reaction sequences leading to the generation of epi-zonarene or β-acoradiene.
It is also noteworthy that the experimentally observed product array of wild-type EIZS can be reconfigured by single-site mutagenesis to convert a minor product into a major product, and vice versa, in accord with previous studies of terpenoid cyclases. For example, we previously reported that F96A EIZS is an (E)-β-farnesene synthase, since (E)-β-farnesene is the predominant sesquiterpene product (70%); wild-type EIZS generates only 5% (E)-β-farnesene.13 By contrast, we now show that F96W EIZS is a zizaene synthase, since zizaene is the predominant sesquiterpene product (65%) (Table 4); wild-type EIZS generates only 9% zizaene. In the absence of an experimentally-determined crystal structure of γ-humulene synthase, Keasling and colleagues used homology modeling to guide the mutagenesis of a limited number of active site hydrophobic residues, similarly remolding the active site to reconfigure the product array.17 Noel and colleagues demonstrated that terpene product ratios can also be reconfigured by mutagenesis of residues remote from the active site, presumably by long-range structural effects that propagate through the protein scaffolding.18
Aromatic Residues are Hot Spots for Reprogramming or Reconfiguring the Cyclization Cascade
Whether the cyclization cascade of EIZS is reprogrammed to generate entirely new products, or whether the proportions of major and minor products are reconfigured, the most striking results are obtained when aromatic residues are mutated. Active site aromatic residues can stabilize carbocation intermediates through cation-π interactions, as suggested for the side chains of the aromatic triad F95, F96, and F198.13 Additionally, aromatic residues provide significant steric bulk and hydrophobic surface area as they define the contour of the active site, as evident in Figure 1.
Interestingly, certain product distributions in Table 4 can be mapped to specific aromatic residues with regard to their influence on a specific carbocation intermediate. For example, consider F96A EIZS. The steric bulk and weakly polar nature of the F96 side chain presumably play a role in stabilizing the farnesyl cation resulting from the initial ionization of FPP, since F96A EIZS generates (E)-β-farnesene as an immediate derailment product. Inspection of Figure 1 reveals that the side chain of F96 is located ~8 Å beneath the plane defined by Mg2+A, Mg2+B, and Mg2+C. Furthermore, the plane formed by Mg2+A, Mg2+C, and F96 is approximately perpendicular to that formed by the three metal ions. We propose that F96 is ideally positioned so as to interact with the initial farnesyl cation intermediate through both its steric bulk and its capacity for cation-π interactions. The diminished progress along the reaction sequence as the product arrays of F96Y, F96L, F96V, and F96A are compared suggests that steric bulk at position 96 is functionally important, presumably for ensuring the close contact between the C1 and C6 atoms enabling (R)-bisabolyl cation formation.
In another more subtle example, the partitioning of the (7S)-homobisabolyl cation intermediate that leads either to normal formation of epi-isozizaene or aberrant formation of zizaene is a function of the folding of the side chain of this intermediate. The conformation of the (7S)-homobisabolyl cation directs electrophilic attack on one face or the other of the trisubstituted double bond of the side chain, and this conformation is presumably enforced by the three-dimensional contour of the active site. Mutants that exhibit significant increases in zizaene formation cluster in the lower region of the active site where they decrease steric bulk: L72V, W325F, V329M, and F332A. Although the methionine side chain in V329M EIZS is slightly longer than the wild-type valine side chain, this substitution reduces the steric bulk around the Cβ atom of the parent branched valine side chain. Alternatively, both F96Y EIZS and F96W EIZS also produce significantly elevated proportions of zizaene. Both mutants retain the original aromatic character of the wild-type phenylalanine side chain, and their larger side chains are located on the opposite side of the active site from the L72/W325/V329/F332 cluster. Thus, the partitioning of the (7S)-homobisabolyl cation toward zizaene formation (Figure 3) can be favored either by pulling the intermediate into the void created by smaller amino acid mutations in the L72/W325/V329/F332 cluster, or by pushing the intermediate toward this cluster by introducing larger amino acids on the opposite side of the active site. This suggests that the L72/W325/V329/F332 cluster may chaperone the conformation of the prenyl side chain of the (7S)-homobisabolyl cation to generate the acorenyl cation leading to epi-isozizaene formation. A model of the (7S)-homobisabolyl cation bound in the active site of EIZS shows how the L72/W325/V329/F332 cluster may achieve this chaperone function (Figure 6).
