

Abstract
Intraductal neoplasms are important precursors to invasive pancreatic cancer and an opportunity to detect and treat pancreatic neoplasia before an invasive carcinoma develops. The diagnostic evaluation of these lesions is challenging as diagnostic imaging and cytological sampling do not provide accurate information on lesion classification, the grade of dysplasia or the presence of invasion. Moreover, the molecular driver gene mutations of these precursor lesions have yet to be fully characterized. Fifty-two intraductal papillary neoplasms, including 48 intraductal papillary mucinous neoplasms (IPMNs) and 4 intraductal tubulopapillary neoplasms (ITPNs), were subjected to the mutation assessment in 51 cancer-associated genes, using Ion Torrent semiconductor-based next-generation sequencing. P16 and Smad4 immunohistochemistry was performed on 34 IPMNs, and 17 IPMN-associated carcinomas. At least one somatic mutation was observed in 46/48 (96%) IPMNs; 29 (60%) had multiple gene alterations. GNAS and/or KRAS mutations were found in 44/48 (92%) of IPMNs. GNAS was mutated in 38/48 (79%) IPMNs, KRAS in 24/48 (50%), and these mutations coexisted in 18/48 (37.5%) of IPMNs. RNF43 was the third most commonly mutated gene and was always associated with GNAS and/or KRAS mutations, as were virtually all the low frequency mutations found in other genes. Mutations in TP53 and BRAF genes (10% and 6%) were only observed in high-grade IPMNs. P16 was lost in 7/34 IPMNs and 9/17 IPMN-associated carcinomas; Smad4 was lost in 1/34 IPMN and 5/17 IPMN-associated carcinomas. In contrast to IPMNs, only one of four ITPN had detectable driver gene (GNAS and NRAS) mutations. Deep sequencing DNA from 7 cyst fluid aspirates identified 10 of the 13 mutations detected in their associated IPMN. Using next-generation sequencing to detect cyst fluid mutations has the potential to improve the diagnostic and prognostic stratification of pancreatic cystic neoplasms.
Keywords: IPMN, pancreatic tumours, NGS, biomarkers
Introduction
Intraductal neoplasms of the pancreas are cystic or mass-forming epithelial neoplasms that characteristically grow primarily within the ductal system [1], and include two main groups: intraductal papillary mucinous neoplasms (IPMNs) and intraductal tubulopapillary neoplasms (ITPNs).
IPMNs are precursor lesions to invasive adenocarcinoma, accounting for 5% of pancreatic neoplasms. Epithelial morphology and mucin expression patterns define four main histologic subtypes of IPMN [2]: intestinal, gastric, oncocytic and pancreaticobiliary. Recent papers support the hypothesis of distinct pathways for carcinogenesis among the different IPMN subtypes [3,4].
Cancer is a fundamentally genetic disease, and IPMNs are no exception. IPMNs commonly harbour activating mutations in KRAS and GNAS, and inactivating mutations in RNF43, CDKN2A/p16, and TP53, and less commonly mutations in BRAF, PIK3CA, STK11 and SMAD4 [5-12]. TP53, CDKN2A/p16 and SMAD4 mutations and/or loss of expression are generally found in higher-grade lesions [13-15]. IPMNs are genetically heterogeneous [16], and different patterns of genetic alterations have been reported in the different morphologic subtypes of IPMNs: Mohri et al. reported KRAS mutations as more prevalent in gastric type than in intestinal type IPMN [4]; Xiao et al. identified KRAS and BRAF mutations and abnormal p53 immunolabeling in the oncocytic type, but less frequently than in the pancreatobiliary IPMN [17]. Yamaguchi et al. analysed 15 gastric type IPMNs, pyloric gland variant, which showed frequent mutations in GNAS and KRAS (60% and 80%, respectively) [18]. ITPN is a rare variant of pancreatic intraductal neoplasms recently recognised as a distinct entity in the WHO classification [1]. A recent mutational analysis of 14 ITPN detected PIK3CA mutations in three cases, and one case each with KRAS and BRAF mutations, while no GNAS mutation was detected, suggesting a different molecular origin for ITPN [12,18].
Massive parallel sequencing, also known as next-generation sequencing (NGS) or deep sequencing can be customized to enable accurate detection of mutations in gene panels with samples with limited DNA [19,20]. Such an approach represents a potent diagnostic complement to histopathological and immunophenotypical diagnosis [21].
In the present study we used targeted NGS to examine the mutational status of 51 cancer-related genes in 52 intraductal neoplasms of the pancreas. We also tested the diagnostic performance of NGS in cystic fluids from 10 patients with IPMN, supporting the use of NGS for classifying cyst type in clinical practice.
Materials and Methods
Cases
A retrospective series of 52 pancreatic intraductal neoplasms from 51 surgically treated patients (Table 1), including 48 IPMN and 4 ITPN with one IPMN and one ITPN coexisting in the same patient, were retrieved from the ARC-Net biobank at Verona University Hospital (www.arc-net.it). The series included 40 fresh frozen (FF) and 12 formalin-fixed paraffin-embedded (FFPE) tissues. IPMNs were classified according to WHO 2010 [1]. Dysplasia was graded as low (LG), intermediate (MG), and high grade (HG). In 10 intestinal-type IPMNs, cystic fluids were harvested from pancreatectomy specimens with a fine needle syringe, and stored at −80°C within 30 min of resection. Normal pancreatic tissues were used to determine the somatic or germline nature of mutations.
Table 1.
Demographic and histopathological data of the series of 52 intraductal neoplasms of the pancreas from 51 patients.*
| Gender | Male | 34 (67.3%) |
| Female | 17 (32.7%) | |
| Age | Average | 64±10 (median 65; range 42-80) |
| Site | Head | 19 (36.5%) |
| Uncinate process | 2 (3.8%) | |
| Head/-Uncinate process | 19 (36.5%) | |
| Body-Tail | 9 (17.3%) | |
| Diffuse | 3 (5.8%) | |
| Distribution | Main-Duct type | 31 (59.6%) |
| Branch-Duct type | 3 (5.8%) | |
| Combined | 18 (34.6%) | |
| Histotype | IPMN-Gastric | 6 (11.5%) |
| IPMN-Intestinal | 36 (69.2%) | |
| IPMN-Pancreaticobiliary | 3 (5.8%) | |
| IPMN-Oncocytic | 3 (5.8%) | |
| ITPN | 4 (7.7%) | |
| Lesion | Low-Grade | 3 (5.8%) |
| Intermediate-Grade | 17 (32.7%) | |
| High-Grade | 28 (53.8%) | |
| Adenocarcinoma | 4 (7.7%) | |
| Associated carcinoma | Colloid Carcinoma | 18 (34.6%) |
| Tubular Carcinoma | 12 (23.1%) | |
| None | 22 (42.3%) |
One male patient had two coexisting lesions, one IPMN and one ITPN; IPMN, intraductal papillary mucinous neoplasm; ITPN, intraductal tubulopapillary neoplasm.
