

Abstract
Proline-rich tyrosine kinase 2 (PYK2), also known as cell adhesion kinase beta (CAKbeta) or protein tyrosine kinase 2b (PTK2B), is a calcium-dependent signaling protein involved in cell migration. Phosphorylation of residue Y402 is associated with activation of PYK2, and leads to the recruitment of downstream signaling molecules. PYK2 was previously implicated in long term potentiation (LTP); however, the role of PYK2 in long term depression (LTD) is unknown. Here we report that PYK2 is activated by NMDA receptor stimulation (chemical LTD) in cultured neurons. Small hairpin RNA (shRNA)-mediated knockdown of PYK2 blocks LTD, but not LTP, in hippocampal slice cultures. We find that the Y402 residue and, to a lesser extent, PYK2 kinase activity contribute to PYK2's role in LTD. Knockdown experiments indicate that PYK2 is required to suppress NMDA-induced ERK phosphorylation. Overexpression of PYK2 depresses NMDA-induced ERK phosphorylation, and inhibits LTP, but not LTD. Our data indicate that PYK2 is critical for induction of LTD, possibly in part by antagonizing ERK signaling in hippocampal neurons.
Keywords: plasticity, PYK2, synapse, phosphorylation, ERK, NMDAR
Introduction
Activity-dependent modification of synapses is widely considered to be a mechanism for learning and memory. In the commonly studied forms of hippocampal synaptic plasticity – NMDA receptor (NMDAR)-dependent LTP and LTD – different levels and/or kinetics of NMDAR-mediated calcium influx are believed to activate distinct signaling pathways. Calcium elevation during LTP promotes activity of calcium/calmodulin-dependent kinase II and AMPA receptor (AMPAR) insertion into the postsynaptic membrane (Malenka et al., 1989; Malinow et al., 1989; Silva et al., 1992), whereas calcium influx during LTD promotes AMPAR endocytosis depending on activity of serine/threonine phosphatases like calcineurin (Mulkey et al., 1993, 1994; Morishita et al., 2001). Src family kinases can contribute to LTP through the phosphorylation and functional enhancement of NMDARs (Grant et al., 1992; Lu et al., 1998). Differential activation of Ras family GTPases and downstream mitogen activated protein kinase (MAPK) pathways may determine the direction of activity-dependent synaptic modification during LTP or LTD (Zhu et al., 2002; Kim et al., 2005; Zhu et al., 2005). Sustained activation of the MAPK ERK, for example, is believed to promote LTP (English and Sweatt, 1997; Zhu et al, 2002). These signaling mechanisms help explain how NMDAR activation can lead to distinct functional outcomes in neurons.
PYK2 is a calcium-dependent tyrosine kinase found in the postsynaptic density (PSD) of central neurons (Huang et al., 2001; Collins et al., 2006; reviewed in Sheng and Hoogenraad, 2007). In non-neuronal cells, PYK2 is involved in osteoclast function (Gil-Henn et al., 2007), macrophage migration (Okigaki et al., 2003), and focal adhesion disassembly (Hashido et al., 2006). While the precise mechanism of PYK2 activation remains unclear, it is believed that a rise in intracellular calcium can directly or indirectly induce dimerization of PYK2 and trans-autophosphorylation at Y402, a residue located N-terminal to the kinase domain (Lev et al., 1995; Park et al., 2004; Kohno et al., 2008, Bartos et al., 2010). In hematopoietic cells, the phosphorylated Y402 and surrounding residues mediate association with and activation of Src family kinases, which in turn phosphorylate additional tyrosine residues in the PYK2 kinase domain to enhance PYK2 kinase activity (Park et al., 2004). Thus Y402 phosphorylation is commonly used as a marker of activated PYK2. Tyrosine phosphorylation of PYK2 also leads to changes in MAPK activity, although the direction of change and the subtype of MAPK affected vary across different cell types (Girault et al., 1999; Zhao et al., 2000).
Using recombinant PYK2 proteins, Huang et al. (2001) found that PYK2 is involved in LTP and the Src-dependent pathway of NMDAR potentiation. The role of PYK2 in synaptic plasticity, however, has not been examined by a molecular genetic approach. Interestingly, PYK2 is reported to bind PSD-95 (Seabold et al., 2003), an abundant scaffold protein of the PSD that promotes synaptic strength and that is required for LTD (Migaud et al., 1998; Stein et al., 2003; Ehrlich and Malinow, 2004; Kim et al., 2007; Xu et al., 2008). Because PYK2 is calcium-regulated, associates with PSD-95, and functions in reorganizing specialized adhesion sites, we hypothesized that PYK2 may play a role in LTD at neuronal synapses. We report here that PYK2 phosphorylation at Y402 is enhanced by stimulation of NMDARs that mimics LTD (chem-LTD) and that PYK2 is required for LTD. PYK2 dampens NMDAR-dependent activation of ERK MAPKs, a function consistent with PYK2's critical role in LTD.
Materials and Methods
Antibodies and chemicals
The following antibodies were used in this study (purchased from Cell Signaling Technology, unless indicated otherwise): rabbit anti-GluA1 (Millipore), rabbit anti-phospho S845 GluA1 (Chemicon), rabbit anti-phospho Y402 PYK2 (Invitrogen), mouse anti-PYK2 (BD Biosciences), rabbit anti-phospho Y416 Src, rabbit anti-Src, mouse anti-phospho T202/Y204 ERK, rabbit anti-ERK, rabbit anti-phospho S9 GSK3beta, rabbit anti-GSK3beta, rabbit anti-phospho T308 Akt, rabbit anti-Akt, rabbit anti-phospho T183/Y185 JNK, rabbit anti-JNK, mouse anti-tubulin (Sigma), anti-NR2A (Covance), anti-NR2B (NeuroMab), anti-PSD95 (NeuroMab), mouse anti-myc (Santa Cruz Biotechnology). The following chemicals were used (purchased from Sigma unless indicated otherwise): NMDA, tetrodotoxin (Tocris), NBQX (Tocris), nimodipine, picrotoxin, chlor-adenosine, FK506 (Tocris), cyclosporine A (Calbiochem).
DNA constructs and Lentivirus shRNA
WT-PYK2 construct (myc-tagged in pCMV vector) was a gift from Dr. Wen-Cheng Xiong, Medical College of Georgia (Xiong and Parsons, 1997). For PYK2 shRNA, the following oligonucleotides were annealed and inserted into the HindIII/BglII sites of pSuper vector (Brummelkamp et al., 2002): 5′-GAT CCC CGC TGT AGC ATA GAG TCA GAT TCA AGA GAT CTG ACT CTA TGC TAC AGC TTT TTA-3′; 5′-AGC TTA AAA AGC TGT AGC ATA GAG TCA GAT CTC TTG AAT CTG ACT CTA TGC TAC AGC GGG-3′. The final target sequence of PYK2 shRNA is 5′-GCT GTA GCA TAG AGT CAG A-3′. Luciferase shRNA (in pSuper vector) was a gift from Dr. Huaye Zhang, University of Virginia (Zhang and Macara, 2006). The target sequence of luciferase shRNA is 5′-CGT ACG CGG AAT ACT TCG A-3′.
Lentiviral transfer vector constructs were modified from the original FUGW vector backbone (Lois et al., 2002). H1 promoter cassettes from pSuper constructs (PYK2 shRNA and luciferase shRNA) were cloned between the HIV-flap and synapsin promoter. For the production of lentiviral vectors, the transfer vector, packaging vector Δ8.2 and VSVG envelope vector were cotransfected into HEK293 cells. Supernatants of culture media were collected 48 hours after transfection, and centrifuged at 50,000 x g to concentrate the lentivirus.