Figure 6.
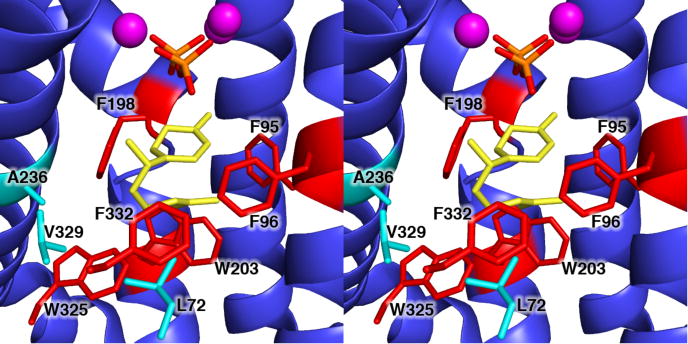
Model of the (7S)-homobisabolyl cation bound in the active site of wild-type EIZS, showing how the L72/W325/V329/F332 cluster could chaperone the precise conformation of the prenyl side chain that would generate the acorenyl cation leading to epi-isozizaene formation as summarized in Figure 3.
Finally, the side chains of F95 and F198 are located in the rear of the active site as viewed in Figure 1. Substitution of these residues with smaller side chains other than alanine may disfavor the 1,2-hydride shift that generates the natural (7S)-homobisabolyl cation intermediate, diverting instead to the enantiomeric (7R)-homobisabolyl cation, which in turn serves as a precursor of β-acoradiene and β-cedrene. Indeed, of the 61% β-cedrene generated by F198L EIZS, 20% is (+)-β-cedrene, which is derived from the (7R)-homobisabolyl cation intermediate (Figure 3). As a further variation of this pathway branching, F95H EIZS generates predominantly β-curcumene by direct deprotonation of the homobisabolyl cation. Whether the (7S)- or (7R)-homobisabolyl cation is formed is presumably a consequence of the precise folding of the preceding (4R)-bisabolyl cation enforced by the active site contour. Thus, bulky side chains in the rear of the active site (as viewed in Figure 1) ensure the formation of the natural (7S)-homobisabolyl cation intermediate that ultimately leads to epi-isozizaene. Smaller side chains in this region may allow the formation of the aberrant (7R)-homobisabolyl cation intermediate, as observed for F198L EIZS.
Product Chemodiversity and Biotechnology Applications
While numerous studies have been published to date describing mutagenesis of class I terpenoid cyclases and resultant changes in products generated, many of these studies have focused on residues outside of the hydrophobic active site pocket, such as residues coordinating to catalytically-obligatory metal ions,10,27–29 residues that hydrogen bond with the substrate diphosphate group,30,31 or residues thought to function in general acid-general base or electrostatic catalysis.10,29,32 Relatively few studies have focused on site-specific mutants specifically prepared so as to remold the hydrophobic active site contour of a class I terpenoid cyclase, and fewer still have been explicitly guided or accompanied by experimentally-determined crystal structures.17,18,33–36 Accordingly, we suggest that our work with EIZS represents the first small step in a journey that will ultimately allow us to correlate the three-dimensional structures of experimentally-verified remolded active site contours with reprogrammed isoprenoid cyclization cascades.