Ethics
The materials have been collected under Program 853 protocol 298CE 15/02/02 and revised Program 1885 protocol 52438 23/11/2010, approved by the Verona University Hospital ethics committee. The protocols include informed consent from the patient.
DNA extraction and quantification
DNA was obtained from tissues after enrichment for neoplastic cellularity using manual microdissection. DNA was extracted using the QiAamp DNA Mini Kit (Qiagen) from frozen tissues and the QIAamp DNA FFPE Tissue Kit from FFPE specimens (Qiagen). DNA from 250 μl of cystic fluid was purified by adding 3 ml of RLTM buffer (Qiagen) and then binding to an AllPrep DNA column (Qiagen) [10]. DNA was quantified and its quality assessed using NanoDrop (Invitrogen) and Qubit (Invitrogen) platforms [22]. The quality of DNA was further evaluated by PCR using the BIOMED 2 PCR multiplex protocol [23].
Deep Sequencing of Multiplex PCR Amplicons of 51 genes
Two multigene panels were used: the 50-gene Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) and an AmpliSeq Custom Panel investigating 7 genes. The first explores selected regions of the following 50 cancer-associated genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, JAK2, JAK3, IDH2, KDR/VEGFR2, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, VHL. The custom panel was designed to target selected regions of 6 genes included in the previous panel (BRAF, CDKN2A, GNAS, KRAS, SMAD4, and TP53) and RNF43 that was not in the first panel. Details of the target regions for both panels are in Supplementary Table 1A&B.
Twenty nanograms of DNA were used for multiplex PCR amplification. Emulsion PCR was performed with the OneTouch DL or OneTouch2 systems (Life Technologies). The quality of the obtained library was evaluated by the Agilent 2100 Bioanalyser on-chip electrophoresis (Agilent Technologies). Sequencing was run on the Ion Torrent Personal Genome Machine (PGM, Life Technologies) loaded with 316 (50-gene panel) or 318 chips (custom panel). Data analysis, including alignment to the hg19 human reference genome and variant calling, was done using the Torrent Suite Software v.3.2 and v.3.6 (Life Technologies). Filtered variants were annotated using the SnpEff software v.3.1 and the IonReporter software v.1.6 (Life Technologies). Alignments were visually verified with the Integrative Genomics Viewer; IGV v.2.2, Broad Institute.
DNA Sanger Sequencing
KRAS (exons 2, 3), GNAS (exons 8, 9), BRAF (exon 15) and TP53 (exons 5, 6, 7, 8), specific PCR fragments were analysed by Sanger sequencing. PCR products were purified using Agencourt AMPure XP magnetic beads (Beckman Coulter), labeled with Big Dye Terminator v3.1 (Applied Biosystems). Agencourt CleanSEQ magnetic beads (Beckman Coulter) were used for post-labeling purification., Sequence analysis was performed on an Applied Biosystems 3130xl Genetic Analyser.
Immunohistochemistry
IPMN tumour phenotypes were investigated according to WHO classification [1] by applying antibodies to mucin core proteins MUC-1 (clone DF3; 1:50; Abcam), MUC2 (clone Ccp58; 1:100; Novocastra Laboratories), MUC5AC (clone CLH2; 1:100; Novocastra Laboratories), and MUC6 (clone CLH5; 1:100; Novocastra Laboratories). The immunohistochemical expression of p53 (clone DO-1; prediluted; Immunotech), β-catenin (clone 15B8; 1:150; Sigma), CDKN2A/p16 (clone JC8; 1:100; Santa Cruz Biotechnology) and Smad4 (clone B-8; 1:200; Santa Cruz Biotechnology) was tested as a surrogate validation of deep sequencing results.
Statistical analysis
Fisher's exact test was used to compare mutational frequencies among IPMN grouped according to either histological subtype or tumour grade. P values of less than 0.05 were considered statistically significant.
Results
Clinico-pathological data
Fifty-two pancreatic intraductal neoplasms, comprising 48 IPMNs and 4 ITPNs, were retrieved from 51 patients as one patient had two separate lesions, an IPMN and an ITPN (Table 1, Figure 1). Of the 52 lesions, 40 (77%) were in the pancreatic head and/or the uncinate process, 9 (17%) in the pancreatic body/tail, and 3 (6%) diffusely involved the gland. Macroscopically, 31 (60%) neoplasms were main-duct type, 3 (6%) branch-duct type, and 18 (34%) combined-type. The 48 IPMNs were classified, according to WHO classification [1], into the following subtypes: 36 intestinal, 6 gastric, 3 pancreaticobiliary and 3 oncocytic type. Invasive carcinoma was associated with the IPMN in 29 of the 51 (57%) patients: 18 of the invasive cancers were colloid carcinomas, and 11 were conventional tubular adenocarcinomas.
Figure 1.

Representative haematoxylin-eosin images and differential mucins immunolabeling of pancreatic intraductal neoplasms (original magnifications 20×). PB, Pancreaticobiliary; ITPN, intraductal tubulopapillary neoplasm.
Tissues and cystic fluid subjected to sequencing
Non-invasive lesions from the 48 IPMN were low-grade (LG) in 3 cases, intermediate-grade (IG) in 17 cases and high-grade (HG) in 28 cases. For three IPMNs (128fp, 129fp and 138fp) sufficient material was obtained from non-invasive and invasive neoplastic areas (Table 2, Supplementary Table 2, Figure 2). One IPMN coexisted with an ITPN in the same patient (case 130fp); the two lesions were microdissected and analysed separately (Supplementary Table 2, Figure 3). All 4 ITPNs showed HG dysplasia. In 10 intestinal-type IPMNs cystic fluid was available and in 7 of these (70%) an adequate library for sequencing was obtained.