PYK2 mutants were constructed by site-directed mutagenesis (Stratagene Quikchange kit). The following primers were used: (1) For Y402F-PYK2, 5′-GCA TAG AGT CAG ACA TCT TTG CAG AGA TTC CTG ATG AG-3′ (2) For K457A-PYK2, 5′-GAA AAA ATT AAT GTG GCC GTC GCG ACC TGT AAG AAA GAT TGT ACC-3′ (3) For WT-PYK2* or K457A-PYK2*, two sequential reactions were performed to introduce 9 point mutations spread throughout the target region (final shRNA-resistant target region: 5′-GCT GCT CTA TTG AAA GCG A-3′). Primer set 1, 5′-CGG TCC CAC CTC TCA GAA AGC TGC TCT ATA GAG TCA GAC ATC TAT GCA GAG-3′; Primer set 2, 5′-CCC ACC TCT CAG AAA GCT GCT CTA TTG AAA GCG ACA TCT ATG CAG AGA TTC C-3′.
Cell culture and chem-LTD
HEK293 cells were maintained in DMEM (Gibco), supplemented with 10% fetal bovine serum (Gibco) and penicillin/streptomycin (Gibco), and transiently transfected with plasmids using Lipofectamine 2000 (Invitrogen). Cells were harvested after 24-48 hours. Hippocampal neurons were cultured as described (Kim et al., 2007), and infected with lentivirus in the media at DIV15 for 7 days in vitro. Mature cultured neurons (DIV 21-26) in 12-well plates were treated for 10 minutes with 20 μM NBQX, 2 μM tetrodotoxin, and 5 μM nimodipine before treatment with 70 μM NMDA for 2.5, 5, or 15 minutes (or untreated). Plates were then placed on ice, and each well was washed with ice-cold PBS before lysing in 2X sample buffer (containing beta-mercaptoethanol). For experiments with calcineurin inhibitors, FK506 (1 μM) or cyclosporine A (1 μM) were added to neurons for 10 min-1 hr prior to NMDA treatment.
Hippocampal slice culture
Organotypic slice cultures were prepared from postnatal day-7 rat hippocampus as described (Sala et al., 2003; Seeburg and Sheng, 2008). Rat brains were dissected in ice-cold buffer containing (in mM): sucrose (238), KCl (2.5), NaHCO3 (26), NaH2PO4 (1), glucose (11), MgCl2 (5) and CaCl2 (1). Hippocampi were cut into 350 μm-thick slices with a McIlwain tissue chopper, and plated on tissue inserts (Millipore) in wells with MEM (Cellgro) culture media containing (in mM): glucose (26), NaHCO3 (5.8), HEPES (30), CaCl2 (2), MgSO4 (2), and supplemented with horse serum (20%), insulin (1 μg/μl), and ascorbic acid (0.0012%). Slices were incubated in 5% CO2 at 35°C.
Electrophysiology
Electrophysiological recordings were performed as described (Kim et al., 2007; Seeburg and Sheng, 2008). Neurons were transfected by biolistic gene gun at DIV 3-5 (total 100 μg DNA; 90% test DNA construct; 10% eGFP marker) and recorded 3 days after transfection. Recordings were carried out in solution containing (in mM): NaCl (119), KCl (2.5), CaCl2 (4), MgCl2 (4), NaHCO3 (26), NaH2PO4 (1), glucose (11), picrotoxin (0.1), and 2-chloroadenosine (0.002-0.004), and bubbled continuously with 5% CO2/95% O2. Patch recording pipettes (2.5-5 M ohms) were filled with internal solution containing (in mM): cesium methanesulfonate (115), CsCl (20), HEPES (10), MgCl2 (2.5), ATP disodium salt (4), GTP trisodium salt (0.4), sodium phosphocreatine (10), and EGTA (0.6), at pH 7.25. Simultaneous whole-cell recordings were obtained from a pair of transfected and neighboring untransfected CA1 pyramidal neurons during stimulation of presynaptic Schaffer collaterals. For basal synaptic transmission experiments, presynaptic fibers were stimulated at 0.2 Hz. AMPAR EPSCs were recorded at -70 mV, and NMDAR EPSCs at +40 mV in the presence of 0.01 mM NBQX. Each data point represents an average of 60 consecutive synaptic responses. For LTD experiments, presynaptic fibers were stimulated at 0.033 Hz during baseline recordings, and synaptic responses were obtained at -70 mV. After at least 10 minutes of baseline recording, LTD was induced by pairing 1 Hz stimulation (200 pulses) with depolarization of the postsynaptic cell to -40 mV. Synaptic responses were then obtained under baseline conditions. For LTP experiments, presynaptic fibers were stimulated at 0.2 Hz during baseline recordings, and synaptic responses obtained at -70 mV. After at least 3 minutes of baseline recording, LTP was induced by pairing 3 Hz stimulation (200 pulses) with postsynaptic depolarization to 0 mV. Synaptic responses were then obtained under baseline conditions. For plasticity experiments, data was collected only if the untransfected cell in a paired recording displayed LTD or LTP.
All recordings were made using a Multiclamp 700A amplifier (Molecular Devices), and data was digitized at 20 kHz with Digidata 1322A (Molecular Devices). Analysis of recordings was performed using Clampfit software (Molecular Devices) for basal transmission experiments, or Igor Pro software (Wavemetrics) for plasticity experiments. Results are expressed as mean ± SEM, and statistical significance was assessed by paired Student's t-test on mean EPSC amplitude for basal transmission experiments, or on mean normalized EPSC (averaged over the last 10 minutes of post-induction recordings) for plasticity experiments.
Immunostaining
Hippocampal cultured neurons were transiently transfected at DIV16-19 with a mix of test DNA construct (80%) and marker (20%; eGFP or beta-galactosidase) using Lipofectamine 2000 (Invitrogen). The control construct used was an empty myc-tag vector. For dendritic spine morphology studies, neurons were fixed three days later in 4% PFA/4% sucrose/PBS, immunostained for eGFP (rabbit anti-GFP from MBL, goat anti-rabbit IgG Alexa 488 conjugate secondary antibody from Invitrogen, all in GDB buffer [30 mM phosphate buffer pH 7.4, 0.1% gelatin, 0.3% Triton-X100, 0.45 M NaCl]), and imaged by confocal microscopy on an LSM510 microscope (Zeiss) as described previously (Tada et al, 2007). Analysis of neuron morphology was performed with Metamorph software (Molecular Devices). For phospho-ERK studies, neurons were pre-treated three days after transfection with 1 μM TTX (1 hr), 20 μM NBQX (10 minutes), and 5 μM nimodipine (10 minutes), followed by exposure to 70 μM NMDA for 2.5 or 5 minutes. Neurons were immediately fixed in 4% PFA/4% sucrose/TBS (made immediately prior to fixation), and immunostained for phospho-ERK (rabbit anti-phospho ERK from Cell Signaling Technologies, goat anti-rabbit IgG Alexa 488 conjugate secondary antibody from Invitrogen) and beta-galactosidase (mouse anti-beta-galactosidase from Promega, goat anti-mouse IgG Alexa 568 conjugate secondary antibody from Invitrogen). The confocal microscope settings were kept the same for all scans within the experiment, and average fluorescence intensity measurements were obtained from cell somas with Metamorph software. Results were all normalized to the untreated empty-vector control, and expressed as mean ± SEM. Analysis was by unpaired Student's t-test, between experimental and control neurons for each time point.