Structure-based reprogramming of terpenoid cyclase activity should have myriad applications in chemistry, biology, and biotechnology. For example, given that plant (E)-β-farnesene synthase has been commercialized for the large-scale production of (E)-β-farnesene in the development of an aviation biofuel,37–39 it is possible that a bacterial or fungal sesquiterpene synthase, such as F96A EIZS13 or the Y92A mutant of aristolochene synthase from Penicillium roqueforti,40 could be similarly adapted for large-scale biofuel generation. The terpenoid cyclase step is typically the bottleneck in microbial biosynthetic engineering experiments,41,42 and plant sesquiterpene synthases can be difficult to express in microbial fermentation systems.43 Thus, the availability of a microbial sesquiterpene synthase engineered to generate a desired sesquiterpene product may provide a useful alternative for the microbial production of advanced biofuels.
It is possible, too, that the use of an engineered bacterial terpenoid cyclase could enhance the flux of sesquiterpene generation by virtue of enhanced catalytic activity relative to a naturally-occurring plant enzyme. For example, bisabolane has recently been identified as a new biosynthetic alternative to D2 diesel fuel, which is utilized in compression ignition engines.43 Bisabolane is generated by hydrogenation of (Z)-α-bisabolene, which in turn is generated by the three-domain plant sesquiterpene cyclase, α-bisabolene synthase from Abies grandis,44 in a microbial expression system (Figure 7). The reported catalytic efficiency (kcat/KM) of this α-bisabolene synthase is 38 M−1s−1. However, bisabolane can also be generated by the hydrogenation of β-curcumene (Figure 7), which is generated by F95H EIZS with kcat/KM = 2600 M−1s−1 (Table 3). Thus, the generation of the bisabolane precursor β-curcumene by F95H EIZS is 68-fold more efficient than the generation of the bisabolane precursor (Z)-α-bisabolene by wildtype α-bisabolene synthase. Since no naturally-occurring β-curcumene synthase has been characterized (apart from a grape sesquiterpene cyclase that generates a mixture of sesquiterpene products, including 22% β-curcumene45), the reprogrammed catalytic activity of F95H EIZS may enable a more efficient approach for economical biofuel generation.
Figure 7.
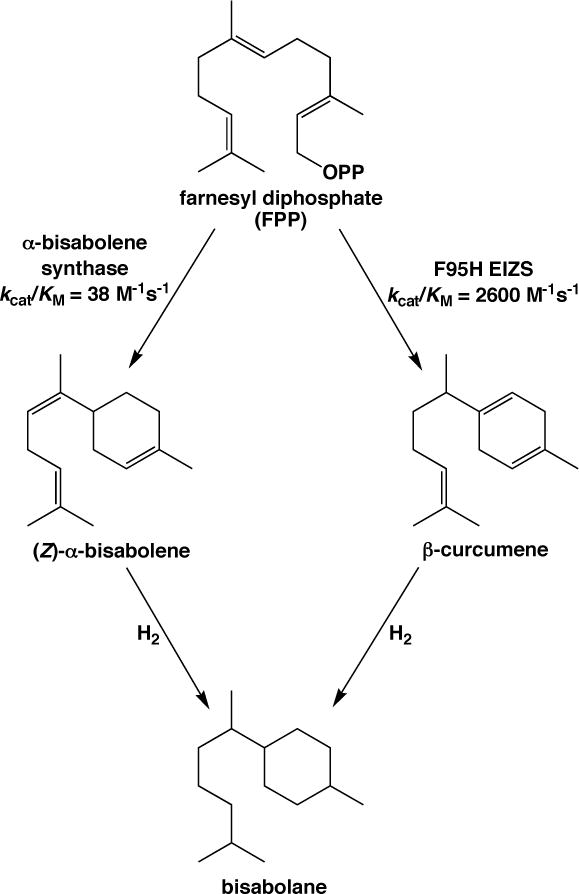
The D2 diesel fuel substitute bisabolane can be generated by the hydrogenation of (Z)-α-bisabolene or β-curcumene. F95H EIZS exhibits 68-fold enhanced catalytic efficiency in the generation of β-curcumene compared with the generation of (Z)-α-bisabolene by α-bisabolene synthase, so it is possible that F95H EIZS could serve as a superior terpenoid synthase for microbial biofuel generation.
