

Abstract
NADPH oxidases (Nox) are established as major sources of reactive oxygen species (ROS). Over the past two decades, Nox-derived ROS have emerged as pivotal in the development of myriad diseases involving oxidative stress. In contrast, Nox are also involved in signaling mechanisms necessary for normal cell function. The study of these enzymes in physiological and pathophysiological conditions is made considerably more complex by the discovery of 7 isoforms: Nox1 through 5 as well as Duox1 and 2, each with its own specific cytosolic components, regulatory control mechanisms, subcellular localization and/or tissue distribution. A clear understanding of the role individual isoforms play in a given system is hindered by the lack of isoform-specific inhibitors. In animal models, knockdown or knockout methodologies are providing definitive answers to perplexing questions of the complex interplay of multiple Nox isoforms in cell and tissue signaling. However, the complex structures and interactions of these heteromeric isozymes predict pleiotropic actions of the Nox subunits and thus suppression of these proteins is almost certain to have untoward effects. Thus, as both therapies and pharmacological tools, molecule-based inhibitors continue to prove extremely useful and rational in design. Unfortunately, many of the available inhibitors have proven non-specific, falling into the category of scavengers or inhibitors of more than one source of ROS. Here, we will review some of the efforts that have been undertaken to develop specific inhibitors of NADPH oxidase over the past decade, from the peptidic inhibitor Nox2ds-tat to more recent small molecule inhibitors that have emerged from high-throughput screening campaigns.
Keywords: NADPH oxidase, Nox isoforms, Inhibitors, Peptides, Small molecules, High-throughput screening
Introduction
Oxidative stress has clearly been implicated in a broad range of pathophysiological conditions including hypertension, diabetes, atherosclerosis, Parkinson’s disease, Alzheimer’s disease, and cancer [1-4]. Oxidative stress results from an imbalance between the production of ROS and the antioxidant capabilities of a given system, culminating in a stimulation of degenerative signaling pathways often leading to unfettered tissue growth and neoplasia, inflammation, and dysfunctional innate immune reactions. By the same token, it is becoming increasingly clear that localized expression of Nox isozymes along with a fine balance between production and metabolism of Nox-derived ROS mediators effect cell functions ranging from basic cell division, migration and differentiation to vestibular balance, neuronal signaling, angiogenesis, and thyroid hormone synthesis [2].
The NADPH oxidases or Nox enzymes are a family of protein complexes that catalyze the transfer of electrons from NADPH to molecular oxygen resulting in the generation of superoxide and/or hydrogen peroxide [5]. Members of this family include Nox1 through 5 as well as Duox1 and 2, each with their own specific cytosolic components, regulatory mechanisms, subcellular localizations, and tissue distribution [1, 6]. In the cardiovascular system, where the existence of various Nox isoforms sharing homology with the enzyme present in neutrophils were first widely studied, the main isoforms present are Nox1, Nox2, Nox4, and Nox5 (Fig. 1). The prototypical member of this family of proteins, Nox2-based oxidase, is the isoform first described in leukocytes (previously defined by its catalytic subunit gp91phox, which is now generally referred to as Nox2). Nox2 oxidase is comprised of two membrane-bound subunits (Nox2 and p22phox) and cytosolic components necessary for its activity including the main organizing and activating subunits, p47phox and p67phox respectively, as well as rac and p40phox. Nox1-based oxidase, on the other hand, requires p22phox for activity and also NOXO1 and NOXA1 cytosolic subunits which are homologs of p47phox and p67phox, respectively. Nox4 is constitutively actively active requiring only p22phox. Nox5 is regulated by calcium through its N-terminal EF-hand domains and does not require other subunits for catalytic activity.
Fig. 1.

Representation of NADPH oxidase family. Noxl and Nox2 require p22phox and the indicated cytosolic regulatory subunits for activation. Nox4 requires only p22phox. Nox5, on the other hand, is regulated by calcium binding through its N-terminal EF motifs
Given the plethora of diseases in which Nox-derived ROS are implicated, the quest for specific inhibitors has intensified in recent years. Despite considerable efforts made in the field, however, the availability and utility of these inhibitors has been limited, in part by the lack of specificity and, more broadly, a lack of clear evidence of the inhibitors’ mechanisms of action. Here, we review two classes of inhibitors, peptide-based and small molecule inhibitors, recently identified by high-throughput screening campaigns.
Peptide-based inhibitors
The majority of chemical Nox inhibitors were identified and characterized based on their abilities to block the catalytic core of the phagocyte NADPH oxidase at a time when only one isoform (gp91phox or Nox2) was known to exist. Moreover, most of these experiments were carried out in cell-free systems. A large number of these compounds lack specificity, e.g., inhibit other flavoproteins enzyme systems (mitochondrial electron-transport chain, nitric oxide synthase, and cytochrome p450 enzymes, among others). Since many, if not most, were not developed by “rational design” or screened for isoform specificity, it should come as no surprise that several or all Nox isoforms would be expected to be blocked. Peptide sequences and peptidomimetics are, by their nature, likely to render superior specificity as they more precisely mimic and thereby disrupt intrinsic protein–protein interactions in the membrane holoenzyme or cytosolic subunits. Indeed, Nox1 shares ~60 % amino acid identity with Nox2, while Nox4 shares only ~39% identity with it [1], allowing for selective targeting of these isoforms with peptide sequences unique between them [7]. That said, the use of peptides as “druggable” therapeutics is often challenged due to their degradation in the gut and limited oral bioavailability. This limitation, however, may be obviated by alternate routes of systemic administration and the emergence of new nanotechnologies aimed at enhancing peptide stability and delivery. Below, we consider briefly the currently available Nox2-, p22phox-, p47phox-, and Rac-derived inhibitor peptides, and discuss their clinical usefulness, specificity, and efficacy.