Western blot
Protein samples were separated on 6% (for PYK2 or GluA1 blotting) or 10% (all others) SDS-PAGE gels in running buffer (25 mM Tris, 200 mM Glycine, 0.1% SDS). Gels were transferred to PVDF membranes (BioRad) in running buffer without SDS. Membranes were blocked with 5% skim milk powder in Tris-buffered saline (TBS, pH 7.6) with 0.1% Tween-20, and probed overnight at 4°C with primary antibodies in TBS-Tween-20 with 5% BSA and 0.01% sodium azide. Membranes were then washed in TBS with 0.2% Triton X-100, and probed with sheep anti-mouse or donkey anti-rabbit HRP-linked secondary antibodies (GE Healthcare) in 5% milk TBS-Tween-20. After a second set of washes in TBS-Triton X-100, membranes were exposed to ECL chemiluminescence reagent (Perkin-Elmer Life Sciences), and developed on an X-OMAT processor (Kodak). For phospho-antibody blots, membranes were subsequently washed in stripping buffer (Thermo Scientific), re-blocked, and then re-probed with antibody against total protein. Quantification was performed using Scion Image software (Scion Corp), and results are expressed as mean normalized ratio (ratio of phospho-signal to total protein, normalized to an untreated control) ± SEM.
For Triton X-100 solubility experiments, cultured neurons in 6 well-plates (DIV 22) were pre-treated for 10 minutes with 20 μM NBQX, 2 μM tetrodotoxin, and 5 μM nimodipine before treatment with 70 μM NMDA for 5 minutes (or untreated), and then placed immediately on ice and homogenized in buffer containing 50 mM HEPES 7.4, 2 mM EDTA, 0.5% Triton X-100, 0.1 mM PMSF, with cocktail protease inhibitors, tyrosine phosphatase inhibitors, and serine/threonine phosphatase inhibitors (Calbiochem). Homogenates were agitated for 20 minutes, then spun at 20,000 g for 20 minutes. Pellet fractions were solubilized in the same buffer with 1% SDS, and boiled for 5 minutes. Homogenate, supernatant, and pellet fractions were quantified by Pierce BCA Protein Assay (Thermo Scientific) prior to loading on SDS-PAGE gels (10 μg protein per well).
Results
Chem-LTD regulates PYK2 phosphorylation at Y402
We first examined whether NMDAR activation regulates PYK2 activity using Y402 phosphorylation as a marker. In cultured hippocampal neurons (DIV 21-26), bath application of NMDA (70 μM for 2.5 or 5 minutes) results in endocytosis of AMPARs, which is associated with dephosphorylation of GluA1 (GluR1) at Ser-845 (Lee et al., 1998; Figure 1A). This chem-LTD protocol also induced a rapid and transient increase in PYK2 Y402 phosphorylation, peaking at ∼1.5-2-fold above basal levels within 5 minutes of NMDAR stimulation (Figure 1B). At this time point, chem-LTD treatment additionally caused a redistribution of total PYK2 protein from Triton-soluble to Triton-insoluble fractions of cultured neurons (Figure S1A, B), although we did not discern any change in PYK2 localization, as assayed by immunostaining (data not shown). PYK2 phosphorylated on Y402 was not specifically enriched in the insoluble fraction after chem-LTD (Figure S1A), implying that Y402 phosphorylation and subcellular redistribution are not directly linked. Together, our data indicate that NMDAR activation results in enhanced activity, as well as altered biochemical distribution, of PYK2 in hippocampal neurons.
Figure 1. Chem-LTD induces PYK2 phosphorylation at Y402.

(A) Time course of GluA1 S845 dephosphorylation after chem-LTD treatment in cultured hippocampal neurons. In this and all other experiments in this study, quantified phospho-protein signal in each lane was normalized to total protein signal, and the final ratio was normalized to untreated (0′) control. Data are quantified in the graph at right, n = 3, ***p < 0.001 compared to untreated control, one-way ANOVA with Dunnett's post-test. (B) Time course of PYK2 Y402 phosphorylation after chem-LTD treatment. n = 3, *p < 0.05 compared to untreated control, one-way ANOVA with Dunnett's post-test.
PYK2 knockdown blocks LTD
Because PYK2 is activated during chem-LTD, we asked whether PYK2 activity is required for LTD. We designed a small hairpin RNA (shRNA) expression construct targeting rat PYK2 (PYK2 shRNA). Expressed via a lentiviral vector, PYK2 shRNA strongly reduced the level of endogenous PYK2 protein in hippocampal cultures, compared to a control shRNA against firefly luciferase (Figure S2). Expression of PSD-95 and other synaptic components were unaffected by either shRNA (Figure S2A), and a “rescue” experiment with an shRNA-resistant PYK2 confirmed on-target specificity of PYK2 shRNA (see below).
PYK2 shRNA was introduced by biolistic transfection into CA1 pyramidal neurons of hippocampal slice cultures, a useful preparation for acute manipulation of protein levels and in-slice comparison with untransfected neighboring neurons (Stein et al., 2003; Seeburg and Sheng, 2008; Xu et al., 2008). LTD – induced by a pairing protocol (see Methods) – was strongly suppressed in neurons transfected with PYK2 shRNA, as compared with nearby untransfected cells in the same slice (average normalized EPSC during the last 10 minutes of recording: PYK2 shRNA 0.91 ± 0.13 of baseline, untransfected control 0.60 ± 0.052, p = 0.031; Figure 2A). As a further control, neurons transfected with luciferase shRNA showed LTD indistinguishable from untransfected neighboring cells (luciferase shRNA 0.71 ± 0.079, untransfected control 0.65 ± 0.053, p = 0.56; Figure 2B). Notably, neither PYK2 shRNA nor control luciferase shRNA had a significant effect on LTP (average normalized EPSC during the last 10 minutes of recording: PYK2 shRNA 2.08 ± 0.17, untransfected control 1.86 ± 0.25, p = 0.48; Figure 2C; luciferase shRNA 1.66 ± 0.32, untransfected control 1.81 ± 0.18, p = 0.64; Figure 2D). Thus PYK2 is specifically necessary for LTD signaling.
Figure 2. Knockdown of PYK2 blocks LTD.

(A, B) PYK2 shRNA blocks LTD (A) but luciferase shRNA does not (B). Groups of four consecutive EPSC measurements were averaged, and then normalized to the average EPSC magnitude of the first 10 minutes of baseline recordings (pre-LTD induction). Statistical significance was assessed by comparing the average normalized EPSC of the last 10 minutes of recording. PYK2 shRNA: n = 12 pairs; p = 0.031, paired Student's t-test. Luciferase shRNA: n = 12 pairs; p = 0.56, paired Student's t-test. Insets show averaged EPSC traces from sample transfected and untransfected (untrans) cells (thick traces from position 1; thin traces from position 2). Scale bars, 50 pA/25 msec. (C, D) Neither PYK2 shRNA (C) nor luciferase shRNA (D) affects LTP. Groups of six consecutive EPSC measurements were averaged, and then normalized to the average EPSC magnitude of the first 3 minutes of baseline recording (pre-LTP induction); the average normalized EPSC of the last 10 minutes of recording was used to assess statistical significance. PYK2 shRNA: n = 9 pairs; p = 0.48, paired Student's t-test. Scale bar, 30 pA/25 msec. Luciferase shRNA: n = 7 pairs; p = 0.64, paired Student's t-test. Scale bar, 50 pA/25 msec. (E) PYK2 shRNA and luciferase shRNA do not significantly alter baseline AMPAR transmission. Each data point represents the average EPSC amplitude of 60 consecutive synaptic responses; EPSC amplitude is shown in pA. PYK2 shRNA paired recordings (transfected cell versus untransfected neighboring cell in the same slice): n = 12 pairs; p = 0.55 versus untransfected control, paired Student's t-test. Luciferase shRNA paired recordings: n = 19 pairs; p = 0.50 versus untransfected control, paired Student's t-test. Insets show averaged EPSC traces from a sample pair of transfected (trans) and neighboring untransfected (untrans) cells. Scale bars, 20 pA/100 msec. (F) PYK2 shRNA and luciferase shRNA have similar effects on NMDAR transmission. Each data point represents the average NMDAR EPSC amplitude of 60 consecutive synaptic responses; EPSC amplitude is shown in pA. PYK2 shRNA paired recordings: n = 9 pairs, p = 0.024 versus untransfected control, paired Student's t-test. Luciferase shRNA paired recordings: n = 18 pairs, p = 0.015 versus untransfected control, paired Student's t-test. Scale bars, 50 pA/100 msec.