Concluding Remarks
In closing, we note that the grand challenge in the engineering of a terpenoid cyclase is predictability – can a terpenoid cyclase be reprogrammed in true a priori fashion using structure-based design? The reprogrammed cyclization cascades of EIZS mutants result from divergent mechanistic sequences that branch from a handful of key carbocation intermediates, such as the farnesyl cation, the (4R)-bisabolyl cation, or the (7S)- or (7R)-homobisabolyl cations. The next step will be to study how the binding of stable analogues of these common intermediates is affected by active site mutations. Such an approach will provide the required foundation for predicting how a carbocation intermediate can be managed and manipulated through alternative mechanistic sequences. Future work in this regard will be reported in due course.
Acknowledgments
We thank the Northeastern Collaborative Access Team for access to beamline 24-ID-C at the Advanced Photon Source, and the National Synchrotron Light Source for access to beamline X-29, for X-ray diffraction data collection experiments.
Funding
Supported by National Institute of Health Grants GM56838 to D.W.C. and GM30301 to D.E.C., an NIH Chemistry-Biology Interface Training Grant to J.A.A., and an NIH Structural Biology and Molecular Biophysics Training Grant to G.G.H.
ABBREVIATIONS
- BME
β-mercaptoethanol
- BTAC
benzyltriethyl ammonium cation
- EIZS
epi-isozizaene synthase
- FPP
farnesyl diphosphate
- GC
gas chromatography
- MS
mass spectrometry
- NSLS
National Synchrotron Light Source
- PEG
polyethylene glycol
- PIPES
piperazine-N,N’-bis(2-ethanesulfonic acid)
- PPi
inorganic pyrophosphate
- r.m.s
root-mean-square
- TCEP
tris(2-carboxyethyl)phosphine
- WT
wild-type
Footnotes
The authors declare no competing financial interest.
ACCESSION CODES
The atomic coordinates and structure factors of unliganded wild-type EIZS, the L72V EIZS-Mg2+3-PPi-BTAC complex, the F95M EIZS-Mg2+3-PPi-BTAC complex, the F95H EIZS-Mg2+3-PPi-BTAC complex, the A236G EIZS-Mg2+3-PPi-BTAC complex, the W325F EIZS-Mg2+3-PPi complex, and the V329A EIZS-Mg2+3-PPi-BTAC complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 4LTV, 4LXW, 4LTZ, 4LZ3, 4LZ0, 4LZC, and 4LUU, respectively.
References
- 1.Sacchettini JC, Poulter CD. Creating isoprenoid diversity. Science. 1997;277:1788–1789. doi: 10.1126/science.277.5333.1788. [DOI] [PubMed] [Google Scholar]
- 2.Christianson DW. Roots of biosynthetic diversity. Science. 2007;316:60–61. doi: 10.1126/science.1141630. [DOI] [PubMed] [Google Scholar]
- 3.Poulter CD, Rilling HC. The prenyl transfer reaction. Enzymatic and mechanistic studies of the 1′–4 coupling reaction in the terpene biosynthetic pathway. Acc Chem Res. 1978;11:307–313. [Google Scholar]
- 4.Poulter CD. Farnesyl diphosphate synthase. A paradigm for understanding structure and function relationships in E-polyprenyl diphosphate synthases. Phytochem Rev. 2006;5:17–26. [Google Scholar]
- 5.Cane DE. Isoprenoid biosynthesis. Stereochemistry of the cyclization of allylic pyrophosphates. Acc Chem Res. 1985;18:220–226. [Google Scholar]
- 6.Cane DE. Enzymatic formation of sesquiterpenes. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 7.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 8.Christianson DW. Unearthing the roots of the terpenome. Curr Opin Chem Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Austin MB, O’Maille PE, Noel JP. Evolving biosynthetic tangos negotiate mechanistic landscapes. Nat Chem Biol. 2008;4:217–222. doi: 10.1038/nchembio0408-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felicetti B, Cane DE. Aristolochene synthase: mechanistic analysis of active site residues by site-directed mutagenesis. J Am Chem Soc. 2004;126:7212–7221. doi: 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- 11.Steele CL, Crock