

Abstract
Rapid mechanical deformation of cells has emerged as a promising, vector-free method for intracellular delivery of macromolecules and nanomaterials. This technology has shown potential in addressing previously challenging applications; including, delivery to primary immune cells, cell reprogramming, carbon nanotube, and quantum dot delivery. This vector-free microfluidic platform relies on mechanical disruption of the cell membrane to facilitate cytosolic delivery of the target material. Herein, we describe the detailed method of use for these microfluidic devices including, device assembly, cell preparation, and system operation. This delivery approach requires a brief optimization of device type and operating conditions for previously unreported applications. The provided instructions are generalizable to most cell types and delivery materials as this system does not require specialized buffers or chemical modification/conjugation steps. This work also provides recommendations on how to improve device performance and trouble-shoot potential issues related to clogging, low delivery efficiencies, and cell viability.
Keywords: Bioengineering, Issue 81, Transfection, microfluidic, vector-free, protein delivery, intracellular delivery, quantum dot delivery, cell reprogramming, siRNA
Introduction
Delivery of macromolecules to the cell cytoplasm is a critical step in therapeutic and research applications. Nanoparticle mediated delivery, for example, has shown potential in gene therapy1,2, while protein delivery is a promising means of affecting cellular function in both clinical3 and laboratory4 settings. Other materials, such as small molecule drugs, quantum dots, or gold nanoparticles, are of interest in applications ranging from cancer therapeutics5,6 to intracellular labeling7,8, and single molecule tracking9.
The cell membrane is largely impermeable to macromolecules. Many existing techniques use polymeric nanoparticles10,11, liposomes12, or chemical modifications13 to facilitate membrane disruption or endocytotic delivery. In these methods, the delivery efficiency and cell viability are often dependent on the structure of the target molecule and the cell type. These methods can be efficient at the delivery of structurally uniform materials, such as nucleic acids, but are often ill-suited for the delivery of more structurally diverse materials, such as proteins14,15 and nanomaterials7. Moreover, the endosome disruption mechanism that most of these methods rely on is often inefficient, hence leaving much material trapped in vesicle structures16. Finally, methods that are often developed for use with established cell lines do not translate well to primary cells.
Membrane poration methods, such as electroporation17,18 and sonoporation19, are an attractive alternative in some applications; however, they are known to cause low cell viability and can be limited by the charge of the target delivery material.
Rapid mechanical deformation of cells, a microfluidic approach to delivery, has recently demonstrated its advantages over current techniques in the context of cell reprogramming20 and nanomaterial delivery21. This method relies on mechanical disruption o the cell membrane to facilitate cytosolic delivery of materials present in the surrounding buffer. The system has demonstrated enabling potential in previously challenging cell types (e.g. primary immune cells and stem cells) and materials (e.g. antibodies and carbon nanotubes). Herein, the general procedure to use these devices for intracellular delivery of target macromolecules is described. The procedure is generalizable to most cell types and delivery materials; however, it is recommended that one conduct a brief optimization of conditions, as detailed in our previously reported design guidelines20, for any previously unreported applications. To date, the system has been used successfully for the delivery of RNA, DNA, gold nanoparticles, quantum dots, carbon nanotubes, proteins, and dextran polymers20,21.
Protocol
1. Storage
-
Store the reservoirs, holders, O-rings and microfluidic devices in 70% ethanol. Use a container (e.g. jar or beaker) that has a lid to prevent evaporation and contamination by dust or outside particles. Place the devices in one container (1), reservoirs and O-rings in a second container (2), and holders in the third (3).
Note: The use of 70% ethanol for storage is to maintain sterility. If the only components of the solution are ethanol and water (i.e. no denaturing agents), all system components should be fully compatible and will not degrade over time.
Change ethanol solution in containers (2) and (3) before each use to prevent cross-contamination across experiments and minimize the presence of unwanted particles that can cause clogging.
2. Experiment Preparation
Place containers (2) and (3) in an ultrasound bath for 5-10 min before each use. This helps remove any contaminating particles from previous experiments.
Clean workspace in biosafety cabinet with 70% ethanol solution.
Spray all materials (3 containers, and tweezers) with a 70% ethanol solution before placing them inside the biosafety cabinet.
3. Assembly
Set down 2-3 low-lint wipes in the work area.
Remove plastic reservoirs from their container with tweezers and set them on the wipes to facilitate evaporation of ethanol solution from inner surfaces. Gently tap the reservoirs on the surface or blow air through them to facilitate removal of the ethanol solution.