We also examined baseline synaptic transmission, and found that neither PYK2 nor luciferase shRNA transfection affected basal AMPAR excitatory postsynaptic currents (AMPAR EPSCs; average transfected/untransfected EPSC amplitude ratio ± SEM: PYK2 shRNA 1.00 ± 0.18, p = 0.55 vs. untransfected control; luciferase shRNA 1.11 ± 0.15, p = 0.50 vs. untransfected control; Figure 2E). Basal NMDAR EPSCs were slightly reduced by PYK2 shRNA, but this effect was small and indistinguishable from that of control luciferase shRNA (PYK2 shRNA 0.80 ± 0.065, p = 0.024 vs. untransfected control; luciferase shRNA 0.82 ± 0.085, p = 0.015 vs. untransfected control; Figure 2F). These results suggest that although PYK2 is required for LTD, it is not critical for the maintenance of basal synaptic function.
Y402F-PYK2 blocks LTD
Overexpression of mutant PYK2 with a phospho-null Y402F substitution has commonly been used to inhibit PYK2 function in a dominant negative manner (Lakkakorpi et al., 2003; Park et al., 2004). In CA1 neurons transfected with Y402F-PYK2, LTD was also completely blocked (Y402F-PYK2 0.96 ± 0.11, untransfected control 0.65 ± 0.055, p = 0.0057; Figure 3A). This result corroborates our earlier loss-of-function result with PYK2 shRNA (see Figure 2A) showing a necessary role for PYK2 in LTD. Overexpression of wild type (WT)-PYK2, on the other hand, had no effect on LTD (WT-PYK2 0.59 ± 0.042, untransfected control 0.54 ± 0.032, p = 0.43; Figure 3B).
Figure 3. PYK2 Y402 is required for LTD.
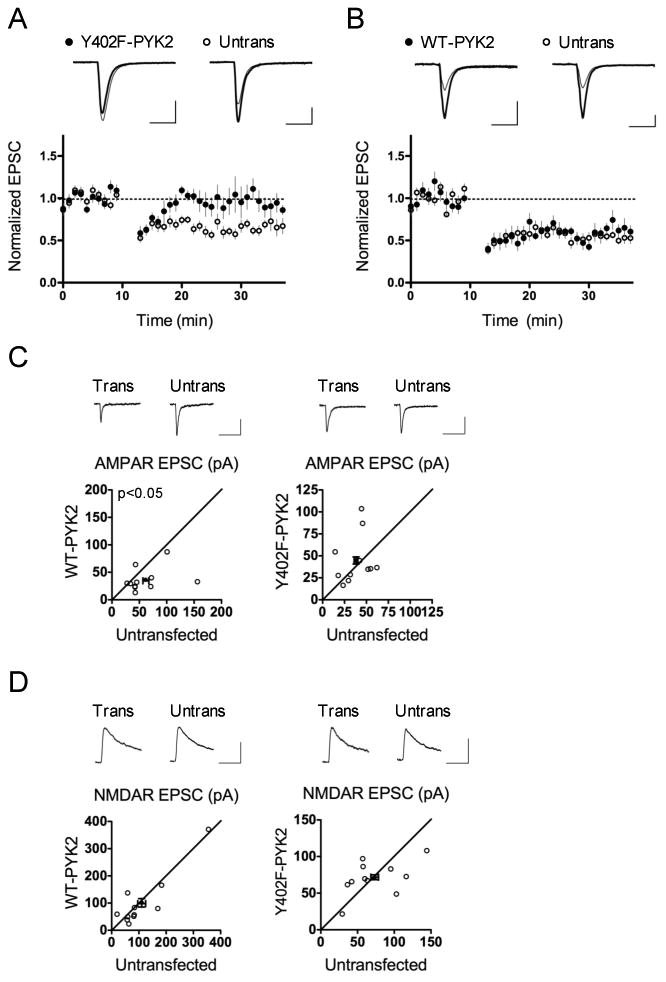
(A) Overexpression of Y402F-PYK2 blocks LTD. n = 10 pairs; p = 0.0057, paired Student's t-test. Scale bars, 50 pA/25 msec. (B) WT-PYK2 overexpression does not affect LTD. n = 9 pairs; p = 0.43, paired Student's t-test. Scale bars, 50 pA/25 msec. (C) Overexpression of WT-PYK2 inhibits AMPAR transmission, whereas expression of Y402F-PYK2 does not. EPSC amplitude is shown in pA. WT-PYK2 paired recordings: n = 11 pairs; p = 0.049 versus untransfected control, paired Student's t-test. Y402F-PYK2 paired recordings: n = 11 pairs; p = 0.45 versus untransfected control, paired Student's t-test. Scale bars, 20 pA/100 msec. (D) Neither WT-PYK2 nor Y402F-PYK2 expression affects NMDAR transmission. EPSC amplitude is shown in pA. WT-PYK2 paired recordings: n = 11 pairs; p = 0.52 versus untransfected control, paired Student's t-test. Y402F-PYK2 paired recordings: n = 11 pairs; p = 0.83 versus untransfected control, paired Student's t-test. Scale bars, 50 pA/100 msec.
In terms of baseline synaptic transmission, neurons overexpressing WT-PYK2 displayed a ∼30% reduction in AMPAR EPSCs (WT-PYK2 0.69 ± 0.11, p = 0.049 vs. untransfected control; Figure 3C), but normal NMDAR EPSCs (WT-PYK2 1.09 ± 0.25; p = 0.52 vs. untransfected control; Figure 3D). Overexpression of Y402F-PYK2, on the other hand, failed to cause synaptic depression. In fact, baseline AMPAR EPSCs were slightly increased by overexpression of Y402F-PYK2, supporting a possible dominant negative effect, although this trend did not reach statistical significance (Y402F-PYK2 1.35 ± 0.30; p = 0.45 vs. untransfected control). These results suggest that under basal conditions, PYK2 overexpression is sufficient to decrease synaptic function in a manner dependent on Y402. This depression did not occlude LTD, however (Figure 3B), suggesting that additional molecular mechanisms are at work during LTD (see Discussion).
Like WT-PYK2 overexpression, Y402F-PYK2 overexpression did not affect NMDAR EPSCs, implying that PYK2 is not required for NMDAR function (Y402F-PYK2 1.10 ± 0.14; p = 0.83 vs. untransfected control; Figure 3D). Unfortunately, there is no available constitutively active PYK2 mutant that can be tested, as the putative phospho-mimic Y402 PYK2 mutant has been found to inhibit PYK2 function like the phospho-null mutant (Li et al., 1999). Together, our results suggest that PYK2 overexpression selectively affects AMPAR, but not NMDAR, transmission.