J, Bohlmann J, Croteau R. Sesquiterpene synthases from grand fir (Abies grandis): comparison of constitutive and wound-induced activities, and cDNA isolation, characterization, and bacterial expression of δ-selinene synthase and γ-humulene synthase. J Biol Chem. 1998;273:2078–2089. doi: 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- 12.Lin X, Hopson R, Cane DE. Genome mining in Streptomyces coelicolor: Molecular cloning and characterization of a new sesquiterpene synthase. J Am Chem Soc. 2006;128:6022–6023. doi: 10.1021/ja061292s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aaron JA, Lin X, Cane DE, Christianson DW. Structure of epi-isozizaene synthase from Streptomyces coelicolor A3(2), a platform for new terpenoid cyclization templates. Biochemistry. 2010;49:1787–1797. doi: 10.1021/bi902088z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin X, Cane DE. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor. Mechanism and stereochemistry of the enzymatic formation of epi-isozizaene. J Am Chem Soc. 2009;131:6332–6333. doi: 10.1021/ja901313v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aaron JA, Christianson DW. Trinuclear metal clusters in catalysis by terpenoid synthases. Pure Appl Chem. 2010;82:1585–1597. doi: 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petsko GA, Kenyon GL, Gerlt JA, Ringe D, Kozarich JW. On the origin of enzymatic species. Trends Biochem Sci. 1993;18:372–376. doi: 10.1016/0968-0004(93)90091-z. [DOI] [PubMed] [Google Scholar]
- 17.Yoshikuni Y, Ferrin TE, Keasling JD. Designed divergent evolution of enzyme function. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 18.Greenhagen BT, O’Maille PE, Noel JP, Chappell J. Identifying and manipulating structural determinates linking catalytic specificities in terpene synthases. Proc Natl Acad Sci USA. 2006;103:9826–9831. doi: 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harangi J. Retention index calculation without n-alkanes: the virtual carbon number. J Chromatogr A. 2003;993:187–195. doi: 10.1016/s0021-9673(03)00320-0. [DOI] [PubMed] [Google Scholar]
- 20.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Cryst. 2005;D61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 21.Collaborative Computational Project, No. 4. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 22.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Picaud S, Brodelius M, Brodelius PE. Expression, purification and characterization of recombinant (E)-β-farnesene synthase from Artemisia annua. Phytochemistry. 2005;66:961–967. doi: 10.1016/j.phytochem.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 25.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau RB, Christianson DW. Bornyl diphosphate synthase: structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc Natl Acad Sci USA. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cane DE, Yang GH, Coates RM, Pyun HJ, Hohn TM. Trichodiene synthase. Synergistic inhibition by inorganic pyrophosphate and aza analogs of the bisabolyl cation. J Org Chem. 1992;57:3454–3462. [Google Scholar]
- 27.Cane DE, Xue Q, Fitzsimons BC. Trichodiene synthase. Probing the role of the highly conserved aspartate-rich region by site-directed mutagenesis. Biochemistry. 1996;35:12369–12376. doi: 10.1021/bi961344y. [DOI] [PubMed] [Google Scholar]
- 28.Seemann M, Zhai G, de Kraker JW, Paschall CM, Christianson DW, Cane DE. Pentalenene synthase. Analysis of active site residues by site-directed mutagenesis. J Am Chem Soc. 2002;124:7681–7689. doi: 10.1021/ja026058q. [DOI] [PubMed] [Google Scholar]