Insert O-rings into their appropriate slot on the reservoirs.
Remove the holder and chips from respective containers and allow ethanol to evaporate (∼1-2 min).
Use tweezers to place the desired chip face-up (i.e. access holes up) in the holder. Raise the holder with the chip to eyelevel to make sure the chip is lying flat in the holder and adjust if necessary with the tweezers. IMPORTANT: If the device does not fit properly in its holder, there is a risk that it will break during the subsequent steps.
Next, gently place the reservoirs on the holder and align them with the clips. Be careful that O-rings do not fall out of their slots during this process.
Gently press down on the reservoirs until they click into place. Ensure that both sides of the reservoirs are secure and that the chip appears to be in the correct position.
4. Cell Preparation
For adherent cells (primary or established lines): Plate cells 1-2 days prior to the experiment such that they are no more than 80% confluent on the day of the experiment.
Place cells in suspension (in PBS or relevant media) and aim for an operating concentration of 1.0 × 106 cells/ml to 1.0 × 107 cells/ml. NOTE: We have not observed significant changes in delivery performance due to cell concentration.
Mix cells and the desired delivery material in a separate tube to obtain the desired material concentration for the experiment. IMPORTANT: Because the described delivery method relies on diffusion to facilitate delivery, a higher material concentration will yield higher delivery. If possible, it is recommended to use a 1 μM solution of the desired material for initial trials. This concentration can then be titrated down in future experiments as needed. The lowest reported concentration used with this device is 10 nM21.
5. Operation
Pipette mixture of cells and delivery material into a reservoir (current design has a max 150 μl capacity). NOTE: Most device designs are fully reversible therefore direction of flow through the channels does not matter. Samples may be loaded into either of the two reservoirs.
Attach pressure tubing to the filled reservoir and tighten the nut to ensure proper sealing (finger tight is often sufficient).
-
Adjust pressure to the desired level on the regulator. This controls the speed at which cells travel through the device. Note that cell flow does not start until button is pressed.
NOTE: In previous work, nitrogen or compressed air have worked equally well as the carrier gas.
Raise device to eye level and orient it so that the liquid in the reservoir is easily visible. NOTE: Track the liquid column to shut off the system before the reservoir is emptied.
Press the button to pressurize the reservoir and begin cell flow.
When the liquid level is approximately 2 mm from the bottom of the reservoir, quickly turn the regulator to 0 psi to stop flow. IMPORTANT: If one fails to stop the flow before the reservoir is emptied, one risks ejecting the sample from the collection reservoir. Also note that if the fluid column is not moving at an appreciable pace, or has slowed substantially relative to its initial flow rate (e.g. 3× slower), the mounted device is probably clogged and needs to be exchanged. Operating a clogged device can lead to higher cell death.
Collect the treated cells from the appropriate reservoir and place them in the desired collection tube/plate. IMPORTANT: Do not dilute the collected cells in any buffers at this stage as the porated cells will continue to uptake material for up to 10 min20. After this window has passed, dilute the cells in the desired media/buffer.
To collect more treated cells or try alternative experimental conditions, repeat steps 5.1 - 5.7 as needed. Recall that the chips are reversible, therefore samples can be mounted in either reservoir. Be sure to exchange chips as needed if they clog. Discard clogged devices.
6. Disassembly
Gently disconnect the reservoirs from the main holder by pushing aside the clip arms.
Place each part in the appropriate storage container (detailed in section 1).
Place used chips in a separate container for disposal.
Representative Results
Figure 1 contains a descriptive schematic of the microfluidic delivery system. Figures 2a-b illustrate typical results from treating HeLa cells with different device designs in the presence of fluorescently conjugated 3 kDa dextran20. If the procedure is followed correctly, system performance will be sensitive to device type and operating speed. Therefore, one should optimize these conditions for a given application before proceeding to more complex experiments. In the range of operating conditions illustrated in the figure, cell viability remains above 80%, however, one must note that suboptimal delivery parameters (e.g. inappropriate chip type) could lead to viabilities below 30%. Thus viability and delivery efficiency must be optimized simultaneously for any previously unreported cell type. If a device design is inappropriate for the target cell type, one will see results ranging from no delivery to high cell death (e.g. 10% viability). If the experimental conditions were inappropriate, for example the target material bound to the cell surface, one may not be able to measure the delivery accurately as the surface binding signal can mask the cytoplasmic signal.