Table 2.
Mutational profile of non-invasive and invasive components in three cases of IPMN with associated carcinoma.
| Sample | Subtype | Lesion | GNAS | KRAS | TP53 | BRAF | STK11 | CDKN2A | PTEN |
|---|---|---|---|---|---|---|---|---|---|
| #128fpB | Intestinal | HG dysplasia | R201H (38%) | V14I (37%) | K601E (33%) | ||||
| #128fpA | Intestinal | Colloid carcinoma | R201H (27%) | V14I (32%) | K601E (28%) | ||||
|
| |||||||||
| #129fpB | Intestinal | IG dysplasia | G12V (42%) | E199D (58%) | V115M (7%) P70L (9%) | ||||
| #129fpA | Intestinal | Colloid carcinoma | G12V (16%) | E199D (57%) | |||||
|
| |||||||||
| #138fpA | Pancreaticobiliary | HG dysplasia | G12D (44%) | V272L (81%) | Q171* (74%) | ||||
| #138fpB | Pancreaticobiliary | Tubular carcinoma | G12D (23%) | V272L (31%) | |||||
Note: IG, intermediate grade; HG, high grade. Mutated alleles percentages are shown in parentheses.
Figure 2. Mutational profiles of matched non-invasive and invasive IPMN components identified by Ion Torrent sequencing.
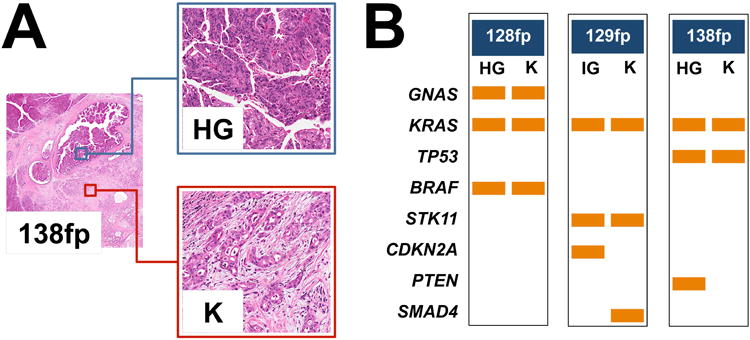
(A) Three cases (128fp, 129fp and 138fp) presented a non-invasive intermediate-grade (IG) or high-grade (HG) dysplastic component and an invasive adenocarcinoma (K) component, both of which were analysed using the 50 genes Ampliseq Hotspot Cancer Panel. Representative H&E images from case 138fp are presented (original magnifications 2× and 20×). (B) For each pair of samples the first column denotes mutations detected in IG or HG dysplastic samples and the second column represents mutations detected in cancer samples. Rows are the genes in which mutations were detected, and the orange bars represent the mutation. Seven mutations observed in the GNAS, KRAS, BRAF, KRAS and STK11 genes were common to both samples; three mutations were observed in only one of the matched lesions.
Figure 3. Concomitant IPMN and ITPN sharing a common GNAS mutation.

Patient 130fp presented two different neoplastic lesions, an intestinal-type IPMN and an ITPN coexisting in pancreatic head. The two phenotypically different lesions showed a common GNAS R201H mutation; the ITPN component presented a NRAS Q61L mutation. Representative H&E images of the lesions are shown (original magnifications, 20×). On the right of each sample is the representation of the reads aligned to the reference genome as provided by the Integrative Genomics Viewer (IGV v.2.1, Broad Institute) software for the hotspots mutations in the GNAS and NRAS genes.
Prevalence of driver gene mutations among the 50 genes of the AmpliSeq panel
An adequate library was obtained from all samples for subsequent sequencing. Mean 100× coverage of 98.9% with a mean read length of 110 bp was obtained in the 40 FF samples and mean 100× coverage of 98.5% with a mean read length of 105 bp was obtained in the 12 FFPE samples.
GNAS and KRAS mutations were the most prevalent alterations, found in 39/52 (75%) and 24/52 (46%) lesions, respectively. GNAS and/or KRAS mutations were identified in 45/52 (87%) cases; concomitant GNAS and KRAS mutations were found in 18/52 (35%) cases. Most GNAS mutations were in codon 201 (R201C and R201H); one Q227L mutation, not previously described in IPMN was also detected; KRAS mutations were in codon 12 in all but five cases, which were one in codon 14 (V14I) (Figure 4), one in codon 22 (Q22K), and three in codon 61 (one Q61R, two Q61H) (Supplementary Table 2).
Figure 4. IPMN showing a KRAS mutation V14I.

The intestinal-type IPMN 128fp presented a V14I KRAS mutation in both the high grade (HG) dysplastic and the invasive carcinomatous (K) component, analysed separately using the 50-gene Ampliseq Hotspot Cancer Panel. Representative H&E image of the lesions is shown (original magnifications, 20×). On the right there is the representation of the reads aligned to the reference genome as provided by the Integrative Genomics Viewer (IGV v.2.1, Broad Institute) software.
The prevalence of mutations in the other genes sequenced was lower. Five (10%) of the 52 lesions harboured a TP53 mutation, and three (6%) a BRAF mutation; mutations in CTNNB1, IDH1, STK11, and PTEN were each found in two tumours (2/52, 4%); ATM, CDH1, CDKN2A, FGFR3, NRAS, SMAD4, and SRC gene mutations were each found in only one of the 52 cases (1/52, 2%). All these genes were mutated in association with GNAS and/or KRAS mutations, with the exception of two IPMNs, one that only had a FGFR3 mutation and another that only had a TP53 mutation (Supplementary Table 2). The five TP53 mutated lesions included a frame-shift deletion (N247_R249del), a stop mutation at codon 306 (R306X), and the non-synonymous variants R175H, I195N, and V272L in exons 5 and 6. BRAF was mutated at the hotspot codons 599-601 (T599delinsIP, VK600E, and K601E) in exon 15. Most samples contained germline non-pathogenic variants in one or more of the following genes: PIK3CA, KIT, TP53, ATM, MET, KDR, FGFR3, and APC (data not shown).