- 29.Vedula LS, Jiang J, Zakharian T, Cane DE, Christianson DW. Structural and mechanistic analysis of trichodiene synthase using site-directed mutagenesis: probing the catalytic function of tyrosine-295 and the asparagine-225/serine-229/glutamate-233-Mg2+B motif. Arch Biochem Biophys. 2008;469:184–194. doi: 10.1016/j.abb.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cane DE, Shim JH, Xue Q, Fitzsimons BC, Hohn TM. Trichodiene synthase. Identification of active site residues by site-directed mutagenesis. Biochemistry. 1995;34:2480–2488. doi: 10.1021/bi00008a011. [DOI] [PubMed] [Google Scholar]
- 31.Vedula LS, Cane DE, Christianson DW. Role of arginine-304 in the diphosphate-triggered active site closure mechanism of trichodiene synthase. Biochemistry. 2005;44:12719–12727. doi: 10.1021/bi0510476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rising AK, Starks CM, Noel JP, Chappell J. Demonstration of germacrene A as an intermediate in 5-epi-aristolochene synthase catalysis. J Am Chem Soc. 2000;122:1861–1866. [Google Scholar]
- 33.Back K, Chappell J. Identifying functional domains within terpene cyclases using a domain-swapping strategy. Proc Natl Acad Sci USA. 1996;93:6841–6845. doi: 10.1073/pnas.93.13.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 35.O’Maille PE, Malone A, Dellas N, Andes Hess B, Jr, Smentek L, Sheehan I, Greenhagen BT, Chappell J, Manning G, Noel JP. Quantitative exploration of the catalytic landscape separating divergent plant sesquiterpene synthases. Nat Chem Biol. 2008;4:617–623. doi: 10.1038/nchembio.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li JX, Fang X, Zhao Q, Ruan JX, Yang CQ, Wang LJ, Miller DJ, Faraldos JA, Allemann RK, Chen XY, Zhang P. Rational engineering of plasticity residues of sesquiterpene synthases from Artemisia annua: product specificity and catalytic efficiency. Biochem J. 2013;451:417–426. doi: 10.1042/BJ20130041. [DOI] [PubMed] [Google Scholar]
- 37.Keasling JD. Manufacturing molecules through metabolic engineering. Science. 2010;330:1355–1358. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]
- 38.Rude MA, Schirmer A. New microbial fuels: a biotech perspective. Curr Op Microbiol. 2009;12:274–281. doi: 10.1016/j.mib.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Bohlmann J. Terpenoid synthases–from chemical ecology and forest fires to biofuels and bioproducts. Structure. 2011;19:1730–1731. doi: 10.1016/j.str.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Deligeorgopoulou A, Allemann RK. Evidence for differential folding of farnesyl pyrophosphate in the active site of aristolochene synthase: a single-point mutation converts aristolochene synthase into an (E)-β-farnesene synthase. Biochemistry. 2003;42:7741–7747. doi: 10.1021/bi034410m. [DOI] [PubMed] [Google Scholar]
- 41.Anthony JR, Anthony LC, Nowroozi F, Kwon G, Newman JD, Keasling JD. Optimization of the mevalonate-based isoprenoid biosynthetic pathway in Escherichia coli for production of the anti-malarial drug precursor amorpha-4,11-diene. Metab Eng. 2009;11:13–19. doi: 10.1016/j.ymben.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. Microbial engineering for the production of advanced biofuels. Nature. 2012;488:320–328. doi: 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- 43.Peralta-Yahya PP, Ouellet M, Chan R, Mukhopadhyay A, Keasling JD, Lee TS. Identification and microbial production of a terpene-based advanced biofuel. Nat Commun. 2011;2483 doi: 10.1038/ncomms1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McAndrew RP, Peralta-Yahya PP, DeGiovanni A, Pereira JH, Hadi MZ, Keasling JD, Adams PD. Structure of a three-domain sesquiterpene synthase: a prospective target for advanced biofuels production. Structure. 2011;19:1876–1884. doi: 10.1016/j.str.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Martin DM, Aubourg S, Schouwey MB, Daviet L, Schalk M, Toub O, Lund ST, Bohlmann J. Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol. 2010;10:226. doi: 10.1186/1471-2229-10-226. [DOI] [PMC free article] [PubMed] [Google Scholar]