Nox2-targeted inhibitory peptides
Despite noted limitations, Nox2ds-tat (Nox2 docking sequence-tat; Nox2ds-tat, previously gp91ds-tat) is generally regarded as the most specific and efficacious Nox inhibitor available. Nox2ds-tat is a chimeric 18-amino acid peptide ([H]-R-K-K-R-R-Q-R-R-R-C-S-T-R-I-R-R-Q-L-[NH2]) that has been shown to inhibit NADPH oxidase activity in vivo and in vitro [8-11]. The Nox2ds portion of the peptide is a 9 amino acid sequence from the cytosolic B-loop of Nox2 (amino acids 86–94) designed to selectively inhibit the interaction between Nox2 and p47phox [9]. The tat portion of the peptide corresponds to a 9 amino acid sequence of the HIV-tat transport region, which allows the peptide to be internalized by cells [12]. The viability of Nox2ds as an inhibitor was predicted by random-sequence peptide phage display library analysis of the human Nox2 and was shown to inhibit NADPH oxidase activity in a neutrophil cell-free system [13].
In vitro assays indicate that Nox2ds-tat inhibits superoxide anion production in endothelial cells in response to various stimuli, including hypoxia [14], nutrient deprivation [15], atrial natriuretic peptide [16], angiopoietin-1 [17], interleukin-4 [18], shear stress [19], and calcineurin inhibitors [20]. Nox2ds-tat also blocked angiotensin II (AngII)-induced superoxide production in human resistance artery smooth muscle cells [10] and collagen-induced NADPH oxidase activity in platelets [21].
These in vitro results are well reflected in vivo. For example, Nox2ds-tat administered by osmotic minipump for 5 days significantly improved acetylcholine-induced endothelium-dependent relaxation in aortic rings from mice with renovascular hypertension [22]. Subcutaneous infusion of Nox2ds-tat attenuated vascular superoxide production, vascular inflammation, and medial hypertrophy in AngII-infused rat model of hypertension [8]. Moreover, targeted delivery of Nox2ds to adventitial fibroblasts attenuated AngII-induced medial hypertrophy and ROS production of the carotid artery [23]. Nox2ds-tat, whether administered subcutaneously or via perivascular delivery by an adenoviral vector, prevented ROS production and neointimal proliferation in response to balloon angioplasty of the carotid artery in rats [11, 24, 25]. Although Nox2ds-tat as a peptide has limited oral bioavailability, parenteral delivery methods, such as subcutaneous infusion, direct application to blood vessels using gene therapy, or intravenous administration are all effective at reducing vascular pathologies associated with increased ROS production.
Nox2ds-tat was designed to specifically inhibit interactions between Nox2 oxidase and p47phox. Nevertheless, there was some concern that sequence homology between Nox1 and Nox2, as well as the fact that Nox1 oxidase may utilize p47phox as its organizer subunit [26], might also predict Nox2ds interference with the assembly of hybrid Nox1 oxidase. Moreover, studies suggested that B-loop peptides bind to the dehydrogenase (DH) domain in the C-terminal tail of Nox4, raising concern for non-isoform-specific inhibition of Nox4 by Nox2ds [27]. To test the specificity of Nox2ds, the potential inhibitory activity in COS cell-free preparations using reconstituted systems expressing the canonical Nox2 (p22phox, Nox2, p47phox, and p67phox), Nox1 (p22phox, Nox1, NOXA1, and NOXO1) or Nox4 (p22phox and Nox4) oxidases, as well as the hybrid Nox1 (p22, Nox1, p47phox, and p67phox) system [28]. The data illustrate that Nox2ds is a potent and efficacious inhibitor of Nox2 NADPH oxidase with an IC50 of 0.74 μM. Furthermore, Nox2ds does not inhibit ROS production in either the COS-Nox1 (canonical or hybrid) or the COS-Nox4 oxidase systems. These results demonstrate that Nox2ds is a specific inhibitor of Nox2 oxidase, support its utility to elucidate the role of Nox2 in organ pathophysiology, and demonstrate its potential as a therapeutic agent.