Validation of mutations in six genes and screen for RNF43 mutations using an AmpliSeq custom panel
Forty-two of the 52 DNA samples (33 FF, 9 FFPE) were re-analysed using a custom gene panel to confirm the mutations in six genes (BRAF, CDKN2A, GNAS, KRAS, SMAD4, TP53) and to evaluate the role of RNF43, a gene that was not included in the first gene panel. An adequate library for subsequent sequencing was obtained for all samples. Mean 100× coverage of 100% with a mean read length of 175 bp was achieved in the 33 FF samples and mean 100× coverage of 99.24% with a mean read length of 150 bp was achieved in the 9 FFPE samples.
All the mutations identified in the first analysis were confirmed, with negligible variations of allelic frequencies (Supplementary Table 3); an additional mutation in CDKN2A (A148T) was detected due to the longer region of gene covered by the custom panel. Mutations in RNF43 gene were detected in six of the 42 samples (14%), and included: one deletion (M18Pfs*31) in exon 2, two insertions (R117Qfs*8 and E258*) in exons 3 and 7, respectively, and two missense variants (N179K) in exon 5 and exon 9 (S321I), and one nonsense (R371*) in exon 9.
Validation of mutations in five genes using Sanger sequencing and immunohistochemistry
Mutations in KRAS (exons 2, 3), GNAS (exons 8, 9), BRAF (exon 15), and TP53 (exons 5, 6, 7, 8) detected by deep sequencing were all confirmed by Sanger sequencing. We used p53 and β-catenin nuclear immunolabeling as a surrogate validation for TP53 and CTNNB1 mutational status. For TP53, cases 114, 939 and 138fp showed abnormal p53 nuclear accumulation of the mutated protein; cases 106 and 108, respectively harbouring the N247_R249del frame-shift deletion and the homozygous R306X mutation, had no immunolabeling (Figure 5). Two CTNNB1 mutated cases had nuclear immunopositivity for β-catenin.
Figure 5. TP53 mutational status corresponds to p53 protein accumulation.

(A) Case 939 showed a heterogeneous pattern of staining which is consistent to TP53 mutational status (21% of mutated alleles, I195N). (B) Case 108 with no evident p53 labelling corresponds to a homozygous stop mutation (83% of mutated alleles, R306*). For each sample a representative p53 immunohistochemical image (original magnifications ×20) and the representation of the reads aligned to the reference genome as provided by the Integrative Genomics Viewer (IGV v.2.1, Broad Institute) software are presented.
Evaluation of CDKN2A/P16 and SMAD4 dysregulation using immunohistochemistry
Immunohistochemistry was used as a surrogate test to assess the involvement of CDKN2A/P16 and SMAD4 genes [7,24,25]. In fact, protein expression may more accurately reflect gene status as both genes are mainly inactivated by homozygous deletion, and CDKN2A/P16 also by promoter methylation.
Material was available for 36 cases: 34 IPMN (23 intestinal-type, 5 gastric-type, 3 pancreaticobiliary, and 3 oncocytic-type) and 2 ITPN. In 17 IPMN the non-invasive lesion coexisted with invasive carcinoma, for a total of 53 lesions analysed (i.e., 34 IPMN, 2 ITPN, 17 invasive carcinoma).
In normal pancreatic parenchyma, p16 was expressed in Langerhans' islets, and in random acinar and duct cells (Figure 6). P16 loss was observed in 19/53 (36%) lesions distributed as follow: 0/1 (0%) LG, 3/13 (23%) IG, 7/22 (31%) HG, and 9/17 (53%) invasive carcinomas. Most p16-loss cases (9/10) were intestinal-type IPMNs. Only 1 of the 2 CDKN2A/P16 mutated IPMNs was available for immunophenotyping, and it lacked p16 expression.
Figure 6. P16 and Smad4 immunoexpression in IPMN.
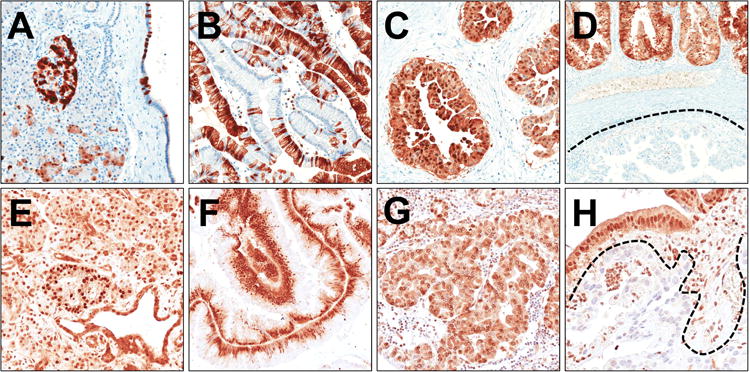
Representative images of p16 (A-D) and Smad4 (E-H) immunoreactions are shown. (A) In normal pancreatic parenchyma, p16 is expressed in Langerhans' islets, and in random acinar and duct cells. (B) A barcode-like p16 positivity in a gastric-type IPMN. (C) Strong nuclear and cytoplasmic p16 expression in an oncocytic-type IPMN. (D) p16 expression heterogeneity in a case of pancreatobiliary-type IPMN (positive and negative p16 components are evident). (E) Normal pancreas shows a strong Smad4 expression in all epithelial and stromal cell types with stronger positivity in Langerhans' islets. Strong Smad4 expression in intestinal-type (F) and oncocytic-type (G) IPMNs. (H) A Smad4 negative invasive carcinoma infiltrating the stroma surrounding a pancreatobiliary-type IPMN. Original magnifications 10× and 20×.
Normal pancreatic parenchyma showed a strong nuclear and cytoplasmic Smad4 expression in all cell types including Langerhans' islets, acinar cells, stromal cells, and duct cells (Figure 6). Only 1 intestinal-type IPMN showed loss of Smad4 expression. Four IPMN-associated invasive carcinomas were Smad4 negative, with the coexisting Smad4-positive non-invasive component. Case 137fp, harbouring a F253C SMAD4 mutation, expressed Smad4. Thus, 5/17 (29%) invasive carcinomas had Smad4 alterations.
The 2 ITPNs showed strong immunoreactions for both p16 and Smad4.
