

Abstract
We recently identified two thiazolidin compounds, 5-[(4-methylphenyl)methylene]-2-(phenylamino)-4(5H)-thiazolone (MMPT) and 5-(2,4-dihydroxybenzylidene)-2-(phenylimino)-1,3-thiazolidin (DBPT), that inhibit the growth of human non-small-cell lung and colon cancer cells independent of P-glycoprotein and p53 status. Here we further investigated the mechanism by which these thiazolidin compounds mediate their anticancer effects. Treatment of cancer cells with MMPT and DBPT led to a time-dependent accumulation of cells arrested in G2/M phase with modulating the protein expression such as cyclin B1, cdc25C, and phosphorylated histone H3. Moreover, treatment with MMPT and DBPT increased M-phase arrest with abnormal spindle formation. DBPT-mediated G2/M-phase arrest and phosphorylation of cdc25C and histone H3 were abrogated when JNK activation was blocked either by SP600125, a specific JNK inhibitor, or by a dominant-negative JNK1 gene. Moreover, DBPT-mediated microtubule disruption was also blocked by SP600125 treatment. Our results demonstrate that thiazolidin compounds can effectively induce G2/M arrest in cancer cells and that this G2/M arrest requires JNK activation.
Keywords: cancer therapy, mitotic arrest, microtubule disruption, non-small-cell lung cancer, cell cycle
Introduction
Microtubules have crucial roles in intracellular transport, morphogenesis, and cell division. Various agents targeting tubulin, the major protein component of microtubules, have been reported to cause cell-cycle arrest at mitosis and induce apoptosis in cancer cells [1, 2]. These microtubule-interacting drugs can be classified into two major groups: the first, which includes drugs such as vinca alkaloids, colchicine, and nocodazole, interferes with mitosis and changes the balance of the tubulin in microtubules to a depolymerized state; the second group, which includes agents such as taxanes, changes the balance of tubulin to a polymerized state. Vinca alkaloids such as vinorelbine and vincristine have been widely used as cytotoxic agents for the treatment of hematopoietic neoplasia and some solid tumors [3, 4]. The taxanes, including paclitaxel and docetaxel, are newer antitumor agents that are used clinically to treat several solid tumors of the head and neck, breast, lung, ovary, and bladder [5–7]. Despite their opposing effects on tubulin polymerization, all of these agents cause cell-cycle arrest at the metaphase-to-anaphase transition [8], and all induce apoptosis.
Although microtubule-interacting drugs have been used clinically to inhibit the progression of some cancers, multiple cycles of treatment usually result in acquired resistance that abolishes the drugs’ efficacy [9]. One of the mechanisms by which neoplastic cells acquire resistance is the overexpression of efflux pumps such as P-glycoprotein, a transmembrane pump that binds to many antitumor drugs and increases their efflux out of cells. Many microtubule-interacting drugs, such as the taxanes and vinblastine, are well-known substrates of P-glycoprotein [10–13]. The development of new compounds that are effective against drug-resistant cells while sparing normal cells is therefore important for cancer therapy.
To this end, we recently identified two thiazolidin compounds, 5-[(4-methylphenyl)methylene]-2-(phenylamino-4(SH)-thiazolone (MMPT) and 5-(2,4-dihydroxybenzylidene)-2-(phenylimino)-1,3-thiazolidin (DBPT), as antineoplastic agents [14, 15]. These compounds have selective cytotoxic effects against human lung and colon cancer cells in vitro and in vivo, and these effects are independent of P-glycoprotein expression. In addition, the compounds have no obvious toxic effects on normal human fibroblasts and mesenchymal stem cells at the 50% inhibitory concentration for cancer cell lines. We also found that MMPT and DBPT induce apoptosis through caspase activation that is dependent on JNK activation. However, JNK activation alone does not induce apoptosis, suggesting that other molecular events are involved in the antitumor activity of these compounds.
The goal of the present study was to investigate the mechanism of the antitumor action of MMPT and DBPT on non-small-cell lung and colon cancer cells. We found that MMPT and DBPT induced G2/M-phase arrest evidenced by up regulation of G2/M checkpoint proteins such as cyclin B1, cdc25C, Bcl-2, and histone H3. Treatment with MMPT and DBPT also caused cell-cycle arrest at mitotic phase with abnormal spindle formation in cancer cells. Moreover, DBPT-mediated G2/M-phase arrest and microtubule disruption were blocked by inhibition of JNK activation, suggesting that JNK activation also plays critical role in G2/M arrest by thiazolidin compounds.
Materials and methods
Cells and culture conditions
The human non-small-cell lung cancer cell lines H1299 and H460 and the human colon cancer cell lines DLD-1 and HCT116 (p53 wild-type and p53−/−; generously provided by Dr. Bert Vogelstein, The Johns Hopkins University, Baltimore, MD) were routinely propagated in monolayer culture in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 25 mM HEPES, 100 units/ml penicillin, and 100 mg/ml streptomycin. Paclitaxel-resistant H460/TaxR cells and vinorelbine-resistant H460/VinR cells were generated from parental H460 cells by initially adding 5 nM paclitaxel or vinorelbine to the culture medium and thereafter escalating the dose of paclitaxel or vinorelbine to 100 nM. The resistance indexes (i.e., IC50 in resistant cells/IC50 in parental cells) were 80.0 in H460/TaxR cells and 213.9 in H460/VinR cells. Both H460/TaxR and H460/VinR expressed high levels of P-glycoprotein [15]. All cell lines were maintained in the presence of 5% CO2 at 37°C.