Several other Nox2-derived peptide sequences worth discussing were shown to inhibit Nox2 oxidase in vitro with varying efficacy. Rotrosen et al. [29] demonstrated that a peptide sequence derived from the C-terminal of Nox2 (SNSESGPRGVHFIFNKENF, amino acids 552–570) inhibited the phagocyte NADPH oxidase in cell-free systems with an IC50 of 32 μM. Nakanishi et al. [30] showed that the peptide sequence NSESGPRGVHFIFNKENF, mimicking amino acids 550–569 of the c-terminal domain of Nox2, binds to p47phox and inhibits superoxide generation in the cell-free assay (IC50 = 4 μM). Leusen et al. [31] reported that a point mutation in the C-terminal of Nox2 (Asp → Gly substitution at residue 500) in a patient with an X-linked chronic granulomatous disease (CGD) is associated with decreased translocation of p47phox/p67phox to the membrane and impaired oxidase activity. Consequently, a synthetic peptide containing this mutation (FAVHHDEEKGVTIG; amino acids 491–504) inhibited NADPH oxidase activity in the cell-free assay (IC50 = 10 μM) and the translocation of p47phox and p67phox in the cell-free translocation assay. It has also been shown that peptides corresponding to residues 27–46, 87–100, 282–296, 304–321, 434–455, and 559–565 inhibited superoxide production in a neutrophil cell-free system with IC50 of 34, 40, 30, 35, 25, and 53 μM, respectively [32]. Finally, another B-loop peptide of Nox2 (amino acids 86–102) that contains a putative p47phox binding site has been shown to inhibit NADPH oxidase activity with an IC50 of 2 μM [33]. In contrast to Nox2ds, the majority of these inhibitors were not tested in experimental disease models. Moreover, their specificity is not yet known.
p22phox-derived inhibitory peptides
Pick and colleagues [34] applied the “peptide walking” strategy to identify peptide sequences in p22phox that bind individual cytosolic NADPH oxidase components and inhibit NADPH oxidase activation in the cell-free system. Their study demonstrated that amino acids 9–23, 31–45, 47–61, 85–99, and 113–127 inhibited NADPH oxidase activity with the IC50 value of 5.7, 3.0, 4.6, 2.0, and 3.0 μM, respectively. A synthetic peptide corresponding to residues 82–95 in p22phox (PFTRNYYVRAVLHL) was reported to dose-dependently inhibit superoxide production (IC50 = 10 μM) in a cell-free system consisting of solubilized membrane and cytosol [35]. Interestingly, AGGPPGGPQVNPIPVTDEVV (amino acids 175–194), a peptide sequence close to the C-terminal end of p22phox, also inhibited superoxide production in a cell-free system, although with less efficacy (IC50 = 36 μM) [30]. It is important to note that p22phox is required for the activation of Nox1, Nox2, Nox3, and Nox4, but not of Nox5. Therefore, it could be expected that p22phox-derived inhibitory peptides inhibit all Noxes except Nox5. Interestingly, a recent study, showing that certain p22phox mutations inhibiting Nox1, Nox2, and Nox3 function and/or maturation did not alter Nox4 activity, suggests distinct p22phox-Nox4 interaction, and thus might provide the basis for the development of selective p22phox-based Nox4 inhibitors [36].
p47phox-derived peptides
Nauseef et al. [37] reported that the p47phox sequence AYRRNSVRFL (amino acids 323–332) inhibited p47phox phosphorylation and translocation and, as a result, superoxide anion generation in a broken cell system (IC50 = 54 μM). A synthetic peptide derived from p47phox (RSRKRLSQDAYRRNSVRF, residues 314–331) inhibited NADPH oxidase in intact neutrophils and inhibited protein kinase C-mediated phosphorylation of p47phox [38]. Moreover, the p47phox residues 315–328, (SRKRLSQDAY RRNS), 323–332 (AYRRNSVRFL), and 334–347 (QRRRQARPGPQSPG) inhibited superoxide production in the broken cell system with respective IC50 values of 18, 57, and 15 μM [39].
Peptidic inhibitors of Nox1 and 4 oxidase
Peptide sequences selectively inhibiting the Nox1- and Nox4-oxidase activity have not yet been developed, although this is an area of intense study. As Nox1 oxidase in large vessel smooth muscle cells has been reported to utilize p47phox as its organizer subunit [10], it is possible that p47phox-derived inhibitory peptides will inhibit Nox1 oxidase in smooth muscle cells, which reportedly contain a hybrid Nox1 system [10]. Interestingly, p47phox-derived peptidic inhibitors are expected to inhibit only the Nox1-oxidase in these smooth muscle cells as they do not express Nox2 [40] and thus may be deemed specific in this milieu. Little or no information exists to our knowledge on peptidic inhibitors of Nox4-oxidase. Functional studies demonstrate that Nox4 requires only p22phox, but not the conventional subunits p47phox and p67phox or their homologues NOXO1 and NOXA1, for ROS production [41]. Importantly, the same study showing unique interaction of Nox4 with p22phox might serve as a basis for the development of Nox4-specific inhibitor. Clearly, this is an area ripe for study, and careful evaluation of these interactions will be necessary to devise potentially selective inhibitors of Nox4.