Mutation prevalence according to histological subtype
Among IPMNs, at least one somatic mutation was observed in 46/48 (96%) cases, while 29 neoplasms (60%) had multiple gene alterations (Table 3, Supplementary Table 2). GNAS and/or KRAS mutations were found in 44/48 (92%) IPMNs: GNAS was mutated in 38/48 (79%) cases, KRAS in 24/48 (50%), and these mutations coexisted in 18/48 (37.5%) cases.
Table 3.
Mutational profiling of the 52 intraductal pancreatic neoplasms according to pathological data.
| Genes | Total (n=52) | Gastric (n=6) | Intestinal (n=36) | Pancreaticobiliary (n=3) | Oncocytic (n=3) | ITPN (n=4) | LG (n=3) | IG (n=17) | HG (n=28) | Carcinoma (n=4) | LG + IG (n=20) | HG + Carcinoma (n=32) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GNAS | 38 (73%) | 6 (100%) | 30 (83%) | 1 (33%) | 1 (33%) | 1 (25%) | 3 (100%) | 15 (88%) | 17 (61%) | 4 (100%) | 18 (90%) | 21 (66%) |
| KRAS | 24 (46%) | 5 (83%) | 14 (39%) | 2 (67%) | 2 (67%) | 0 | 2 (67%) | 11 (65%) | 9 (32%) | 2 (50%) | 13 (65%) | 11 (34%) |
| TP53 | 5 (10%) | 0 | 3 (8%) | 2 (67%) | 0 | 0 | 0 | 0 | 5 (18%) | 0 | 0 | 5 (15%) |
| BRAF | 3 (6%) | 0 | 3 (8%) | 0 | 0 | 0 | 0 | 0 | 2 (7%) | 1 (25%) | 0 | 3 (9%) |
| CTNNB1 | 2 (4%) | 0 | 2 (6%) | 0 | 0 | 0 | 0 | 1 (6%) | 1 (4%) | 0 | 1 (5%) | 1 (3%) |
| IDH1 | 2 (4%) | 0 | 2 (6%) | 0 | 0 | 0 | 0 | 1 (6%) | 1 (4%) | 0 | 1 (5%) | 1 (3%) |
| STK11 | 2 (4%) | 0 | 2 (6%) | 0 | 0 | 0 | 0 | 1 (6%) | 1 (4%) | 1 (5%) | 1 (3%) | |
| PTEN | 2 (4%) | 1 (17%) | 0 | 1 (25%) | 0 | 0 | 0 | 1 (6%) | 1 (4%) | 0 | 1 (5%) | 1 (3%) |
| ATM | 1 (2%) | 0 | 1 (3%) | 0 | 0 | 0 | 0 | 0 | 1 (4%) | 0 | 0 | 1 (3%) |
| CDH1 | 1 (2%) | 0 | 1 (3%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25%) | 0 | 1 (3%) |
| CDKN2A | 1 (2%) | 0 | 1 (3%) | 0 | 0 | 0 | 0 | 1 (6%) | 0 | 0 | 1 (5%) | 0 |
| FGFR3 | 1 (2%) | 0 | 1 (3%) | 0 | 0 | 0 | 0 | 0 | 1 (4%) | 0 | 0 | 1 (3%) |
| NRAS | 1 (2%) | 0 | 0 | 0 | 0 | 1 (25%) | 0 | 0 | 1 (4%) | 0 | 0 | 1 (3%) |
| SMAD4 | 1 (2%) | 1 (17%) | 0 | 0 | 0 | 0 | 0 | 1 (6%) | 0 | 0 | 1 (5%) | 0 |
| SRC | 1 (2%) | 0 | 1 (3%) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25%) | 0 | 1 (3%) |
Note: LG, low-grade dysplasia; IG, intermediate grade dysplasia; HG, high grade dysplasia. ITPN, intraductal tubulo-papillary neoplasm
All 6 gastric type IPMNs harboured a GNAS mutation and five of these cases also had a KRAS mutation (Table 3). Among intestinal type IPMNs, most harboured GNAS (30/36, 83%) and/or KRAS (14/36, 39%) mutations. The prevalence of GNAS mutation among gastric and intestinal IPMN subtypes was similar (P=0.568). Conversely, KRAS alterations were less frequent in intestinal than in gastric IPMNs, although not reaching statistical significance (P=0.075). TP53 and BRAF alterations were each observed in 3 of the 36 (8%) intestinal-type IPMNs and in none of the gastric IPMNs. Among the less represented IPMN subtypes (pancreaticobiliary, n=3, oncocytic, n=3), GNAS and KRAS variants were present in the pancreaticobiliary and oncocytic IPMNs (1 of 3 GNAS, and 2 of 3 KRAS). Notably, TP53 was mutated only in the pancreaticobiliary-type IPMNs (2 of 3). RNF43 mutations were detected in 4 (12%) of 32 intestinal-type and one of two pancreaticobiliary-type IPMNs, and were always associated with GNAS and/or KRAS mutations. None of the gastric, oncocytic or ITPN had RNF43 mutations.
The only mutations detected in the four ITPNs, were detected in case 130fp, where the coexisting IPMN and ITPN lesions shared the same GNAS R201H mutation; this ITPN also had an additional NRAS Q61L mutation (Supplementary Table 2, Figure 3).
Mutation prevalence according to tumour grade
Low- or intermediate-grade dysplastic samples were characterized by recurrent mutations in GNAS (90%) and KRAS (65%); while the prevalence of these mutations was significantly lower in high grade/invasive carcinoma samples (66% of the high-grade lesions harboured a GNAS and 34% a KRAS mutation, P=0.0001 and P=0.046, respectively) (Table 3). We had significantly more intestinal-type IPMNs than in the series by Wu et al. [10], and this was likely a factor in the lower prevalence of mutant KRAS in the IPMNs with high-grade dysplasia (Supplementary Table 2). Notably, TP53 and BRAF mutations were found in high-grade lesions (15% and 9%, respectively), but not in any of the low or intermediate-grade lesions.
Mutations in matched non-invasive and invasive tumours
Three cases (128fp, 129fp and 138fp) with matched non-invasive and invasive components had similar mutational profiles (Table 2, Figure 2). Seven mutations observed in the GNAS, KRAS, BRAF, KRAS and STK11 genes were shared between the pre-invasive and invasive lesions; three point mutations in CDKN2A, SMAD4, and PTEN were observed in only one of the matched lesions.