Changes in synaptic structure often correlate with changes in synaptic function (reviewed in Tada and Sheng, 2006; Alvarez and Sabatini, 2007); however, none of the above modifications we introduced (PYK2 knockdown, WT-PYK2 overexpression, or Y402F-PYK2 overexpression) significantly affected dendritic spine morphology or dendritic branching in mature cultured neurons (Figure S3). Thus PYK2 does not play a major role in determining basal-state neuronal morphology.
PYK2 kinase activity contributes to LTD
Is the kinase activity of PYK2 required for LTD? Alanine substitution at K457 in PYK2 abolishes kinase activity (Li et al., 1999; Lakkakorpi et al., 2003; Park et al., 2004). If PYK2 kinase activity were important, we would expect overexpression of K457A-PYK2 in neurons to have effects similar to overexpression of Y402F-PYK2. When we examined basal synaptic transmission, however, we found that K457A-PYK2 overexpression significantly depressed baseline AMPAR EPSCs, to the same extent seen with WT-PYK2 overexpression (K457A-PYK2 0.75 ± 0.11; p = 0.036 vs. untransfected control; Fig. 4A). Because kinase-dead PYK2 proteins could still be trans-autophosphorylated by endogenous wild-type PYK2 (as occurs in osteoclasts; Lakkakorpi et al., 2003), we turned to a “molecular replacement” approach in which endogenous PYK2 is suppressed by PYK2 shRNA while kinase-dead PYK2 is overexpressed by co-transfection of shRNA-resistant K457A-PYK2. We altered nine nucleotides in the shRNA target region of K457A- and WT-PYK2 to render these constructs resistant to suppression by PYK2 shRNA, without changing the amino acid sequence (Figure S4). Transfection of shRNA-resistant WT-PYK2 (WT-PYK2*) together with PYK2 shRNA resulted in significantly decreased baseline AMPAR EPSCs (WT-PYK2* + shRNA 0.76 ± 0.14; p = 0.036 vs. untransfected control; Figure 4B), which is, as expected, similar to overexpression of WT-PYK2 in the absence of PYK2 shRNA. Overexpression of shRNA-resistant kinase-dead K457A-PYK2 (K457A-PYK2*) in conjunction with PYK2 shRNA also reduced basal AMPAR EPSC amplitude (K457A-PYK2* + shRNA 0.73 ± 0.15; p = 0.026 vs. untransfected control; Figure 4B), similar to overexpression of the K457A-PYK2 mutant alone. Thus overexpression of PYK2 reduces basal synaptic strength, independent of its kinase activity. These data are consistent with a scaffold function of PYK2 that inhibits synaptic strength.
Figure 4. PYK2 kinase activity is required for LTD.

(A) Overexpression of K457A-PYK2 inhibits AMPAR transmission. WT-PYK2 overexpression data from Figure 3C is repeated here for comparison. EPSC amplitude is shown in pA. K457A PYK2 paired recordings: n = 13 pairs; p = 0.036 versus untransfected control, paired Student's t-test. Scale bars, 20 pA/100 msec. (B) Both WT-PYK2* and K457A-PYK2*, in the presence of PYK2 shRNA, depress baseline AMPAR transmission. EPSC amplitude is shown in pA. PYK2 shRNA and shRNA-resistant PYK2 constructs were mixed 1:1 for transfections. WT-PYK2* + shRNA paired recordings: n = 13 pairs; p = 0.036 versus untransfected control, paired Student's t-test. K457A-PYK2* + shRNA paired recordings: n = 12 pairs; p = 0.026 versus untransfected control, paired Student's t-test. Scale bars, 20 pA/100 msec. (C, D) WT-PYK2* fully restores LTD (C), whereas K457A-PYK2* only partially rescues LTD (D), in the presence of PYK2 shRNA. WT-PYK2* + shRNA: n = 10 pairs; p = 0.91, paired Student's t-test. K457A-PYK2* + shRNA: n = 11 pairs; p = 0.031, paired Student's t-test. Scale bars, 50 pA/25 msec.
Although unimportant for the regulation of basal synaptic strength, could PYK2 kinase activity play a role in LTD? We tested LTD in CA1 neurons that had undergone “molecular replacement” with WT or kinase-dead PYK2. WT-PYK2* fully rescued the LTD defect caused by PYK2 shRNA expression (WT-PYK2* + shRNA 0.61 ± 0.054, untransfected control 0.60 ± 0.043; p = 0.91; Figure 4C), a result that also confirms the on-target specificity of the shRNA effect. K457A-PYK2*, however, only partially rescued LTD in neurons cotransfected with PYK2 shRNA (K457A-PYK2* + shRNA 0.73 ± 0.079, untransfected control 0.50 ± 0.041; p = 0.031; Figure 4D). These data imply that PYK2 kinase activity is involved in the function of PYK2 in LTD; however, kinase-independent functions also seem to contribute.
PYK2 is required for inhibition of ERK phosphorylation in chem-LTD
PYK2 is a large scaffold protein (∼116 kDa) as well as a kinase, and it is likely that PYK2 recruits and modulates multiple signaling proteins at the synapse. To gain an understanding of how PYK2 influences postsynaptic signaling during NMDAR activation, we screened the activation profile of several signaling proteins after chem-LTD stimulation in cultured neurons where PYK2 expression had been suppressed by PYK2 shRNA lentivirus. Compared to luciferase shRNA control, PYK2 knockdown significantly enhanced the phosphorylation of ERK T202/Y204 at 5 minutes following chem-LTD, which corresponds to the peak of PYK2 phosphorylation at Y402 (Figure 5A). There was also a slight, but not significant, enhancement of basal ERK phosphorylation with PYK2 knockdown. We did not find significant alteration in the phosphorylation-activation time courses of Src (Y416), GSK3beta (S9), Akt (T308), or JNK (T183/Y185) (Figure S5). These data suggest that PYK2 is required for dampening ERK activation in response to NMDAR stimulation.
Figure 5. PYK2 knockdown enhances NMDA-dependent ERK phosphorylation and delays NMDA-dependent GluA1 S845 dephosphorylation.

(A) Representative blots of phosphorylation time course of PYK2 and ERK following chem-LTD. Graph at right shows quantification of the phospho-ERK signal intensity ratio (average of phospho-p42 and phospho-p44 intensity ratios, normalized to untreated luciferase shRNA-transfected samples). n = 4, **p < 0.01, two-way ANOVA with Bonferroni's post-test between time-matched samples. (B) Representative blot of GluA1 S845 dephosphorylation following chem-LTD, in luciferase shRNA- and PYK2 shRNA-expressing neurons. Quantification of phospho-GluA1 signal intensity ratios (normalized to the corresponding untreated samples) is shown on the right. n = 4; *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA with Dunnett's post-test. (C) Representative blot and quantification of phospho-GluA1 from neurons treated with the calcineurin inhibitor FK506 (1 μM), or vehicle (DMSO). n = 6; *p < 0.05, ***p < 0.001, one-way ANOVA with Dunnett's post-test. (D) Representative blot and quantification of phospho-GluA1 from neurons treated with the calcineurin inhibitor cyclosporine A (1 μM) or vehicle (DMSO). n = 5; *p < 0.05, ***p < 0.001, one-way ANOVA with Dunnett's post-test.