Chemicals
MMPT and DBPT were obtained from ChemBridge Corporation (San Diego, CA), dissolved in dimethylsulfoxide (DMSO) to a concentration of 10 mM, and stored at 4°C as described previously [14, 15]. Nocodazole and vinorelbine were purchased from Sigma-Aldrich (St. Louis, MO) and dissolved in DMSO to a concentration of 10 mM. Paclitaxel was purchased from Bristol-Myers Squibb (Princeton, NJ). The JNK-specific inhibitor SP600125 was purchased from Calbiochem (La Jolla, CA), dissolved in dimethyl sulfoxide, stored at −20°C, and protected from light. An equal volume of DMSO (<0.2%) had no obvious effects on cell viability and was used as a control.
Flow cytometry analysis
For analysis of intracellular DNA content, lung cancer and colon cancer cells were treated with various concentrations (3–10 μM) of MMPT or DBPT, collected in 0.125% trypsin, washed twice in PBS, and fixed in 70% methanol overnight at −20°C. The cells were then resuspended in PBS containing 10 μg/ml propidium iodide (Roche Diagnostics, Indianapolis, IN) and 10 μg/ml RNase A (Sigma-Aldrich) at 37°C for 30 min. Cell-cycle analysis was performed using an EPICS Profile II flow cytometer (Coulter, Hialeah, FL) with MultiCycle Phoenix Flow Systems software (Phoenix Flow Systems, San Diego, CA). All experiments were repeated three times.
For detection of cells in mitosis, fixed cells were resuspended in PBS containing 5 μg/ml mouse monoclonal antibody against MPM-2 (Upstate Biotechnology, Lake Placid, NY). The cells were then incubated with fluorescein isothiocyanate-conjugated goat anti-mouse secondary antibody (BD Biosciences PharMingen, San Diego, CA), 10 μg/ml propidium iodide, and 10 μg/ml RNase A at 37°C for 1 h in the dark. Stained cells were analyzed with the EPICS Profile II flow cytometer using MultiCycle Phoenix Flow Systems software. All experiments were repeated at least twice.
Western blot analysis
Western blot analysis was performed with primary antibodies against cyclin A, cdc2p34, cdc25C, and Bcl-2 (Santa Cruz Biotechnology, Santa Cruz, CA); cyclin B1 (NeoMarkers, Fremont, CA); phosphorylated histone H3 (Upstate Biotechnology); and β-actin (Sigma). Cells were washed twice in cold PBS, collected, and lysed in lysis buffer (62.5 mM Tris [pH 6.8], 2% sodium dodecyl sulfate, and 10% glycerol) containing 1× proteinase-inhibitor cocktail (Roche Diagnostics). The lysates were spun at 14,000 × g in a microcentrifuge at 4°C for 10 min, and the supernatants were used as whole-cell extracts. Protein concentrations were determined using a BCA protein assay kit (Pierce, Rockford, IL). Equal amounts (30–50 μg) of proteins were subjected to electrophoresis under reducing conditions on 10–12.5% (w/v) polyacrylamide gels and then electrophoretically transferred to nitrocellulose transfer membranes (Amersham Biosciences, Piscataway, NJ). The membranes were incubated with primary antibody followed by peroxidase-linked secondary antibody. An electrochemiluminescence Western blotting system (Amersham) was used to detect secondary probes.
Immunofluorescence microscopy
Cells were grown on a glass chamber slide (Becton Dickinson, Franklin Lakes, NJ) and treated with the indicated chemicals for 14 h. The cells were then washed with PBS and permeabilized with microtubule-stabilizing buffer [80 mM PIPES-KOH (pH 6.8), 5 mM egtazic acid, 1 mM MgCl2, and 0.5% Triton X-100] for 5 min at room temperature. The permeabilized cells were fixed with chilled absolute methanol for 10 min at −20°C as described previously [16]. After being washed, cells were incubated with a mouse anti-α-tubulin monoclonal antibody (Sigma) for 1 h at ambient temperature and then incubated with a fluorescein isothiocyanate-conjugated anti-mouse secondary antibody (BD Biosciences PharMingen). After extensive washing, cells were rinsed once in water, mounted with Vectashield (Vector Laboratories, Burlingame, CA), and examined under a Nikon BX61 microscope (Melville, NY).
Statistical Analysis
Differences among the treatment groups were assessed by analysis of variance using StatSoft statistical software (Tulsa, OK). p< 0.05 was considered significant.