NGS can identify IPMN specific mutations in cystic fluid
DNAs from cystic fluids were available for 10 intestinal-type IPMNs, and were deep sequenced with the 50-gene AmpliSeq Cancer Panel. An adequate library was obtained in 7 cyst fluids and sequencing detected 10 of the 13 mutations present in the matched IPMN, including six of seven GNAS mutations, three of three KRAS mutations, and one of two TP53 mutations.
Discussion
Intraductal papillary mucinous neoplasms are important precursors to invasive pancreatic cancer and represent an opportunity for detection and treatment of pancreatic neoplasia before an invasive carcinoma develops [2,26-28]. While the phenotypic classification of IPMNs is well established, the molecular drivers of this precursor lesion have yet to be fully characterized [4,12,17,29]. In this study, we explored the mutational status of 51 cancer related genes in 52 intraductal lesions, including 48 IPMN and 4 ITPN, and tested the diagnostic performance of the test on 10 cystic fluid samples.
The 48 IPMNs harboured frequent somatic mutations in GNAS and KRAS, and at least one of the two genes was mutated in 44/48 (92%) of the IPMNs. GNAS mutations were identified in 79% of tumours and most of them involved codon 201. KRAS was mutated in 50% of IPMNs, and mutations were located in codons 12, 14, 22, and 61. Our literature review finds that KRAS codon 14 mutations have never been reported for colorectal or pancreatic cancer. Although KRAS codon 14 mutations are rare, they are considered as driver in nature [30,31]. The high prevalence of GNAS and KRAS mutations found in our series is similar to that of previous studies, and suggests that these genes may be ideal targets for early detection efforts [11,32].
RNF43 was the gene with the third most frequent intragenic mutations detected by our sequencing analysis. These RNF43 mutations were always associated with GNAS and/or KRAS mutations. Five of the six mutations detected are newly described, as only one had been previously reported for IPMN [11,32]. Our series adds to the limited literature regarding the prevalence and distribution of RNF43 in this class of tumours, as only 8 IPMNs have been evaluated for RNF43 mutations in the literature [10].
For other genes, mutations were detected in a small percentage (<5%) of samples in CDKN2A, CTNNB1, IDH1, STK11, PTEN (each one in two of the 52 tumours) and ATM, CDH1, FGFR3, NRAS, SMAD4, SRC (each one in one of the 52 neoplasms). These genes were mutated in association with GNAS and/or KRAS mutations, with the exception of two IPMNs, one IPMN that only had a FGFR3 mutation and another that only had a TP53 mutation. Prior studies have reported STK11 mutations in a similar percentage of IPMNs (9%) [8]. Prior studies have found ATM and CDH1 somatic mutations in a small percentage of pancreatic ductal adenocarcinomas but these mutations have not been yet reported in IPMNs [33,34]. Somatic mutations in IDH1, PTEN and FGFR3, NRAS, SRC have not been reported in IPMNs or in pancreatic ductal adenocarcinomas [33,34]. CTNNB1 mutations are characteristic of pancreatic solid and pseudopapillary tumours, but nuclear expression of β-catenin has been described in some IPMNs [17]. The mutations we found in IDH1, NRAS and CTNNB1 arose in known hotspots for these genes. Although PTEN mutations have not been previously reported in IPMNs or ITPNs [12], loss of PTEN expression in IPMNs has been reported [35], and mutations in PIK3CA, another gene in this pathway also occur in a small percentage of cases [5,16,35]. The pathogenetic role of CDKN2A/P16 and SMAD4 was further investigated by immunohistochemical analysis. Our results demonstrated that 36% of IPMNs show loss of p16 and 24% of IPMN-related invasive carcinomas loss of Smad4, which is in accordance to previous studies [4,7].
The different histological subtypes of IPMNs showed a partial overlap in the mutational spectrum, as well as some differences. GNAS gene mutations were common in all types, consistent with previous reports [10,11,32,36]. KRAS mutations were more common in gastric-type compared to intestinal-type IPMNs (83% vs. 39%, P=0.07), as has been reported by Mohri et al. (82% vs .27%) [4]. Other authors have reported that oncocytic-type IPMNs are less likely to have KRAS gene mutations than pancreaticobiliary type-IPMNs [17], while in our study all three pancreaticobiliary and two of three oncocytic cases harboured KRAS mutations.
Three of four ITPNs had no mutations in the 50-gene panel. The ITPN that coexisted with an IPMN had mutations in both GNAS and NRAS. The mutation observed in GNAS is of note, because Yamaguchi and colleagues have recently reported that this gene was not mutated in a series of 14 ITPNs [18]. Previous reports found activating PIK3CA mutations in 3 of 14 (21%) of ITPNs [12,18]. Two different groups identified PIK3CA mutations in 11% (4/36) and 9% (2/21) of IPMNs, respectively [5,16]; noteworthy, the missense hot-spot mutation H1047R in exon 20 was found in 4 of 6 invasive cancers. In our series, PIK3CA mutations were absent in both ITPN and IPMN; given the good quality of sequences we obtained, this discrepancy is possibly due to the small number of cases analysed by each of the three studies. Although we only analysed four ITPN cases, the absence of KRAS and BRAF mutations in ITPN supports the hypothesis of a distinct molecular profile and possibly an independent origin for ITPN lesions. TP53 and BRAF mutations were only found in high-grade lesions (15% and 9%, respectively), suggesting that these mutations could be a potential diagnostic marker for high-grade dysplasia.
Recent guidelines emphasize conservative management of patients with IPMNs unless there are stigmata of cancer [37], particularly for elderly patients who often have comorbid conditions that increase surgical risk. Because pancreatic imaging does not provide sufficient information about the characteristics of pancreatic cysts, fine needle is used for further evaluation, but current tests do not provide sufficient diagnostic information. Therefore, the identification of novel diagnostic approaches based on genetic analyses of cyst fluid aspirated at the time of endoscopic ultrasound is particularly appealing. In the present study, we demonstrate that targeted next-generation sequencing is feasible with cystic fluid samples, and can provide information about the genetic profile of these lesions. Notably cyst fluid analysis identified only 10 of the 13 somatic mutations found in the corresponding IPMNs. We suspect that this was because of a low concentration of mutations in the cyst fluid, below the limit of detection of our assay and this could be related to how the cyst fluids were sampled. Further study is needed to evaluate the concentration of mutations in IPMN cyst fluids. There are other approaches that can be employed to increase the limit of detection of next-generation sequencing such as BEAMing or Safe-Seqs analysis [11,38].