LTD is associated with dephosphorylation of GluA1 S845, and elevated Ras-ERK signaling correlates with GluA1 S845 phosphorylation in hippocampal slice cultures (Qin et al., 2005). Control neurons virally infected with luciferase shRNA showed rapid increasing dephosphorylation of GluA1 S845, reaching significance at 5 min after NMDAR stimulation (Figure 5B). In contrast, neurons infected with PYK2 shRNA showed no significant loss of S845 phosphorylation till 15 minutes (Figure 5B). The delay in S845 GluA1 dephosphorylation by PYK2 knockdown was similar to the effect of calcineurin inhibitors (FK506 and cyclosporine A), which are known to block LTD (Figure 5C, D). Thus PYK2's effect on GluA1 dephosphorylation is consistent with its effects on LTD.
Does overexpression of WT-PYK2 decrease activity-dependent ERK activation? We immunostained for phospho-ERK in neurons transfected with plasmids expressing WT-PYK2 or Y402F-PYK2, and measured fluorescence intensities in cells at 2.5 and 5 minutes chem-LTD treatment, as described previously (Kim et al., 2005). Consistent with the PYK2-knockdown experiments above, overexpression of Y402F-PYK2 enhanced NMDA-induced ERK phosphorylation, relative to control neurons transfected with an empty vector (Figure 6). This enhancement was significant in unstimulated neurons and at 2.5 minutes post-chem-LTD treatment (Figure 6). Overexpression of WT-PYK2, on the other hand, significantly suppressed NMDA-induced ERK phosphorylation at 5 minutes chem-LTD treatment (Figure 6). Thus overexpression of PYK2 is sufficient to dampen NMDA-induced ERK activation, just as PYK2 knockdown enhances it.
Figure 6. WT-PYK2 overexpression depresses NMDA-dependent ERK phosphorylation.

(A) Representative images of transfected neurons (DIV16+3) immunostained for phospho-ERK (green) and beta-gal marker (red) untreated (0′) or after chem-LTD treatment (2.5′ and 5′). Scale bar, 30 μm. (B) Quantification of data from (A). Average fluorescence intensities of phospho-ERK signal in cell somas were all normalized to untreated empty-vector control. Each experimental group was compared to the corresponding empty-vector control for that time point. n = 26-53 neurons per group, *p < 0.05, **p < 0.01, ***p < 0.001, unpaired Student's t-test.
Overexpression of PYK2 blocks LTP
Because it further inhibited NMDA-dependent ERK activation, we hypothesized that WT-PYK2 overexpression would inhibit LTP or even enhance LTD. While there was no effect of WT-PYK2 overexpression on LTD (Figure 3B), we did observe that WT-PYK2 significantly reduced LTP relative to untransfected controls (WT-PYK2 1.53 ± 0.10, untransfected control 2.56 ± 0.29, p = 0.011; Figure 7A). In contrast, overexpression of Y402F-PYK2 did not affect LTP (Y402F-PYK2 2.13 ± 0.28, untransfected control 1.93 ± 0.19, p = 0.64; Figure 7B). Thus PYK2 overexpression inhibits LTP, in a manner dependent on Y402 and consistent with effects on ERK phosphorylation.
Figure 7. WT-PYK2 overexpression inhibits LTP.
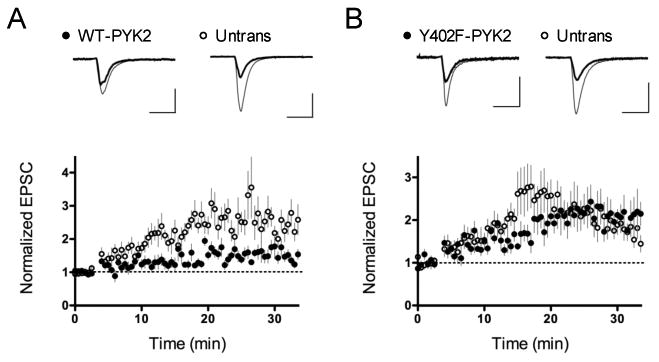
(A, B) Overexpression of WT-PYK2 (A), but not Y402F-PYK2 (B) reduces LTP. WT-PYK2: n = 8 pairs; p = 0.011, paired Student's t-test. Scale bar, 20 pA/25 msec (WT-PYK2), 50 pA/25 msec (untransfected). Y402F-PYK2: n = 7 pairs; p = 0.64, paired Student's t-test. Scale bar, 20 pA/25 msec (Y402F-PYK2), 30 pA/25 msec (untransfected).
Discussion
We report here that PYK2 plays a critical role in LTD. PYK2 is phosphorylated at Y402 by chem-LTD activation of NMDARs, and shRNA and dominant negative experiments show that it is required for LTD in hippocampal slice cultures. We find that non-kinase (presumably scaffold) and, to a lesser extent, kinase functions of PYK2 contribute to LTD. We propose the following model: Following NMDAR stimulation, the rise in postsynaptic calcium activates PYK2, resulting in phosphorylation at Y402. Y402 is autophosphorylated primarily by the PYK2 kinase domain, as proposed previously (Lev et al., 1995; Park et al., 2004; Kohno et al., 2008; Bartos et al., 2010), but other tyrosine kinases could also contribute to Y402 phosphorylation (Lakkakorpi et al., 2003). We consider it unlikely that Src mediates activity-dependent Y402 phosphorylation, however, because we find that Src phosphorylation is not activated by chem-LTD (Figure S5). Phospho-Y402 PYK2 then recruits additional signaling proteins to mediate downstream effects essential for LTD, one of which could be the dampening of postsynaptic activation of ERK.
Overexpression of PYK2 is sufficient to depress basal synaptic strength, and like PYK2's role in LTD, this effect requires Y402 (Figure 3). There are a few mechanistic distinctions, however, between the basal and activity-dependent roles of PYK2. PYK2 kinase activity is required in part for LTD, but not at all for depression of basal synaptic strength (Figure 4). Moreover, PYK2 overexpression suppresses ERK phosphorylation following NMDAR stimulation, but not in the basal state (Figure 6). These mechanistic differences could explain how WT-PYK2 overexpression or WT-PYK2* “molecular replacement,” both of which depress basal synaptic strength, failed to occlude LTD (Figure 3, 4). NMDAR-dependent recruitment of PYK2 through Y402-independent mechanisms (Figure S1) could additionally gate PYK2's role in LTD, much like how NMDAR stimulation gates phosphatase action in LTD (Morishita et al., 2001).
Two properties of PYK2 make it an attractive component of LTD signaling. First, PYK2 is activated by a rise in intracellular calcium. One possible mechanism is that calcium-calmodulin complexes bind PYK2 directly, and induce dimerization and transphosphorylation at Y402 (Kohno et al., 2008). It has also been proposed that calcium-calmodulin binding to PSD-95 induces PYK2 association and clustering, which favors Y402 transphosphorylation in the protein complex (Bartos et al., 2010). In agreement with our findings, this study also reported that chemical activation of NMDARs leads to PYK2 Y402 phosphorylation and redistribution in cultured hippocampal neurons (Bartos et al., 2010). Another possibility is that PYK2 Y402 phosphorylation-activation occurs via the calcium-dependent serine/threonine phosphatase calcineurin (Faure et al., 2007). In PC12 cells, calcineurin enhances PYK2 activity and Y402 phosphorylation through serine/threonine dephosphorylation of multiple PYK2 residues (Faure et al., 2007). The link between calcineurin and PYK2 is intriguing, because calcineurin is well-established to be required for LTD (Mulkey et al., 1993, 1994). Forebrain-specific knockout of calcineurin function abolishes LTD, and causes schizophrenia-like behavioral deficits in working memory, prepulse inhibition, and social interaction (Zeng et al., 2001; Miyakawa et al., 2003). The behavioral effects of loss of function of PYK2 remain to be determined.