Peptide delivery
Peptide-based inhibitors by nature have the advantage of being targeted to specific regions and interactions of the protein of interest and, thus, have the potential to be more specific and engender fewer off-target effects than other inhibitory moieties. Peptide use as therapeutic agents, however, is limited by diminished oral bioavailability, since most peptides are rapidly inactivated by gastrointestinal enzymes. In consideration of this limitation, intense focus is now being placed on alternative routes of delivery. The most viable of these appears to be by i.v. or patch administration, but may also include aerosolization techniques depending on the desired distribution of the drug. With the help of recent technological developments, drugencapsulating polymeric microparticles (1–1,000 μm) or nanoparticles (1–1,000 nm) are becoming a drug delivery platform that can enhance the efficacy of active pharmaceutical ingredients, including poorly water soluble compounds, ionic drugs, proteins, peptides, and siRNA and DNA therapeutics [42]. Among the advantages of these formulations are that they may: (1) be adaptable to various routes of administration; (2) protect the peptidic inhibitor from enzymatic degradation; (3) allow cell internalization, i.e., custom tailoring to a target cell by the size and surface chemistry of the particles; and (4) target the peptide to the site of action via interaction with a specific receptor or marker or in response to a specific physico-chemical stimulus [43]. In summary, it is expected that innovative advances in the burgeoning field of nanotechnology will continue to enhance prospects for peptide delivery and promote their use in the design of novel therapies.
Small molecule inhibitors
Given the limitations of peptidic inhibitor strategies, the search for specific Nox inhibitors has expanded to include small molecule inhibitors. Historically, Nox inhibitors such as diphenyl iodonium (DPI) and apocynin have proven to lack Nox-specificity [44, 45]. Thus, these compounds as well as AEBSF [46], S17834 [47], and others, will not be discussed here as they have been extensively reviewed elsewhere [48-51]. In the last few years, however, multiple academic research laboratories and pharmaceutical entities have invested substantial effort into high-throughput screening of small molecule libraries in the hopes of finding specific Nox inhibitors. The fruits of these efforts are now coming into view. Here, we will focus on the most recent compounds that have emerged in the literature as a result of high-throughput screening (HTS) campaigns (Table 1).
Table 1.
Isoform specificity of small molecule inhibitors of NADPH oxidase
| Name | Structure | IC50s (μM) |
References | |||
|---|---|---|---|---|---|---|
| Nox1 | Nox2 | Nox4 | XO | |||
| VAS2870 |
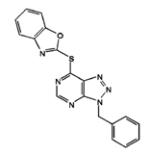
|
↓ | 10.6 (Neutrophils) 2 (HL-60) | ↓ | ? | [52-58] |
| VAS3947 |
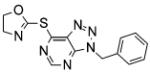
|
↓ | 1–2 (HL-60 cells) | ↓ | - | [60] |
| Fulvene-5 |
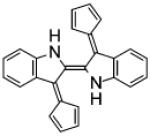
|
? | ↓ | ↓ | ? | [63] |
| ML171 |
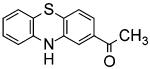
|
0.25 | 5.00 | 5.00 | 5.50 | [70] |
| GKT 136901 |
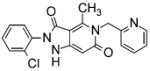
|
0.160a | 1.530a | 0.165a | 30a | [64-66, 79, 80] |
| Celastrol |
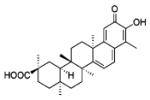
|
0.41 | 0.59 | 2.7 | - | [74] |
↓ inhibits, - no effect, ? not determined
Ki in μM
VAS2870 and VAS3947
In recent years, a novel Nox inhibitor VAS2870 (3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine) has received considerable attention. In a cell-free system containing membranes and cytosolic fractions from human neutrophils, VAS2870 was shown to inhibit superoxide anion production with an IC50 of 10.6 μM. In whole-cell assays of HL-60 cells, the IC50 observed was about fivefold lower (2 μM). Incidentally, both human neutrophils and HL-60 cells predominantly express Nox2. Importantly, VAS2870 has been shown not to be a superoxide anion scavenger or a xanthine oxidase inhibitor, as evidenced by its inability to affect superoxide anion levels in the xanthine/xanthine oxidase assay. Moreover, it was found to be non-cytotoxic [52]. The ability of VAS2870 to inhibit ROS generation and PDGF-BB-mediated migration of vascular smooth muscle cells from rat thoracic aorta has also been demonstrated [52]. PDGF-BB’s pro-vasculogenic effects on differentiating mouse embryonic stem cells, thought to be a ROS-dependent process, are also blocked by the use of VAS2870 [53]. VAS2870 has also been used to inhibit ROS generation induced by oxidized low-density lipoprotein in human umbilical vein endothelial cells [54], in which the predominant Nox isoform is reportedly Nox4. Moreover, in aged spontaneously hypertensive rats (SHR), a model of hypertension and endothelial dysfunction in which there is an increased expression of Nox1 and Nox2 but not Nox4, VAS2870 inhibits ROS production and restores endothelium-dependent aorta relaxation [55]. Furthermore, VAS2870 has been successfully used to explore the role of Nox-dependent ROS generation in α1-adrenoreceptor-activated vasoconstriction [56], post-stroke oxidative stress and neurodegeneration [57], where it was as effective in attenuating ROS as was deleting Nox4, and also in thalidomide-induced ROS generation [58]. VAS2870 impairs cell growth and enhances TGF-β-induced apoptosis of FaO rat hepatoma cells via a Nox-dependent signaling pathway, which in this case corresponds to Nox1, the only Nox these cells express [59]. Taken together, multiple studies prove the effectiveness of VAS2870 to inhibit the development of Nox-dependent cell dysfunction. However, the data are conflicting as to which Nox is being targeted.