Results
Treatment with thiazolidin compounds causes induction of G2/M-phase arrest in cancer cells
We recently found that MMPT and DBPT effectively induce JNK-dependent apoptosis in non-small-cell lung and colon cancer cells through caspase activation [14, 15]. To further elucidate the anticancer mechanisms of thiazolidin compounds, we examined the effects of MMPT and DBPT on cell-cycle progression. H1299 and H460 cells were treated with 0.3–10 μM MMPT and analyzed for cell-cycle distribution by flow cytometry. Unexpectedly, G2/M-phase arrest was elicited by 3 μM MMPT after 12 h of treatment (Fig. 1A). The accumulation of cells in G2/M phase was accompanied by synchronous decreases in the numbers of cells in the G1 and S phases. Treatment with 10 μM MMPT also resulted in accumulation of cells in G2/M phase, beginning as early as 6 h and peaking around 80% after 12 h of treatment (Fig. 1B). Because p53 is the wild type in H460 cells but homologously deleted in H1299 cells, this result suggested that the action of MMPT on G2/M-phase arrest is p53 independent. Treatment of DLD-1, HCT116 wild-type, and HCT116 p53-deficient colon cancer cells with 3 or 5 μM DBPT, respectively, also resulted in a time-dependent increase in cells arrested in G2/M phase (Fig. 1C). Accumulation of cells in G2/M phase started as early as 6 h and peaked around 80% at 12 h after treatment in all three cell lines (Fig. 1D), indicating that DBPT-mediated G2/M-phase arrest is also p53 independent. In contrast, G2/M-phase arrest was not observed in normal human fibroblasts treated with MMPT or DBPT at the 50% inhibitory concentration for cancer cells (data not shown).
Fig. 1.
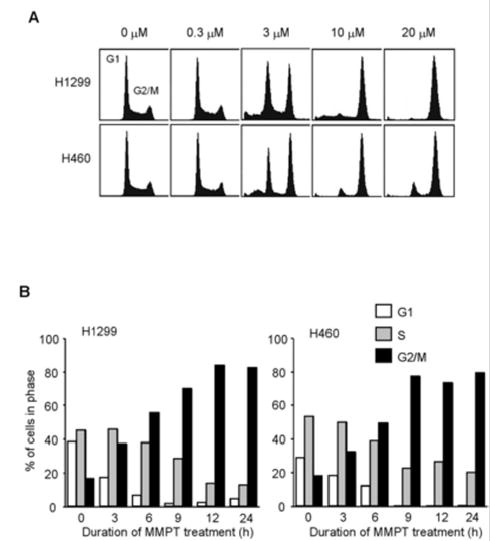
Effect of thiazolidin compounds on cell-cycle progression in cancer cells. A: Flow cytometry analysis of propidium iodide-stained H1299 and H460 cells treated with various concentrations of MMPT for 12 h. B: Percentages of H1299 and H460 cells in G1, S, and G2/M phases after treatment with 10 μM MMPT for the indicated times. C: Flow cytometry analysis of propidium iodide-stained DLD-1 cells, wild-type HCT116 cells (HCT116+/+), and p53-deficient HCT116 cells (HCT116−/−) were treated with 3 or 5 μM DBPT, respectively, for the indicated time periods. D: Percentages of DLD-1, HCT116+/+, and HCT116−/− cells in G1, S, and G2/M phases after treatment with 3 or 5 μ M DBPT for the indicated times.
Thiazolidin compounds affect the regulation of G2/M checkpoint proteins
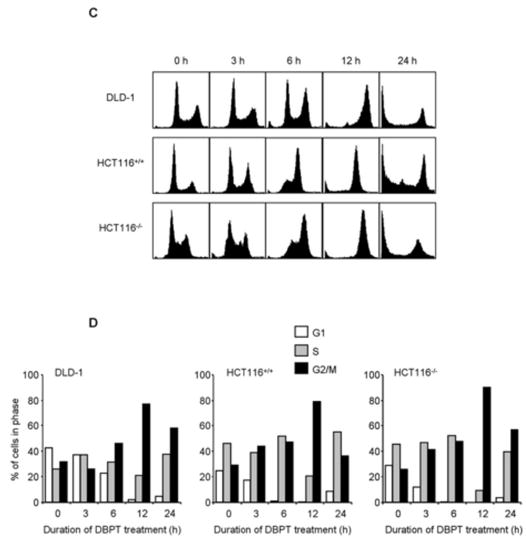
We next used western blotting to examine the effects of MMPT and DBPT on the expression of G2/M checkpoint proteins in cancer cells. Cyclin A expression was noticeably reduced after 24 h of treatment with MMPT and DBPT, and cyclin B1 expression was transiently increased after 6–12 h of treatment (Fig. 2A, 2B). No dramatic change was observed for level of cdc2p34. We also detected a slower-migrating form of the cdc25C phosphatase and a phosphorylated form of histone H3 after 6–24 h of MMPT treatment. Similar changes in the migration of cdc25C and phosphorylation of histone H3 were observed in DLD-1 cells 6–24 h after treatment with 3 μM DBPT (Fig. 2B).
Fig. 2.
Thiazolidin compounds regulate G2/M checkpoint proteins. H1299 and H460 cells (A) and DLD-1 cells (B) were treated with 10 μM MMPT or 3 μM DBPT, respectively, for the indicated time periods. Expression and phosphorylation status of G2/M checkpoint proteins in whole-cell lysates were analyzed by immunoblotting with the indicated antibodies. Arrowheads indicate phosphorylated form of cdc25C. β-Actin was used as the loading control. p-, phosphorylated. C: H460, H460/TaxR, and H460/VinR cells were treated with 10 μM MMPT for the indicated time periods. Phosphorylation status of Bcl-2 in whole-cell lysates was analyzed by immunoblotting with anti-Bcl-2 antibody. Arrowhead indicates phosphorylated form of Bcl-2. β-actin was used as the loading control.