Second, PYK2 is known to bind PSD-95 (Seabold et al., 2003), which is required for LTD (Migaud et al., 1998). Furthermore, a study has shown that mutation of the SH3-GK domain in PSD-95 blocks LTD in hippocampal neurons (Xu et al., 2008); the SH3-GK region is where PYK2 appears to bind PSD-95 (Seabold et al., 2003). It is possible that PYK2 association with PSD-95 plays a role in LTD, and alteration of PYK2 signaling may be one explanation for the LTD defect observed in the presence of PSD-95 SH3-GK mutants. This is consistent with a recent study proposing that PYK2 binding to PSD-95 is required for PYK2 activation and phosphorylation at Y402 (Bartos et al., 2010).
There is much evidence linking ERK signaling to synaptic plasticity and learning and memory (reviewed in Thomas and Huganir, 2004). ERK pathway inhibitors reduce spatial learning and fear conditioning (Blum et al., 1999; Selcher et al., 1999; Atkins et al., 1998). These inhibitors also block LTP in acute hippocampal slices (English and Sweatt, 1997) and in vivo hippocampal LTD (Thiels et al., 2002), leading to the view that differences in the magnitude or duration of ERK activation between LTP and LTD could be important, with more modest elevations in ERK activity favoring LTD (Thiels et al., 2002; Coba et al., 2008). In fact, different ERK activation time-courses are known to couple to distinct processes in other cell types: modest, transient ERK activity induces proliferation in PC12 cells, whereas prolonged ERK activity induces differentiation (Marshall, 1995). Our data support the idea that alteration of ERK signaling dynamics could affect the recruitment of LTP versus LTD pathways, with PYK2 playing a role in determining that balance. In fact, we found that an intermediate level of stimulation (2 Hz, 200 pulses, at 0 mV holding potential) induced LTP in neurons expressing PYK2 shRNA but not in control neurons transfected with luciferase shRNA (H.H. and M.S., unpublished observations), consistent with the idea that loss of PYK2 favors LTP induction. Further studies, however, are needed to confirm this conclusion.
What mechanisms might mediate PYK2's effect on ERK signaling in synapses? In some non-neuronal cell types, PYK2 has been linked to enhancement of ERK activity through the recruitment of Grb2 (adaptor) and Sos (guanine nucleotide exchange factor [GEF] for Ras) proteins (Lev et al., 1995; Dikic et al., 1996). In NIH3T3 cells, however, PYK2 has been reported to inhibit ERK activity (Zhao et al., 2000). In neurons, ERK signaling is regulated by Ras GEFs and GTPase activating proteins (GAPs), several of which are represented in the PSD (Thomas and Huganir, 2004; Sheng and Hoogenraad, 2007). PYK2's presence in the PSD, and in particular the NMDAR complex (Seabold et al., 2003), could enable interactions with GTPase regulators in the PSD, and thus provide another coupling mechanism between calcium influx and MAPK activity.
Although chem-LTD and electrophysiological LTD share overlapping molecular mechanisms (Lee et al., 1998), they are quite different experimental paradigms. Thus an important caveat is that the results we obtained with chem-LTD in dissociated neurons cultures (bath NMDAR stimulation) may not fully explain the role of PYK2 in electrophysiological LTD in hippocampal slices.
Our conclusion that PYK2 is important for LTD is in apparent contradiction to Huang et al (2001), who reported that micropipette infusion of recombinant PYK2 into CA1 cells induced Src-dependent NMDAR potentiation and occluded LTP. It is interesting to note that this blockade of LTP by PYK2 infusion is consistent with our finding that WT-PYK2 overexpression inhibits LTP (Figure 7). One explanation for why we did not see an increase in NMDAR strength with WT-PYK2 overexpression is that the differential post-translational modification, assembly, and trafficking of injected recombinant protein versus transfected cDNA of PYK2 could lead to different results in neurons. Injection of recombinant PYK2 could lead to abnormal PYK2 function at extrasynaptic sites, for example. It is also possible that acute versus chronic time courses of PYK2 manipulation have different effects. Lastly, our measurements of NMDAR EPSCs at positive holding potentials, a practice commonly used in this preparation, may not completely reflect the full breadth of changes in NMDAR function.
We consider PYK2 to be a necessary component of the collective signaling network that underlies LTD induction. As a large scaffold protein, PYK2 could interact with multiple factors at the synapse. For example, PYK2 has been linked to the small GTPases RhoA and Rap2 (Lim et al., 2008; McLeod et al., 2004), which cause loss of spines, the dendritic protrusions containing synaptic sites (Nakayama et al., 2000; Ryu et al., 2008). LTD is associated with spine shrinkage in hippocampal neurons (Okamoto et al., 2004; Zhou et al., 2004). While we did not find a significant difference in steady-state spine morphology with PYK2 shRNA or WT-PYK2 expression in dissociated neurons (Figure S3), it would be interesting to see if PYK2 plays a role in activity-dependent spine shrinkage or elimination.
PYK2 activation has been reported in pathological conditions of neurotoxicity, cerebral ischemia, and seizures (Cheung et al., 2000; Tian et al., 2000; Wright et al., 2007), raising the possibility that PYK2 could contribute to synapse failure during stroke or neurotoxic conditions via LTD-like mechanisms. Further studies on the signaling proteins involved in LTD may guide future therapeutic approaches for diseases of synapse loss or neurodegeneration.
Supplementary Material
Acknowledgments
We thank Wen-Cheng Xiong and Huaye Zhang for reagents, and Kelly Foster and Daniel Seeburg for data acquisition and analysis programs. We thank Dieter Edbauer, Kenny Futai, Nelly Khidekel, and Baris Bingol for critical comments on the manuscript. MS was Investigator of the Howard Hughes Medical Institute.