Figure 1. Schematic of the full system and its interface to the microfluidic devices.
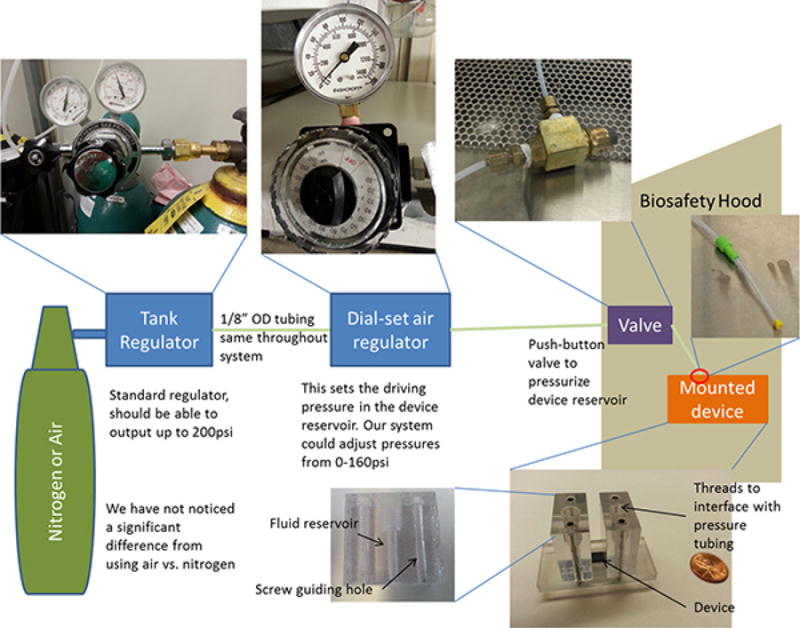
Figure 2. Representative results with HeLa cells20.
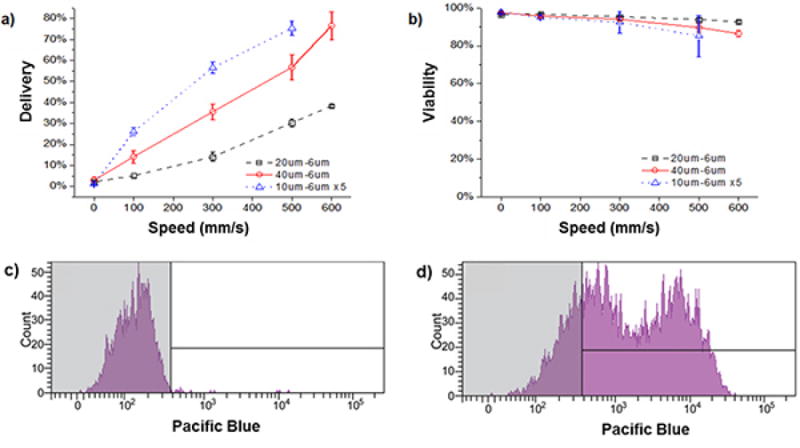
a) Delivery efficiency and b) cell viability of HeLa cells treated by 3 different device designs in the presence of cascade blue conjugated 3 kDa dextran (0.2 mg/mL). In these experiments, the cell solution was at a concentration of 106 cells per mL in PBS containing 3% fetal bovine serum and 1% F68 pluronics. Viability was measured by propidium iodide staining of dead cells and delivery results are shown for live cells only. All data points were run in triplicate, and error bars represent 2 SDs. c) A representative histogram for an experimental control where HeLa cells were exposed to cascade blue conjugated 3 kDa dextran for at least the same amount of time as a device treated case. d) The corresponding case where the cells were passed through the device to facilitate delivery. The fraction of the live cell population that is in the unshaded region of the histogram is defined as the ‘delivered’ population.
Figures 2c-d illustrate our preferred method for measuring delivery efficiency within the live cell population. The control case in Figure 2c is meant to reflect all surface binding and endocytotic effects as the cells are exposed to the delivery material for at least as long as the treated cases. The delivered cell population is defined as the unshaded region such that only 1-5% of the control population falls within that region. Note that previous work has established that delivery in this approach is not mediated by endocytosis20. The double-peak distribution observed in the delivered example is not necessarily characteristic of the system as we have observed more and less uniform distributions for different experimental conditions.
Discussion
Certain aspects of the described experimental procedure (i.e. factors other than chip design and operating speed) may need to be optimized depending on the cell type and delivery material the system is applied to. The discussion that follows addresses some of the most common factors to consider when designing experiments.