Bcl-2 phosphorylation is a common event during M phase [17, 18]. To determine whether thiazolidin compounds use this mechanism to induce G2/M-phase arrest, we examined time-dependent changes in Bcl-2 phosphorylation in H460, H460/TaxR, and H460/VinR cells treated with 10 μM MMPT. Western blotting revealed an obvious increase in Bcl-2 phosphorylation after 12–24 h of MMPT treatment (Fig. 2C). This result indicated that the effect of MMPT on Bcl-2 phosphorylation is independent on P-glycoprotein expression.
Induction of M-phase arrest by thiazolidin compounds in cancer cells
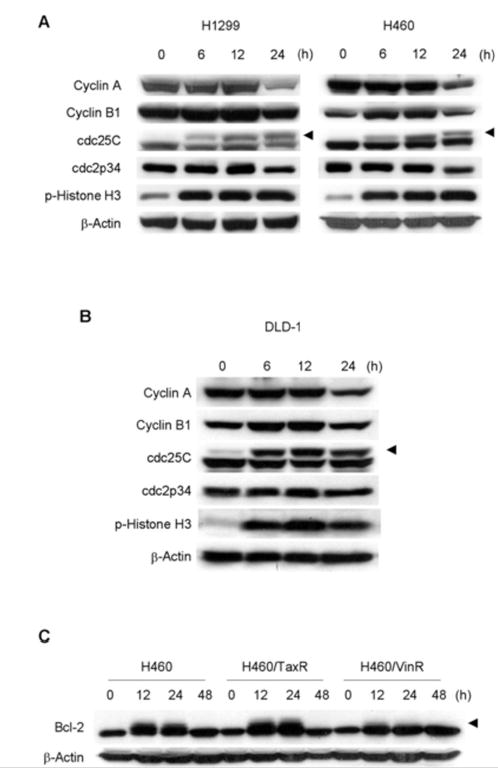
To further pinpoint whether cell-cycle arrest by thiazolidin compounds occurred at the G2 or M phase, we conducted a flow cytometry analysis of MMPT-treated H1299 and H460 cells stained with propidium iodide and an antibody against MPM-2, a marker for the onset of mitosis [19]. Anti-mouse secondary antibody was used as a control, and H460 cells treated with 50 nM paclitaxel were used as a positive control for MPM-2 (data not shown). We observed a marked accumulation of MPM-2-positive cells 12 h after treatment with 10 μM MMPT (Fig. 3A, 3B). A similar accumulation of MPM-2-positive cells was observed in DLD-1 cells treated with 3 μM DBPT (Fig. 3C). These results indicate that treatment of cancer cells with thiazolidin compounds results in an accumulation of cells arrested in M phase.
Fig. 3.
Induction of M-phase arrest by thiazolidin compounds. H1299 (A) and H460 (B) cells were treated with 10 μM MMPT and DLD-1 (C) cells were treated with 3 μM DBPT for the indicated time periods. Cells were then stained with anti-MPM-2 antibody and propidium iodide (PI) and analyzed by flow cytometry. Histograms show DNA content of cells; dot plots show proportions of mitotic and 4N cells after MMPT or DBPT treatment. Boxes indicate cells staining positively for MPM-2. Percentages of MPM-2-positive cells are shown.
Effect of thiazolidin compounds on mitotic spindles in cancer cells
In addition to inducing M-phase arrest and Bcl-2 phosphorylation, most microtubule-interacting agents inhibit mitosis by disrupting the organization of mitotic spindles. To examine whether thiazolidin compounds share this ability, we evaluated the status of the mitotic spindles in H460, H460/TaxR, and H460/VinR cells after 14 h of treatment with DMSO, vinorelbine, MMPT, or paclitaxel (Fig. 4A). Normal spindle formation was confirmed in dividing cells that had been treated with DMSO. Vinorelbine treatment of parental H460 cells resulted in the accumulation of many mitotic cells with depolymerized mitotic spindles. A similar accumulation was not observed in vinorelbine-treated H460/TaxR or H460/VinR cells. Monoaster spindles were observed in all three cell lines after treatment with MMPT, and some of the H460/TaxR and H460/VinR cells had two- or three-spindle polar organization. This result suggested that the effect of MMPT on microtubule disruption is independent on P-glycoprotein expression. Similar changes were observed in DLD-1 cells after 14 h of treatment with 3 μM DBPT (Fig. 4B). In contrast, the increased microtubule polymerization caused by paclitaxel treatment resulted in destruction of the mitotic spindles and aggregated spindle formation in H460 and DLD-1 cells. This phenomenon was, however, not observed in paclitaxel-treated H460/TaxR and H460/VinR cells (Fig. 4A, 4B).
Fig. 4.