VAS3947, a triazolo pyrimidine derivative of VAS2870 has been shown to inhibit Nox activity in various cell systems [60]. VAS3947 does not display antioxidant scavenging effects and does not inhibit other flavoproteins, as determined by its lack of effect on xanthine oxidase and eNOS activities [60]. Comparative analysis of VAS3947 and other molecules commonly used to inhibit Nox activity (apocynin, AEBSF, and DPI) demonstrate a more effective and specific inhibition of Nox for this second generation VAS compound. Three cell lines were used which jointly express all Nox isoforms present in the vasculature (Nox1, Nox2, Nox4, and Nox5) and also Nox3. The IC50 values of VAS3947 varied depending on the cell model tested, with values of 12 μM for CaCo-2 cells (mainly expressing Nox1 and Nox2 but some Nox4 and Nox5), 2 μM for HL-60 cells (mainly Nox2 and Nox5) and 13 μM for A7r5 cells (mainly Nox4, but also Nox1 and Nox3) indicating that this compound, although specific for the Nox class of enzymes, does not appear to be an isoform-specific inhibitor. As such, both VAS2870 and VAS3947 appear to hold significant promise to be excellent pan-Nox inhibitors until other assays are performed to test Nox specificity.
Fulvene-5
As a consequence of their chemical similarity to DPI, and a long history of animal and human exposure and approval for human use by the Food and Drug Administration, triphenylmethane dyes, such as brilliant green and gentian violet, have recently been tested for Nox inhibition. Studies show these compounds to be potent and efficacious inhibitors of Nox2 and Nox4 activity [61]. A structure-based approach to develop more specificity led to the synthesis of the fulvene derivatives class of inhibitors [62]. Fulvenes are aromatic molecules that have the advantage of being highly water soluble. One of these derivatives, Fulvene-5, which was shown in vitro to be able to equally inhibit Nox2 and Nox4, was successfully applied in vivo to block the growth of endothelial tumors of mice [63]. There, fulvene-5’s mechanism of action seems to involve its ability to inhibit Nox4 activity, since the effect of fulvene treatment is consistent with the results obtained using shRNA for Nox4. Further efforts appear necessary to test fulvene-5’s Nox specificity, mechanism of action, and full pharmacological profile for potency, efficacy, and cytotoxicity.
Gkt136901
A HTS campaign using a cell-free assay of ROS production on human Nox4 recently led to the discovery of several pyrazolopyridine dione inhibitors with nanomolar potency against Nox4 and Nox1. Further improvement of the physical properties and pharmacological profiles led to the identification of a potent, orally bioavailable, lead compound, GKT136901. This molecule and some of its derivatives proved highly potent in in vitro assays of human fibroblast differentiation, epithelial cell apoptosis, and epithelial-mesenchymal transition (EMT), as well as in preventive and curative murine models of bleomycin-induced pulmonary fibrosis [64]. The role that glucose-induced upregulation of Nox4 plays in the molecular mechanisms underlying oxidative stress and fibrosis in type2 diabetic nephropathy has also been implicated with the help of GKT136901. GKT136901 demonstrated relative non-specificity for Nox4 (Ki = 165 ± 5 nM) versus Nox1 (Ki = 160 ± 10 nM) [65].
Notably, however, its Ki for Nox1/4 was tenfold higher than for Nox2 (Ki = 1,530 ± 90 nM). A recent study by Garrido-Urbani et al. [66] highlights the use of GKT13690 as an inhibitor of Nox1. In this case, the role of Nox1 in tumor angiogenesis through a PARPα-mediated mechanism was demonstrated using Nox1-deficient mice as well as by treatment with GKT13690. Nox1-deficient mice, but not Nox2- or Nox4-deficient, have impaired angiogenesis, and Nox1 silencing or treatment with GKT13690 decreased endothelial cell migration and tube-like structure formation. Thus, the nanomolar IC50 for Nox1 and Nox4 and its lack of inhibition of other sources of ROS makes GKT13690 a promising dual Nox1/4 inhibitor. Demonstrations that (1) Nox 1 and 4 are oppositely regulated by agonists in rat VSMCs [67], (2) Nox4 enhances endothelium-dependent relaxation and lowers blood pressure in marked contrast to Nox1 and Nox2 [68], and (3) Nox4 is protective in pressure-overload hypertrophy of the heart [69], indicate that particular caution will likely be necessary in considering the clinical outcome of therapeutic treatment with GKT136901.