The prevalence of KRAS mutations in our series (50%) of IPMNs is lower than that observed by Wu and colleagues, who reported 79% prevalence of KRAS mutations in IPMNs [11]. This difference is reflected in differences in the prevalence of the histological subtypes of IPMNs in the two series: Wu et al. studied 48 IPMNs, the majority of which were gastric-type (27 cases) and included 10 intestinal-type, 5 pancreaticobiliary-type, and 6 undetermined. Our series included 6 gastric-type and 36 intestinal-type IPMNs (see Table 3). For gastric-type IPMNs, KRAS mutations were found in 25/27 (93%) cases of Wu et al. and in 5/6 (83%) of our series; for intestinal-type IPMNs, they found mutations in 4/10 (40%) of cases whereas we found mutations in 14/36 (39%) of cases.
In conclusion, gastric and intestinal IPMNs commonly harbour GNAS and KRAS gene mutations that likely precede mutations in other genes that are less common in IPMNs and when found are generally more common in higher-grade lesions. Moreover, our data also confirm the suggestion that ITPN appears to be a pancreatic intraductal disease different from IPMN [12,18]. TP53 gene mutations have been only found in higher-grade lesions, suggesting that these mutations could be a specific marker for high-grade dysplasia or invasive cancer. Next-generation sequencing of cystic fluid samples can identify the majority of mutations arising in IPMNs potentially improving the diagnostic and prognostic stratification of IPMNs.
Supplementary Material
(A) Targeted regions of the 50-genes Ampliseq Cancer Hotspot Panel v2. (B) Targeted regions of the Ampliseq custom panel.
Mutations found in 52 intraductal pancreatic neoplasms sequenced for 50 cancer-related genes.
Validation of mutations found in 42 intraductal pancreatic neoplasms by deep sequencing with Ampliseq custom panel.
Acknowledgments
We thank Dr. Samantha Bersani for her valuable help in performing immunohistochemistry. The study was supported by Italian Cancer Genome Project (FIRB RBAP10AHJB), Associazione Italiana Ricerca Cancro (AIRC grants n. 12182 and 6421), Fondazione Italiana Malattie Pancreas – Ministero Salute (CUP_J33G13000210001), National Institutes of Health CA62924, Susan Wojcicki and Dennis Troper, and the Michael Rolfe Foundation. The funding agencies had no role in the collection, analysis and interpretation of data and in the writing of the manuscript.
Abbreviations
- IPMN
intraductal papillary mucinous neoplasms
- ITPN
intraductal tubulopapillary neoplasms
- WHO
World Health Organisation
Footnotes
Potential competing interests: The authors have no competing interests to declare.
Author Contribution statement: AS, MF, RHH and AMaitra conceived the study. EA and MdM designed the study. MG, JY, and MdM designed the validation experiment. MG supervised the validation experiment. RTL coordinated patients and sample data management and supervised ethical protocols. GM, RS and CB collected materials and clinical data. BR, MF, PCapelli, GZ, AS analysed histopathological data. MF, BR, PCastelli microdissected samples. EA, MdM, JY, DA and YA carried out deep sequencing and raw data analysis. AMafficini and JY performed bioinformatic analysis. MF, PCastelli and GZ analysed immunohistochemistry. EA, AMafficini, MF and MdM drafted the manuscript. AMafficini, MG, RHH, AMaitra and RTL revised the manuscript. AS finalised the paper. All authors approved the submitted version.
References
- 1.Adsay NV, Fukushima N, Furukawa T, et al. Intraductal neoplasms of the pancreas. In: Bosman FT, Carneiro F, Hruban RH, et al., editors. WHO classification of tumours of the digestive system. 4th. IARC; Lyon: 2010. [Google Scholar]
- 2.Luttges J, Zamboni G, Longnecker D, et al. The immunohistochemical mucin expression pattern distinguishes different types of intraductal papillary mucinous neoplasms of the pancreas and determines their relationship to mucinous noncystic carcinoma and ductal adenocarcinoma. Am J Surg Pathol. 2001;25:942–948. doi: 10.1097/00000478-200107000-00014. [DOI] [PubMed] [Google Scholar]
- 3.Chadwick B, Willmore-Payne C, Tripp S, et al. Histologic, immunohistochemical, and molecular classification of 52 IPMNs of the pancreas. Appl Immunohistochem Mol Morphol. 2009;17:31–39. doi: 10.1097/PAI.0b013e31817c02c6. [DOI] [PubMed] [Google Scholar]
- 4.Mohri D, Asaoka Y, Ijichi H, et al. Different subtypes of intraductal papillary mucinous neoplasm in the pancreas have distinct pathways to pancreatic cancer progression. J Gastroenterol. 2012;47:203–213. doi: 10.1007/s00535-011-0482-y. [DOI] [PubMed] [Google Scholar]
- 5.Schonleben F, Qiu W, Ciau NT, et al. PIK3CA mutations in intraductal papillary mucinous neoplasm/carcinoma of the pancreas. Clin Cancer Res. 2006;12:3851–3855. doi: 10.1158/1078-0432.CCR-06-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abe K, Suda K, Arakawa A, et al. Different patterns of p16INK4A and p53 protein expressions in intraductal papillary-mucinous neoplasms and pancreatic intraepithelial neoplasia. Pancreas. 2007;34:85–91. doi: 10.1097/01.mpa.0000240608.56806.0a. [DOI] [PubMed] [Google Scholar]
- 7.Biankin AV, Biankin SA, Kench JG, et al. Aberrant p16(INK4A) and DPC4/Smad4 expression in intraductal papillary mucinous tumours of the pancreas is associated with invasive ductal adenocarcinoma. Gut. 2002;50:861–868. doi: 10.1136/gut.50.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato N, Rosty C, Jansen M, et al. STK11/LKB1 Peutz-Jeghers gene inactivation in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol. 2001;159:2017–2022. doi: 10.1016/S0002-9440(10)63053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schonleben F, Qiu W, Allendorf JD, et al. Molecular analysis of PIK3CA, BRAF, and RAS oncogenes in periampullary and ampullary adenomas and carcinomas. J Gastrointest Surg. 2009;13:1510–1516. doi: 10.1007/s11605-009-0917-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011;108:21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Kuboki Y, Hatori T, et al. Somatic mutations in PIK3CA and activation of AKT in intraductal tubulopapillary neoplasms of the pancreas. Am J Surg Pathol. 2011;35:1812–1817. doi: 10.1097/PAS.0b013e31822769a0. [DOI] [PubMed] [Google Scholar]