References
- Alvarez V, Sabatini B. Anatomical and physiological plasticity of dendritic spines. Ann Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Atkins C, Selcher J, Petraitis J, Trzaskos J, Sweatt J. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Bartos J, Ulrich J, Li H, Beazely M, Chen Y, MacDonald J, Hell J. Postsynaptic clustering and activation of Pyk2 by PSD-95. J Neurosci. 2010;30:449–463. doi: 10.1523/JNEUROSCI.4992-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum S, Moore A, Adams F, Dash P. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J Neurosci. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp T, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- Cheung H, Takagi N, Teves L, Logan R, Wallace N, Gurd J. Altered association of protein tyrosine kinases with postsynaptic densities after transient cerebral ischemia in the rat brain. J Cereb Blood Flow Metab. 2000;20:505–512. doi: 10.1097/00004647-200003000-00009. [DOI] [PubMed] [Google Scholar]
- Coba M, Valor L, Kopanitsa M, Afinowi N, Grant S. Kinase networks integrate profiles of N-methyl-D-aspartate receptor-mediated gene expression in hippocampus. J Biol Chem. 2008;283:34101–34107. doi: 10.1074/jbc.M804951200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M, Husi H, Yu L, Brandon J, Anderson C, Blackstock W, Choudhary J, Grant S. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem. 2006;1(97 Supp):16–23. doi: 10.1111/j.1471-4159.2005.03507.x. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge S, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English J, Sweatt J. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Faure C, Corvol J, Toutant M, Valjent E, Hvalby O, Jensen V, Messari S, Corsi J, Kadare G, Girault J. Calcineurin is essential for depolarization-induced nuclear translocation and tyrosine phosphorylation of PYK2 in neurons. J Cell Sci. 2007;120:3034–3044. doi: 10.1242/jcs.009613. [DOI] [PubMed] [Google Scholar]
- Gil-Henn H, Destaing O, Sims N, Aoki K, Alles N, Neff L, Sanjay A, Bruzzaniti A, De Camili P, Baron R, Schlessinger J. Defective microtubule-dependent podosome organization in osteoclasts lead to increased bone density in Pyk2 (-/-) mice. J Cell Biol. 2007;178:1053–1064. doi: 10.1083/jcb.200701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault J, Costa A, Derkinderen P, Studler J, Toutant M. FAK and PYK2/CAKbeta in the nervous system: a link between neuronal activity, plasticity, and survival? Trends Neurosci. 1999;22:257–263. doi: 10.1016/s0166-2236(98)01358-7. [DOI] [PubMed] [Google Scholar]
- Grant S, O'Dell T, Karl K, Stein P, Soriano P, Kandel E. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- Hashido M, Hayashi K, Hirose K, Iino M. Ca2+ lightning conveys cell-cell contact information inside cells. EMBO Reports. 2006;7:1117–1123. doi: 10.1038/sj.embor.7400821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali D, Pelkey K, Pitcher G, Lu Y, Aoto H, Roder J, Sasaki T, Salter M, MacDonald J. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–496. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- Kim M, Dunah A, Wang Y, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kim M, Futai K, Jo J, Hayashi Y, Cho K, Sheng M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron. 2007;56:488–502. doi: 10.1016/j.neuron.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kohno T, Matsuda E, Sasaki H, Sasaki T. Protein-tyrosine kinase CAKbeta/PYK2 is activated by binding Ca2+/calmodulin to FERM F2 alpha2 helix and thus forming its dimer. Biochem J. 2008;410:513–523. doi: 10.1042/BJ20070665. [DOI] [PubMed] [Google Scholar]
- Lakkakorpi P, Bett A, Lipfert L, Rodan G, Duong L. PYK2 autophosphorylation, but not kinase activity, is necessary for adhesion-induced association with c-Src, osteoclast spreading, and bone resorption. J Biol Chem. 2003;278:11502–11512. doi: 10.1074/jbc.M206579200. [DOI] [PubMed] [Google Scholar]
- Lee H, Kameyama K, Huganir R, Bear M. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musachhio J, Plowman G, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- Li X, Dy R, Cance W, Graves L, Earp H. Interactions between two cytoskeleton-associated tyrosine kinases: calcium-dependent tyrosine kinase and focal adhesion tyrosine kinase. J Biol Chem. 1999;274:8917–8924. doi: 10.1074/jbc.274.13.8917. [DOI] [PubMed] [Google Scholar]
- Lim Y, Lim S, Tomar A, Gardel M, Bernard-Trifilo J, Chen X, Uryu S, Canete-Soler R, Zhai J, Lin J, Schlaepfer W, Nalbant P, Bokock G, Ilic D, Waterman-Storer C, Schlaepfer D. Pyk2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong E, Pease S, Brown E, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lu Y, Roder J, Davidow J, Salter M. Src activation in the induction of long-term potentiation in CA1 hippocampal neurons. Science. 1998;279:1363–1367. doi: 10.1126/science.279.5355.1363. [DOI] [PubMed] [Google Scholar]
- Malenka R, Kauer J, Perkel D, Mauk D, Kelly P, Nicoll R, Waxham M. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien R. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- Marshall C. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- McLeod S, Shum A, Lee R, Takei F, Gold M. The Rap GTPases regulate integrin-mediated adhesion, cell spreading, actin polymerization, and Pyk2 tyrosine phosphorylation in B lymphocytes. J Biol Chem. 2004;279:12009–12019. doi: 10.1074/jbc.M313098200. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster L, Watabe A, Makhinson M, He Y, Ramsay M, Morris R, Morrison J, O'Dell T, Grant S. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Leiter L, Gerber D, Gainetdinov R, Sotnikova T, Zeng H, Caron M, Tonegawa S. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA. 2003;100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Connor J, Xia H, Quinlan E, Shenolikar S, Malenka R. Regulation of synaptic strength by protein phosphatase 1. Neuron. 2001;32:1133–1148. doi: 10.1016/s0896-6273(01)00554-2. [DOI] [PubMed] [Google Scholar]
- Mulkey R, Herron C, Malenka R. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- Mulkey R, Endo S, Shenolikar S, Malenka R. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Nakayama A, Harms M, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Okigaki M, Davis C, Falasca M, Harroch S, Felsenfeld D, Sheetz M, Schlessinger J. Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proc Natl Acad Sci USA. 2003;100:10740–10745. doi: 10.1073/pnas.1834348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Avraham H, Avraham S. RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. J Biol Chem. 2004;279:33315–33322. doi: 10.1074/jbc.M313527200. [DOI] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart J, Stornetta R, Seidenman K, Mack V, van Aelst L, Zhu J. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 2005;19:2000–2015. doi: 10.1101/gad.342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu K, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178–8188. doi: 10.1523/JNEUROSCI.1944-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Futai K, Yamamoto K, Worley P, Hayashi Y, Sheng M. Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer 1a. J Neurosci. 2003;23:6327–6337. doi: 10.1523/JNEUROSCI.23-15-06327.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabold G, Burette A, Lim I, Weinberg R, Hell J. Interaction of the tyrosine kinase Pyk2 with the N-methyl-D-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J Biol Chem. 2003;278:15040–15048. doi: 10.1074/jbc.M212825200. [DOI] [PubMed] [Google Scholar]
- Seeburg D, Sheng M. Activity-induced Polo-Like Kinase 2 is required for homeostatic plasticity of hippocampal neurons during epileptiform activity. J Neurosci. 2008;28:6583–6591. doi: 10.1523/JNEUROSCI.1853-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher J, Atkins C, Trzaskos J, Paylor R, Sweatt J. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad C. The postsynaptic architecture of excitatory synapses: a more quantitative view. Ann Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Silva A, Paylor R, Wehner J, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Stein V, House D, Bredt D, Nicoll R. Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci. 2003;23:5503–5506. doi: 10.1523/JNEUROSCI.23-13-05503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Tada T, Simonetta A, Batterton M, Kinoshita M, Edbauer D, Sheng M. Role of Septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr Biol. 2007;17:1752–1758. doi: 10.1016/j.cub.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E, Kanterewicz B, Norman E, Trzaskos J, Klann E. Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J Neurosci. 2002;22:2054–2062. doi: 10.1523/JNEUROSCI.22-06-02054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Huganir R. MAPK cascade signaling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tian D, Litvak V, Lev S. Cerebral ischemia and seizures induce tyrosine phosphorylation of PYK2 in neurons and microglial cells. J Neurosci. 2000;20:6478–6487. doi: 10.1523/JNEUROSCI.20-17-06478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S, Malinin N, Powell K, Yednock T, Rydel R, Griswold-Prenner I. Alpha2beta1 and alphaVbeta1 integrin signaling pathways mediate amyloid-beta-induced neurotoxicity. Neurobiol Aging. 2007;28:226–237. doi: 10.1016/j.neurobiolaging.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Xiong W, Parsons J. Induction of apoptosis after expression of PYK2, a tyrosine kinase structurally related to focal adhesion kinase. J Cell Biol. 1997;139:529–539. doi: 10.1083/jcb.139.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter O, Steiner P, Czervionke B, Sabatini B, Malenka R. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot B, Miyakawa T, Bear M, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Zhang H, Macara I. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zheng C, Guan J. Pyk2 and FAK differentially regulate progression of the cell cycle. J Cell Sci. 2000;113:3064–3072. doi: 10.1242/jcs.113.17.3063. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma K, Poo M. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zhu J, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack S, Kim M, Baumgart J, Velamoor V, Auberson Y, Osten P, van Aelst L, Sheng M, Zhu J. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.