ML171
ML171 was identified by a cell-based HTS using the human colon cancer cell line HT29, which only expresses Nox1. Structure-activity relationship analysis led to the modification of the primary hit, trifluoromethyl-phenothiazine, into 2-acetylphenothiazine (ML171). ML171 demonstrated an IC50 for Nox1 in the nanomolar range, 20-fold lower than those for Nox2, 3, 4, and xanthine oxidase. Furthermore, ML171 did not inhibit mitochondrial ROS production. This inhibitor was successfully used to elucidate the relevance of Nox1 in mechanisms of cancer invasion, where it specifically blocked the formation of functional invadopodia in human colon cancer cells by decreasing ROS-dependent ECM degradation [70]. These effects were counteracted by overexpression of Nox1 protein, supporting the specificity and selectivity of ML171 to block Nox1 actions. Thus, ML171 is probably the best example of a well-characterized isoform-specific Nox inhibitor in the literature to date.
Celastrol
Celastrol is a triterpenoid antioxidant compound isolated from the Chinese Thunder of God vine (T. wifordii), which has been used in traditional Chinese medicine for its beneficial and curative effects on various inflammatory diseases and cancer [71]. The mechanism of action of celastrol is not completely understood, as many potential targets have been implicated including NFκB, Hsp90, and others [72]. In fact, Nox1 and iNOS seem to play a role in the protective effects of celastrol on endothelial barrier function [73]. More recently, Jaquet et al. [74] focused their interest on the direct effect of celastrol on various Nox isoforms. Their data show that celastrol is a potent inhibitor of Nox isoforms in general. Close examination of tabulated results show that celastrol is relatively non-specific when comparing Nox1 versus Nox2 (IC50s = 0.41 ± 0.20 and 0.59 ± 0.34 μM, respectively). Nevertheless, those IC50s are approximately fivefold lower than for Nox 4 and Nox5 (IC50s = 2.79 ± 0.79 and 3.13 ± 0.85 μM, respectively), indicating a higher potency for inhibition of Nox1 and 2 versus Nox4 and 5. Interestingly, analysis of non-Michaelis–Menten kinetics of the Nox1 and Nox2 reactions with celastrol shows Hill slopes higher than 1, suggesting positive cooperativity in the inhibition of Nox1 and Nox2 by this drug, and gives insights into its mechanisms of action. Celastrol purportedly binds directly to p47phox without interfering with its phosphorylation-dependent translocation to the membrane. Instead, it appears to block interactions between the tandem SH3 domain of p47phox, or NOXO1, and the proline-rich region of p22phox.
Taking together, celastrol demonstrates potential as a pharmacological tool to test the involvement of the Nox enzymes in given pathways; however, its therapeutic potential will require extensive validation since celastrol has been reported to have a broad range of actions that may or may not involve Nox activity [75-77].
Other agents
In a recent study, Borbely et al. [78] showed the methodical development of derivatives of primary hits obtained by a HTS campaign searching for small molecule inhibitors of Nox4. Primary screening consisted of a cell-based assay using Nox4-transfected HEK293 cells and a small-molecule library of approximately 1,000 compounds, from which a total of 73 proved to be effective. The most potent compounds belong to the following core structures: oxalyl hydrazides, flavonoids, oxindols, benzoquinolines, and benzothiophenes. Within each group, modifications of functional groups were made at different positions of the core and their structure–activity relationships analyzed. The best hit molecules shared a 3D structure consisting of four pharmacophore points featuring hydrogen bond donors, acceptors, and 2 aromatic rings independent of the core structure. Although these studies shed light on the structural requirements for inhibitors under the conditions tested, there is no information to our knowledge on the specificity of these compounds for Nox4 as compared to other Noxes or other ROS-generating enzymes. Nevertheless, this work is an insightful demonstration of the power of medicinal chemistry and rational drug design that will be necessary for the development of isoform-specific Nox inhibitors.
Conclusions and perspectives
Although considerable efforts are being invested in the development of isoform-specific inhibitors of Nox enzymes, only two compounds, one peptidic, Nox2ds, and the other small molecule, ML171, have thus far proved specific for one Nox isoform. Our laboratory has expanded its scope to include small molecule inhibitors of Nox2, for which no specific chemical inhibitors are, to our knowledge, available. Using cell-based HTS of unique small molecule libraries, a strategic and rigorous series of secondary screens and counterscreens are applied including analysis of the effects of primary “hits” on mitochondrial function. A set of lead chemotypes have been identified, which are now being rigorously evaluated in collaboration with medicinal chemists. New compounds are being assessed in terms of functional group arrays and reactive functionalities to eliminate potential redox active agents, detergent-like and promiscuous binders, and synthetically difficult to “tract” compounds.
The difficulty of isoform-specific Nox inhibitor discovery is clear, given the high degree of homology among the various Nox isoforms in terms of catalytic activity and structure. Thus, NADPH oxidase remains an elusive yet important target for the development of therapeutic agents for the various pathologies in which this family of enzymes is involved. Without a doubt, inhibitors described here, despite not being isoform-specific, hold potential as pharmacological tools for the understanding of the roles of Nox enzymes, especially in pathways in which one Nox isoform is expressed. Although these compounds are promising candidates for the development of useful therapeutic strategies, their clinical potential is yet to be demonstrated. Nevertheless, the available Nox inhibitors and those being developed are a much needed step forward in our understanding of mechanisms of action and functions of Noxes both in signal transduction and pathology. Finally, although HTS provides the opportunity to rapidly assess thousands of compounds, through multiple primary and secondary screenings, the successful identification of isoform-specific inhibitors is in no way definite. Indeed, peptides or peptidomimetics targeting specific protein–protein interactions may hold the greatest promise in this regard.