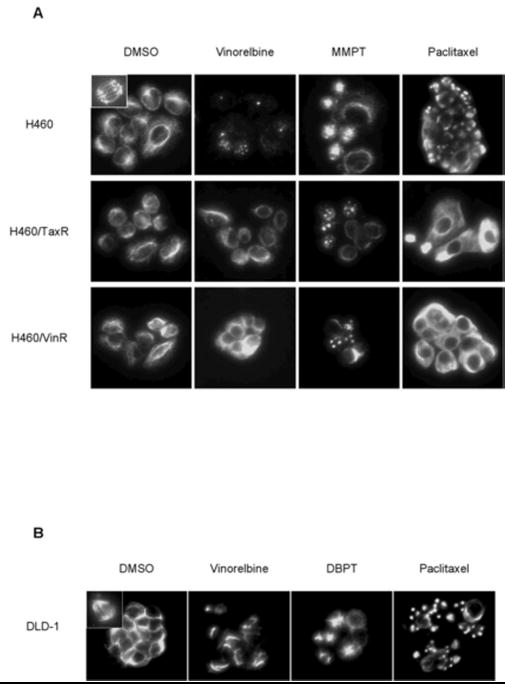
Effect of thiazolidin compounds on cellular microtubule organization. H460, H460/TaxR, and H460/VinR cells (A) and DLD-1 cells (B) were treated with DMSO (control), 100 nM vinorelbine, 15 μM MMPT/3 μM DBPT, or 100 nM paclitaxel. After 14 h, cellular microtubules were visualized by indirect immunofluorescence with anti-α-tubulin antibody. Normal spindle formation was observed in control (inside left upper box). Abnormal monoaster spindles were observed in cells treated with MMPT or DBPT but not in control cells.
JNK activation is required for DBPT-induced G2/M-phase arrest and microtubule disruption
Our previous study showed that activation of JNK but not ERK or p38 is required for MMPT- and DBPT-induced apoptosis [14, 15]. To further investigate the possible role of JNK activation on induction of G2/M-phase arrest, we treated DLD-1 cells with DBPT in the presence or absence of the JNK inhibitor SP600125 for 12 h and then evaluated the cell-cycle progression by flow cytometry. Treatment with 20 μM SP600125 alone, which was enough to block DBPT-mediated JNK activation [14], resulted in a slight increase of cells at G2/M phase. Interestingly, SP600125 dramatically blocked DBPT-induced G2/M-phase arrest (Fig. 5A). Whereas the percentage of G2/M-phase cells was 40.8% with 20 μM SP600125 alone and 86.0% with 3 μM DBPT alone, only 44.5% of cells were in G2/M-phase with the combination of SP600125 and DBPT. This result indicates that SP600125 inhibits DBPT-induced G2/M-phase arrest (p< 0.001). Our previous data showed that JNK1 activation is crucial for DBPT-mediated apoptosis and that different JNK isoforms may have different roles in DBPT-induced apoptosis [14]. To further test the role of JNK1 and JNK2 activation in DBPT-mediated G2/M-phase arrest, we evaluated the effect of DBPT in DLD-1 cells stably transfected with plasmid expressing the dominant-negative mutants, dnJNK1 or dnJNK2 [14]. Cells were treated with 3 μM DBPT for 6 h, and analyzed cell-cycle progression by flow cytometry. The expression of dnJNK1 in DLD-1 cells significantly blocked DBPT-mediated G2/M-phase arrest after 6 h (p< 0.001). In contrast, the expression of dnJNK2 enhanced DBPT-mediated G2/M-phase arrest when compared with DBPT-treated parental DLD-1 cells (p< 0.01) (Fig. 5B). These results indicate that JNK1 and JNK2 may also have different roles in DBPT-mediated G2/M-phase arrest. We also evaluated the expression of cdc25C and phosphorylated histone H3 in DLD-1 cells treated with DBPT in the presence of SP600125 for 6 h. Treatment with 20 μM SP600125 alone had no effect on phosphorylated form of histone H3 and cdc25C expression when compared with control. Interestingly, SP600125 dramatically blocked DBPT-mediated changes in the migration of cdc25C and phosphorylation of histone H3 (Fig. 5C). To further examine whether inhibition of JNK affects DBPT-caused microtubule disruption, DLD-1 cells were treated with DBPT or paclitaxel in the presence or absence of 20 μM SP600125 for 14 h and the mitotic spindles were visualized by immunofluorescence microscopy (Fig. 5D). Central spindle formation was detected in DLD-1 cells treated with DMSO and SP600125. Interestingly, although abnormal monoaster spindle formations were observed in DLD-1 cells treated with DBPT alone, they were not seen in DLD-1 cells treated with DBPT in the presence of SP600125. In contrast, SP600125 had little impact on microtubule depolymerization of DLD-1 cells treated with paclitaxel. These results suggest that JNK1 activation is required for DBPT-mediated microtubule disruption.
Fig. 5.