- 13.Iacobuzio-Donahue CA, Klimstra DS, Adsay NV, et al. Dpc-4 protein is expressed in virtually all human intraductal papillary mucinous neoplasms of the pancreas: comparison with conventional ductal adenocarcinomas. Am J Pathol. 2000;157:755–761. doi: 10.1016/S0002-9440(10)64589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kanda M, Sadakari Y, Borges M, et al. Mutant TP53 in duodenal samples of pancreatic juice from patients with pancreatic cancer or high-grade dysplasia. Clin Gastroenterol Hepatol. 2013;11:719–730. e715. doi: 10.1016/j.cgh.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato N, Ueki T, Fukushima N, et al. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology. 2002;123:365–372. doi: 10.1053/gast.2002.34160. [DOI] [PubMed] [Google Scholar]
- 16.Lubezky N, Ben-Haim M, Marmor S, et al. High-throughput mutation profiling in intraductal papillary mucinous neoplasm (IPMN) J Gastrointest Surg. 2011;15:503–511. doi: 10.1007/s11605-010-1411-8. [DOI] [PubMed] [Google Scholar]
- 17.Xiao HD, Yamaguchi H, Dias-Santagata D, et al. Molecular characteristics and biological behaviours of the oncocytic and pancreatobiliary subtypes of intraductal papillary mucinous neoplasms. J Pathol. 2011;224:508–516. doi: 10.1002/path.2875. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi H, Kuboki Y, Hatori T, et al. The discrete nature and distinguishing molecular features of pancreatic intraductal tubulopapillary neoplasms and intraductal papillary mucinous neoplasms of the gastric type, pyloric gland variant. J Pathol. 2013;231:335–341. doi: 10.1002/path.4242. [DOI] [PubMed] [Google Scholar]
- 19.Scarpa A, Sikora K, Fassan M, et al. Molecular typing of lung adenocarcinoma on cytological samples using a multigene next generation sequencing panel. PLoS One. 2013;8:e80478. doi: 10.1371/journal.pone.0080478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beadling C, Neff TL, Heinrich MC, et al. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 2013;15:171–176. doi: 10.1016/j.jmoldx.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Luchini C, Capelli P, Fassan M, et al. Next-generation histopathological diagnosis: a lesson from a hepatic carcinosarcoma. J Clin Oncol. 2014 doi: 10.1200/JCO.2012.47.5855. in press. [DOI] [PubMed] [Google Scholar]
- 22.Simbolo M, Gottardi M, Corbo V, et al. DNA qualification workflow for next generation sequencing of histopathological samples. PLoS One. 2013;8:e62692. doi: 10.1371/journal.pone.0062692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamo A, Bertolaso A, van Raaij AW, et al. Application of microfluidic technology to the BIOMED-2 protocol for detection of B-cell clonality. J Mol Diagn. 2012;14:30–37. doi: 10.1016/j.jmoldx.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas : a new marker of DPC4 inactivation. Am J Pathol. 2000;156:37–43. doi: 10.1016/S0002-9440(10)64703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilentz RE, Geradts J, Ma ynard R, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58:4740–4744. [PubMed] [Google Scholar]
- 26.Furukawa T, Kloppel G, Volkan Adsay N, et al. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Arch. 2005;447:794–799. doi: 10.1007/s00428-005-0039-7. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura A, Horinouchi M, Goto M, et al. New classification of pancreatic intraductal papillary-mucinous tumour by mucin expression: its relationship with potential for malignancy. J Pathol. 2002;197:201–210. doi: 10.1002/path.1109. [DOI] [PubMed] [Google Scholar]
- 28.Yonezawa S, Taira M, Osako M, et al. MUC-1 mucin expression in invasive areas of intraductal papillary mucinous tumors of the pancreas. Pathol Int. 1998;48:319–322. doi: 10.1111/j.1440-1827.1998.tb03913.x. [DOI] [PubMed] [Google Scholar]
- 29.Fassan M, Simbolo M, Bria E, et al. High-throughput mutation profiling identifies novel molecular dysregulation in high-grade intraepithelial neoplasia and early gastric cancers. Gastric Cancer. 2013 doi: 10.1007/s10120-013-0315-1. [DOI] [PubMed] [Google Scholar]
- 30.Schubbert S, Zenker M, Rowe SL, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 31.Tyner JW, Erickson H, Deininger MW, et al. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749–1755. doi: 10.1182/blood-2008-04-152157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furukawa T, Kuboki Y, Tanji E, et al. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Carracedo D, Turk A, Fine S, et al. Loss of PTEN expression predicts poor prognosis in patients with intraductal papillary mucinous neoplasms of the pancreas. Clin Cancer Res. 2013;19:6830–6841. doi: 10.1158/1078-0432.CCR-13-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dal Molin M, Matthaei H, Wu J, et al. Clinicopathological Correlates of Activating GNAS Mutations in Intraductal Papillary Mucinous Neoplasm (IPMN) of the Pancreas. Ann Surg Oncol. 2013;20:3802–3808. doi: 10.1245/s10434-013-3096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka M, Fernandez-del Castillo C, Adsay V, et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology. 2012;12:183–197. doi: 10.1016/j.pan.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Kinde I, Wu J, Papadopoulos N, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Targeted regions of the 50-genes Ampliseq Cancer Hotspot Panel v2. (B) Targeted regions of the Ampliseq custom panel.
Mutations found in 52 intraductal pancreatic neoplasms sequenced for 50 cancer-related genes.
Validation of mutations found in 42 intraductal pancreatic neoplasms by deep sequencing with Ampliseq custom panel.