References
- 1.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 2.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambeth JD, Krause KH, Clark RA. NOX enzymes as novel targets for drug development. Semin Immunopathol. 2008;30:339–363. doi: 10.1007/s00281-008-0123-6. [DOI] [PubMed] [Google Scholar]
- 4.Csanyi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med. 2009;47:1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3249–3277. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- 7.El-Benna J, Dang PM, Perianin A. Peptide-based inhibitors of the phagocyte NADPH oxidase. Biochem Pharmacol. 2010;80:778–785. doi: 10.1016/j.bcp.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23:776–782. doi: 10.1161/01.ATV.0000066684.37829.16. [DOI] [PubMed] [Google Scholar]
- 9.Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 10.Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL. Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries. Regulation by angiotensin II. Circ Res. 2002;90:1205–1213. doi: 10.1161/01.res.0000020404.01971.2f. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- 12.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeLeo FR, Yu L, Burritt JB, Loetterle LR, Bond CW, Jesaitis AJ, Quinn MT. Mapping sites of interaction of p47-phox and flavocytochrome b with random-sequence peptide phage display libraries. Proc Natl Acad Sci USA. 1995;92:7110–7114. doi: 10.1073/pnas.92.15.7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Shabrawey M, Bartoli M, El-Remessy AB, Platt DH, Matragoon S, Behzadian MA, Caldwell RW, Caldwell RB. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopes NHM, Vasudevan SS, Gregg D, Selvakumar B, Pagano PJ, Kovacic H, Goldschmidt-Clermont PJ. Rac-dependent monocyte chemoattractant protein-1 production is induced by nutrient deprivation. Circ Res. 2002;91:798–805. doi: 10.1161/01.res.0000040421.54108.81. [DOI] [PubMed] [Google Scholar]
- 16.Furst R, Brueckl C, Kuebler WM, Zahler S, Krotz F, Gorlach A, Vollmar AM, Kiemer AK. Atrial natriuretic peptide induces mitogen-activated protein kinase phosphatase-1 in human endothelial cells via Rac1 and NAD(P)H oxidase/Nox2-activation. Circ Res. 2005;96:43–53. doi: 10.1161/01.RES.0000151983.01148.06. [DOI] [PubMed] [Google Scholar]
- 17.Harfouche R, Malak NA, Brandes RP, Karsan A, Irani K, Hussain SN. Roles of reactive oxygen species in angiopoietin-1/tie-2 receptor signaling. FASEB J. 2005;19:1728–1730. doi: 10.1096/fj.04-3621fje. [DOI] [PubMed] [Google Scholar]
- 18.Walch L, Massade L, Dufilho M, Brunet A, Rendu F. Proatherogenic effect of interleukin-4 in endothelial cells: modulation of oxidative stress, nitric oxide and monocyte chemoattractant protein-1 expression. Atherosclerosis. 2006;187:285–291. doi: 10.1016/j.atherosclerosis.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 19.Duerrschmidt N, Stielow C, Muller G, Pagano PJ, Morawietz H. NO-mediated regulation of NAD(P)H oxidase by laminar shear stress in human endothelial cells. J Physiol. 2006;576:557–567. doi: 10.1113/jphysiol.2006.111070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krotz F, Keller M, Derflinger S, Schmid H, Gloe T, Bassermann F, Duyster J, Cohen CD, Schuhmann C, Klauss V, Pohl U, Stempfle HU, Sohn HY. Mycophenolate acid inhibits endothelial NAD(P)H oxidase activity and superoxide formation by a Rac1-dependent mechanism. Hypertension. 2007;49:201–208. doi: 10.1161/01.HYP.0000251162.14782.d4. [DOI] [PubMed] [Google Scholar]
- 21.Krotz F, Sohn HY, Gloe T, Zahler S, Riexinger T, Schiele TM, Becker BF, Theisen K, Klauss V, Pohl U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood. 2002;100:917–924. doi: 10.1182/blood.v100.3.917. [DOI] [PubMed] [Google Scholar]
- 22.Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109:1795–1801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 23.Liu J, Ormsby A, Oja-Tebbe N, Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res. 2004;95:587–594. doi: 10.1161/01.RES.0000142317.88591.e6. [DOI] [PubMed] [Google Scholar]
- 24.Dourron HM, Jacobson GM, Park JL, Liu J, Reddy DJ, Scheel ML, Pagano PJ. Perivascular gene transfer of NADPH oxidase inhibitor suppresses angioplasty-induced neointimal proliferation of rat carotid artery. Am J Physiol Heart Circ Physiol. 2005;288:H946–H953. doi: 10.1152/ajpheart.00413.2004. [DOI] [PubMed] [Google Scholar]