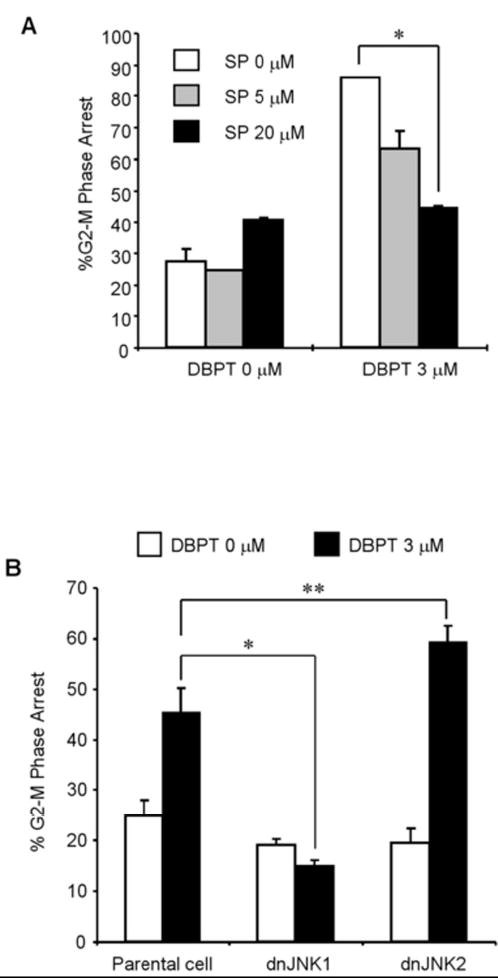
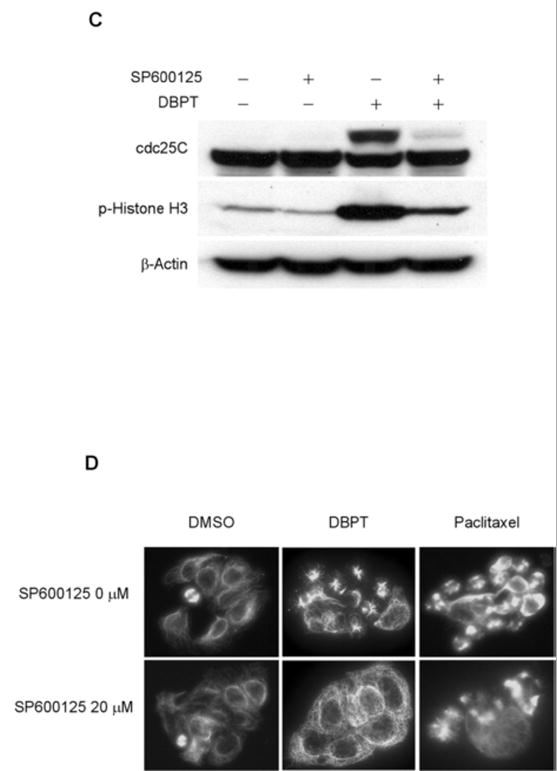
The effects of JNK inhibition on DBPT-mediated G2/M-phase arrest and microtubule disruption. A: Inhibition of JNK activation blocks DBPT-mediated G2/M-phase arrest. DLD-1 cells were treated with 3 μM DBPT in the presence or absence of 5 or 20 μM SP600125 for 12 h. The cell-cycle progression of stained cells with PI was determined by flow cytometry. Data represent mean ± SD from three independent experiments performed in triplicate. *p< 0.001, as compared with DBPT treatment alone. B: Effect of JNK1 activation on DBPT-mediated G2/M-phase arrest. DLD-1 cells stably transfected with dnJNK1, dnJNK2 or an empty vector (control) were treated with 3 μM DBPT for 6 h. The cell-cycle progression of stained cells with PI was determined by flow cytometry. Data represent mean ± SD of two independent experiments. *p< 0.001 and **p< 0.01, as compared with DBPT-treated parental DLD-1 cells. C: SP600125 blocks DBPT-mediated histone H3 phosphorylation. DLD-1 cells were treated with 3 μM DBPT in the presence or absence of 20 μM SP600125 for 6 h. Whole-cell lysates were analyzed by immunoblotting with cdc25C and phosphorylated histone H3 antibodies. β-actin was used as the loading control. D: Effect of JNK inhibition on DBPT-mediated microtubule disruption. DLD-1 cells were treated with DMSO (control), 3 μM DBPT, or 100 nM paclitaxel in the presence or absence of 20 μM SP600125 for 14 h. Cellular microtubules were visualized by indirect immunofluorescence with anti-α-tubulin antibody. The data represent one of two experiments with similar results.
Discussion
In the present study, we found that thiazolidin compounds can induce G2/M arrest and disruption of mitotic spindles in non-small-cell lung and colon cancer cells. Our flow cytometry and immunoblotting analyses showed that thiazolidin compounds elicit changes in the expression of G2/M checkpoint proteins and increase the phosphorylation of MPM-2, cdc25C, and histone H3 in non-small-cell lung and colon cancer cells. Consistently, DBPT also induced G2/M-phase arrest with increased phosphorylation of MPM-2, cdc25C, and histone H3 in prostate cancer cell lines (unpublished data, Teraishi F et al). These changes in expression and phosphorylation are consistent with mitotic arrest [19–21]. In addition, the result of immunofluorescence microscopy showed that thiazolidin compounds interfere with normal spindle formation like nocodazole and vinorelbine. Together, these data indicate that thiazolidin compounds induce mitotic arrest with microtubule disruption. A recent report showed that p53 activation has a key role in the induction of G2/M-phase arrest by several chemotherapeutic agents [22, 23]. However, microtubule-interacting agents such as paclitaxel, nocodazole, and vinorelbine cause G2/M-phase arrest independent of p53 status, despite increasing levels of transcriptionally active p53 [24, 25]. Similarly, we found that although MMPT and DBPT upregulate p53 expression (unpublished data, Teraishi F et al), they are still able to cause G2/M-phase arrest in p53-deficient H1299 and HCT116−/− cells.