To improve the delivery signal for fluorescently labeled compounds, one needs to address sources of background fluorescence. Surface binding and endocytosis, for example, can be addressed by washing cells 5-10 min after delivery to remove excess material before culture or further processing. If electing to forgo a wash step, it would be best to dilute the treated cells by 5-10x in the desired media to lower the concentration of target material in the surrounding solution and hence reduce endocytotic rates. In some cases, for example protein delivery, one may find it necessary to actively block the cell surface (e.g. using unlabeled bovine serum albumin) to minimize non-specific binding of the delivery material. In a given application, if the fluorescence intensity of the untreated case, as measured by flow cytometry, is over one decade lower than the endocytosis control then surface binding/endocytosis may be an issue. Note that issues with background signal are only relevant for direct fluorescent detection of the delivery material, functional assays (e.g. cell reprogramming) are usually unaffected as endocytosed or surface bound material are often inactive.
To improve the viability of cells after treatment, one needs to minimize the amount of processing post-delivery. It is recommended that one avoid excessive centrifugation or wash steps and transfer cells into their desired media in an incubator 5-10 min after delivery is complete. In adherent lines, cells are often fully adhered and dividing within 24 hr after treatment. As discussed previously20, we have not observed any long-term side-effects from the use of this technique.
The microfluidic devices used in this delivery system are prone to clogging over time. Clogs often form at the constriction as it is the narrowest feature on the device. Clogging can be caused by damaged cells or by environmental contaminants. The latter's effect can be minimized by ensuring a clean, dust-free environment for operating the devices (e.g. a well maintained biosafety cabinet). For cell lines, we recommend passaging cells 1-2 days prior to delivery so as to minimize the presence of cell debris in the resulting suspension. When operating the device, one must be mindful of the volumetric flow rate of the fluid in the reservoir. If the flow rate drops to under a third of its initial rate, the device may be causing excessive cell death and should be exchanged, or the direction of flow reversed. Potential clogging issues can also be mitigated by passing the cell suspension through a 40 μm cell strainer mesh. This process ensures a single cell suspension thereby preventing potential clogs from forming in the wider, low flow rate sections of the chip.
When designing experiments for different delivery materials, one must account for the size of the target material and its concentration. As this technology relies on the passive diffusion of material across the disrupted cell membrane, the molar concentration gradient and the hydrodynamic radius of the delivery material will govern the rate of mass transfer - assuming the membrane disruptions are large enough to accommodate the material. It is thus advisable to use high molar concentrations (e.g. 1 μM) of large target materials relative to their smaller counter parts. The technique has demonstrated effective delivery at concentrations as low as 10 nM in the case of quantum dot delivery21. Moreover, the technique is not thought to be limited by the nature of the target delivery material except in the context of material size and diffusivity (i.e. low diffusivity materials will have lower delivery efficiencies and material larger than the size of the disruptions probably cannot enter the cytoplasm).
Studies in cell reprogramming and quantum dot delivery have illustrated the strengths of the cell squeezing approach relative to electroporation. Based on the current understanding of the membrane disruption mechanisms in these two techniques, it would appear that cell squeezing has a significant advantage in applications involving proteins, small molecules, and nanomaterials20,21. The contrast between the two methods in applications involving nucleic acids (which are highly charged compared to the aforementioned materials) is unclear.
This microfluidic approach to delivery relies on mechanical disruption of the cell membrane to facilitate passive diffusion of material into the cell and has been shown to be applicable to a range of cell types20. The system is thus in principal appropriate for the cytosolic delivery of almost any material to almost any cell type. We believe the system's advantages are most pronounced in cases involving traditionally difficult to transfect primary cells (e.g. immune and stem cells) and conventionally challenging materials (e.g. proteins, quantum dots, carbon nanotubes, and RNA). The aforementioned potential in immune and stem cell applications is emphasized by the shortcomings of traditional delivery methods in addressing these cell types. However, the system may require further improvement to facilitate gene delivery by plasmid vectors as these applications require delivery to the nucleus. In summary, we believe the microfluidic deformation approach to delivery can potentially address many existing challenges to intracellular delivery. The system's independence from chemical vectors or electric fields enables its robust application to a range of studies. The instructions provided herein should enable any interested researchers to operate such a system independently and adapt it for their research purposes.