Most microtubule-interacting agents induce apoptosis in cancer cells, however, the mechanisms by which they do so remain uncharacterized. Nevertheless, a previous report showed that endonucleolytic cleavage of DNA, a characteristic of apoptosis, is observed only in cells arrested in G2/M phase after treatment with epothilone and paclitaxel [26]. It has also been reported that paclitaxel-induced cell death in cancer cells might result from apoptosis directly after mitotic arrest [2, 27, 28]. Our data showed that both MMPT- and DBPT-mediated G2/M-phase arrest occurred as early as 6 h and peaked around 80% after 12 h of treatment, whereas MMPT- and DBPT-induced apoptosis mainly started after 12 h of treatment [14, 15]. Thus, G2/M-phase arrest by thiazolidin compounds occurred earlier than that of apoptosis. Moreover, DBPT-mediated G2/M-phase arrest and phosphorylation of cdc25C and histone H3 were both blocked by inhibition of JNK activation with either a specific JNK inhibitor or a dominant-negative JNK1 gene. Because DBPT-induced G2/M-phase arrest precedes apoptosis and because our previous study showed that DBPT-induced apoptosis was also blocked by inhibition of JNK activation [14], it is very likely that these two events (G2/M arrest and apoptosis) are coupled and that persistent G2/M arrest leads to apoptosis. Indeed, several reports have shown that Bcl-2 phosphorylation may be involved in one of the pathways of antimitotic drug-induced apoptosis [29–31]. Our study showed that Bcl-2 phosphorylation increased in H460, H460/TaxR, and H460/VinR cells after MMPT treatment. Phosphorylated Bcl-2 appeared to accumulate at the same time as the phosphorylated forms of MPM-2, cdc25C, and histone H3. However, it is also possible that G2/M-phase arrest and apoptosis-induction by thiazolidin compounds occurred independently, although both events are blocked by inhibition of JNK activation. Therefore, molecular mechanisms of apoptotic induction and G2/M arrest caused by thiazolidin compounds remain to be further delineated.
Although this study has revealed the effects of thiazolidin compounds in cell cycle arrest and microtubule disruption, the direct targets of thiazolidin compounds remain to be identified. Whereas inhibition of JNK activation blocked DBPT-caused G2/M-phase arrest and microtubule disruption, the mechanism by which the JNK signaling pathway triggers G2/M arrest and microtubule disruption are not yet clear. Further characterization of biological function of thiazolidin compounds may shed light on roles of JNK activation in G2/M arrest and apoptosis-induction.
Acknowledgments
We thank Pierrette Lo for editorial review and Karen M. Ramirez for technical assistance with flow cytometry analysis. This study was supported by grants from National Cancer Institute grants RO1 CA 092487-01A1 (B. Fang), RO1 CA 098582-01A1 (B. Fang), Core Grant CA 16672, and Lung SPORE (CA 70907) development award.
References
- 1.Correia JJ, Lobert S. Physiochemical aspects of tubulin-interacting antimitotic drugs. Current Pharmaceutical Design. 2001;7:1213–1228. doi: 10.2174/1381612013397438. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–825. [PubMed] [Google Scholar]
- 3.Masters GA, Hoffman PC, Drinkard LC, Samuels BL, Golomb HM, Vokes EE. A review of ifosfamide and vinorelbine in advanced non-small cell carcinoma of the lung. Seminars in Oncology. 1998;25:8–14. [PubMed] [Google Scholar]
- 4.Ngan VK, Bellman K, Panda D, Hill BT, Jordan MA, Wilson L. Novel actions of the antitumor drugs vinflunine and vinorelbine on microtubules. Cancer Research. 2000;60:5045–5051. [PubMed] [Google Scholar]
- 5.Fossella FV, Lee JS, Murphy WK, Lippman SM, Calayag M, Pang A, et al. Phase II study of docetaxel for recurrent or metastatic non-small-cell lung cancer. J Clin Oncol. 1994;12:1238–1244. doi: 10.1200/JCO.1994.12.6.1238. [DOI] [PubMed] [Google Scholar]
- 6.Rowinsky EK. The development and clinical utility of the taxane class of antimicrotubule chemotherapy agents. Annu Rev Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 7.Sandercock J, Parmar MK, Torri V. First-line chemotherapy for advanced ovarian cancer: paclitaxel, cisplatin and the evidence. Br J Cancer. 1998;78:1471–1478. doi: 10.1038/bjc.1998.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorger PK, Dobles M, Tournebize R, Hyman AA. Coupling cell division and cell death to microtubule dynamics. Curr Opin Cell Biol. 1997;9:807–814. doi: 10.1016/s0955-0674(97)80081-6. [DOI] [PubMed] [Google Scholar]