Acknowledgments
We thank T. Shatova for helpful discussion on experimental design and data analysis. The assistance and expertise of G. Paradis, the personnel of the flow cytometry core at the Koch Institute, and the staff of the Microsystems Technology Laboratory at Massachusetts Institute of Technology are gratefully acknowledged. This work was supported by National Institutes of Health Grants RC1 EB011187-02, DE013023, DE016516, EB000351, and partially by National Cancer Institute Cancer Center Support (Core) Grants P30-CA14051 and MPP-09Call-Langer-60.
Footnotes
Video Link: The video component of this article can be found at http://www.jove.com/video/50980/
Disclosures: The authors Armon Sharei, Robert Langer, and Klavs F. Jensen are shareholders of SQZ Biotechnologies Company that produces the microfluidic devices used in this article.
References
- 1.Schaffert D, Wagner E. Gene therapy progress and prospects: synthetic polymer-based systems. Gene Ther. 2008;15:1131–1138. doi: 10.1038/gt.2008.105. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 4.Kim D, et al. Generation of Human Induced Pluripotent Stem Cells by Direct Delivery of Reprogramming Proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. Polyvalent Oligonucleotide Gold Nanoparticle Conjugates as Delivery Vehicles for Platinum(IV) Warheads. J Am Chem Soc. 2009;131:14652–14653. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang ZX, Zhang ZY. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer and Metastasis Reviews. 2008;27:263–272. doi: 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derfus AM, Chan WCW, Bhatia SN. Intracellular Delivery of Quantum Dots for Live Cell Labeling and Organelle Tracking. Adv Mater. 2004;16:961–966. doi: 10.1002/adma.200306111. [DOI] [Google Scholar]
- 8.Michalet X, et al. Quantum Dots for Live Cells, in Vivo Imaging, and Diagnostics. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahan M, et al. Diffusion Dynamics of Glycine Receptors Revealed by Single-Quantum Dot Tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 10.Slowing II, Trewyn BG, Lin VSY. Mesoporous Silica Nanoparticles for Intracellular Delivery of Membrane-Impermeable Proteins. J Am Chem Soc. 2007;129:8845–8849. doi: 10.1021/ja0719780. [DOI] [PubMed] [Google Scholar]
- 11.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 12.Joseph Z. Cationic lipids used in gene transfer. Advanced Drug Delivery Reviews. 1997;27:17–28. doi: 10.1016/s0169-409x(97)00019-7. [DOI] [PubMed] [Google Scholar]
- 13.Verma A, et al. Surface-structure-regulated cell-membrane penetration by monolayer-protected nanoparticles. Nat Mater. 2008;7:588, 595. doi: 10.1038/nmat2202. http://www.nature.com/nmat/journal/v7/n7/suppinfo/nmat2202_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan M, et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat Nano. 2010;5:48–53. doi: 10.1038/nnano.2009.341. [DOI] [PubMed] [Google Scholar]
- 15.Shi Kam NW, Jessop TC, Wender PA, Dai H. Nanotube Molecular Transporters: Internalization of Carbon Nanotube-Protein Conjugates into Mammalian Cells. J Am Chem Soc. 2004;126:6850–6851. doi: 10.1021/ja0486059. [DOI] [PubMed] [Google Scholar]
- 16.Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. Journal of Controlled Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 17.li S. Electroporation Gene Therapy: New Developments In Vivo and In Vitro. Current Gene Therapy. 2004;4:309–316. doi: 10.2174/1566523043346336. [DOI] [PubMed] [Google Scholar]
- 18.Fox M, et al. Electroporation of cells in microfluidic devices: a review. Anal Bioanal Chem. 2006;385:474–485. doi: 10.1007/s00216-006-0327-3. [DOI] [PubMed] [Google Scholar]
- 19.Miller DL, Pislaru SV, Greenleaf JF. Sonoporation: Mechanical DNA Delivery by Ultrasonic Cavitation. Somatic Cell and Molecular Genetics. 2002;27:115–134. doi: 10.1023/a:1022983907223. [DOI] [PubMed] [Google Scholar]
- 20.Sharei A, et al. A vector-free microfluidic platform for intracellular delivery. Proc Natl Acad Sci USA. 2013;110:2082–2087. doi: 10.1073/pnas.1218705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J, et al. Nonendocytic Delivery of Functional Engineered Nanoparticles into the Cytoplasm of Live Cells Using a Novel, High-Throughput Microfluidic Device. Nano Lett. 2012;12:6322–6327. doi: 10.1021/nl303421h. [DOI] [PMC free article] [PubMed] [Google Scholar]