- 9.Drukman S, Kavallaris M. Microtubule alterations and resistance to tubulin-binding agents (review) Int J Oncol. 2002;21:621–628. [PubMed] [Google Scholar]
- 10.Bosch I, Croop J. P-glycoprotein multidrug resistance and cancer. Biochim Biophys Acta. 1996;1288:F37–F54. doi: 10.1016/0304-419x(96)00022-4. [DOI] [PubMed] [Google Scholar]
- 11.Goldman B. Multidrug resistance: can new drugs help chemotherapy score against cancer? J Natl Cancer Inst. 2003;95:255–257. doi: 10.1093/jnci/95.4.255. [DOI] [PubMed] [Google Scholar]
- 12.Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/s0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 13.Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proc Natl Acad Sci U S A. 1998;95:7024–7029. doi: 10.1073/pnas.95.12.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teraishi F, Wu S, Zhang L, Guo W, Davis JJ, Dong F, et al. Identification of a Novel Synthetic Thiazolidin Compound Capable of Inducing c-Jun NH2-Terminal Kinase-Dependent Apoptosis in Human Colon Cancer Cells. Cancer Res. 2005;65:6380–6387. doi: 10.1158/0008-5472.CAN-05-0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teraishi F, Wu S, Sasaki J, Zhang L, Zhu HB, Davis JJ, et al. P-Glycoprotein-Independent Apoptosis Induction by a Novel Synthetic Compound, MMPT [5-[(4-Methylphenyl)methylene]-2-(phenylamino)-4(5H)-thiazolone] J Pharmacol Exp Ther. 2005;314:355–362. doi: 10.1124/jpet.105.085654. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki J, Ramesh R, Chada S, Gomyo Y, Roth JA, Mukhopadhyay T. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Molecular Cancer Therapeutics. 2002;1:1201–1209. [PubMed] [Google Scholar]
- 17.Ling YH, Tornos C, Perez-Soler R. Phosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosis. J Biol Chem. 1998;273:18984–18991. doi: 10.1074/jbc.273.30.18984. [DOI] [PubMed] [Google Scholar]
- 18.Scatena CD, Stewart ZA, Mays D, Tang LJ, Keefer CJ, Leach SD, et al. Mitotic phosphorylation of Bcl-2 during normal cell cycle progression and Taxol-induced growth arrest. J Biol Chem. 1998;273:30777–30784. doi: 10.1074/jbc.273.46.30777. [DOI] [PubMed] [Google Scholar]
- 19.Vandre DD, Borisy GG. Anaphase onset and dephosphorylation of mitotic phosphoproteins occur concomitantly. J Cell Sci. 1989;94 (Pt 2):245–258. doi: 10.1242/jcs.94.2.245. [DOI] [PubMed] [Google Scholar]
- 20.Giet R, Glover DM. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J Cell Biol. 2001;152:669–682. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris MC, Heitz A, Mery J, Heitz F, Divita G. An essential phosphorylation-site domain of human cdc25C interacts with both 14-3-3 and cyclins. J Biol Chem. 2000;275:28849–28857. doi: 10.1074/jbc.M002942200. [DOI] [PubMed] [Google Scholar]
- 22.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science . 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 23.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 24.Stewart ZA, Tang LJ, Pietenpol JA. Increased p53 phosphorylation after microtubule disruption is mediated in a microtubule inhibitor- and cell-specific manner. Oncogene. 2001;20:113–124. doi: 10.1038/sj.onc.1204060. [DOI] [PubMed] [Google Scholar]
- 25.Tishler RB, Lamppu DM, Park S, Price BD. Microtubule-active drugs taxol, vinblastine, and nocodazole increase the levels of transcriptionally active p53. Cancer Res. 1995;55:6021–6025. [PubMed] [Google Scholar]
- 26.Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88:2619–2628. doi: 10.1002/1097-0142(20000601)88:11<2619::aid-cncr26>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 27.Basu A, Haldar S. Microtubule-damaging drugs triggered bcl2 phosphorylation-requirement of phosphorylation on both serine-70 and serine-87 residues of bcl2 protein. Int J Oncol. 1998;13:659–664. doi: 10.3892/ijo.13.4.659. [DOI] [PubMed] [Google Scholar]
- 28.Woods CM, Zhu J, McQueney PA, Bollag D, Lazarides E. Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway. Mol Med. 1995;1:506–526. [PMC free article] [PubMed] [Google Scholar]
- 29.Blagosklonny MV, Giannakakou P, el Deiry WS, Kingston DG, Higgs PI, Neckers L, et al. Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death. Cancer Res. 1997;57:130–135. [PubMed] [Google Scholar]
- 30.Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A. 1999;96:3775–3780. doi: 10.1073/pnas.96.7.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, Wang Z, Boise L, Dent P, Grant S. Loss of the bcl-2 phosphorylation loop domain increases resistance of human leukemia cells (U937) to paclitaxel-mediated mitochondrial dysfunction and apoptosis. Biochem Biophys Res Commun. 1999;259:67–72. doi: 10.1006/bbrc.1999.0669. [DOI] [PubMed] [Google Scholar]