- 25.Weaver M, Liu J, Pimentel D, Reddy DJ, Harding P, Peterson EL, Pagano PJ. Adventitial delivery of dominant-negative p67phox attenuates neointimal hyperplasia of the rat carotid artery. Am J Physiol Heart Circ Physiol. 2006;290:H1933–H1941. doi: 10.1152/ajpheart.00690.2005. [DOI] [PubMed] [Google Scholar]
- 26.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 27.Jackson HM, Kawahara T, Nisimoto Y, Smith SM, Lambeth JD. Nox4 B-loop creates an interface between the transmembrane and dehydrogenase domains. J Biol Chem. 2010;285:10281–10290. doi: 10.1074/jbc.M109.084939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csanyi G, Cifuentes-Pagano E, Al Ghouleh I, Ranayhossaini DJ, Egana L, Lopes LR, Jackson HM, Kelley EE, Pagano PJ. Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic Biol Med. 2011;51(6):1116–1125. doi: 10.1016/j.freeradbiomed.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rotrosen D, Kleinberg ME, Nunoi H, Leto T, Gallin JI, Malech HL. Evidence for a functional cytoplasmic domain of phagocyte oxidase cytochrome b558. J Biol Chem. 1990;265:8745–8750. [PubMed] [Google Scholar]
- 30.Nakanishi A, Imajoh-Ohmi S, Fujinawa T, Kikuchi H, Kanegasaki S. Direct evidence for interaction between COOH-terminal regions of cytochrome b558 subunits and cytosolic 47-kDa protein during activation of an O(2−)-generating system in neutrophils. J Biol Chem. 1992;267:19072–19074. [PubMed] [Google Scholar]
- 31.Leusen JH, de Boer M, Bolscher BG, Hilarius PM, Weening RS, Ochs HD, Roos D, Verhoeven AJ. A point mutation in gp91-phox of cytochrome b558 of the human NADPH oxidase leading to defective translocation of the cytosolic proteins p47-phox and p67-phox. J Clin Invest. 1994;93:2120–2126. doi: 10.1172/JCI117207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park MY, Imajoh-Ohmi S, Nunoi H, Kanegasaki S. Synthetic peptides corresponding to various hydrophilic regions of the large subunit of cytochrome b558 inhibit superoxide generation in a cell-free system from neutrophils. Biochem Biophys Res Commun. 1997;234:531–536. doi: 10.1006/bbrc.1997.6672. [DOI] [PubMed] [Google Scholar]
- 33.Yu L, Cross AR, Zhen L, Dinauer MC. Functional analysis of NADPH oxidase in granulocytic cells expressing a delta488-497 gp91(phox) deletion mutant. Blood. 1999;94:2497–2504. [PubMed] [Google Scholar]
- 34.Dahan I, Issaeva I, Gorzalczany Y, Sigal N, Hirshberg M, Pick E. Mapping of functional domains in the p22(phox) subunit of flavocytochrome b(559) participating in the assembly of the NADPH oxidase complex by “peptide walking”. J Biol Chem. 2002;277:8421–8432. doi: 10.1074/jbc.M109778200. [DOI] [PubMed] [Google Scholar]
- 35.Park MY, Imajoh-Ohmi S, Nunoi H, Kanegasaki S. Peptides corresponding to the region adjacent to His-94 in the small subunit of cytochrome b558 inhibit superoxide generation in a cell-free system from human neutrophils. Biochem Biophys Res Commun. 1994;204:924–929. doi: 10.1006/bbrc.1994.2548. [DOI] [PubMed] [Google Scholar]
- 36.von Lohneysen K, Noack D, Jesaitis AJ, Dinauer MC, Knaus UG. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J Biol Chem. 2008;283:35273–35282. doi: 10.1074/jbc.M804200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nauseef WM, McCormick S, Renee J, Leidal KG, Clark RA. Functional domain in an arginine-rich carboxyl-terminal region of p47phox. J Biol Chem. 1993;268:23646–23651. [PubMed] [Google Scholar]
- 38.Labadia ME, Zu YL, Huang CK. A synthetic peptide containing a predominant protein kinase C site within p47phox inhibits the NADPH oxidase in intact neutrophils. J Leukoc Biol. 1996;59:116–124. doi: 10.1002/jlb.59.1.116. [DOI] [PubMed] [Google Scholar]
- 39.DeLeo FR, Nauseef WM, Jesaitis AJ, Burritt JB, Clark RA, Quinn MT. A domain of p47phox that interacts with human neutrophil flavocytochrome b558. J Biol Chem. 1995;270:26246–26251. doi: 10.1074/jbc.270.44.26246. [DOI] [PubMed] [Google Scholar]
- 40.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–R297. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 41.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 42.D’Addio SM, Prud’homme RK. Controlling drug nanoparticle formation by rapid precipitation. Adv Drug Deliv Rev. 2011;63(6):417–426. doi: 10.1016/j.addr.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 43.De Koker S, De Cock LJ, Rivera-Gil P, Parak WJ, Auzely Velty R, Vervaet C, Remon JP, Grooten J, De Geest BG. Polymeric multilayer capsules delivering biotherapeutics. Adv Drug Deliv Rev. 2011;63(9):748–761. doi: 10.1016/j.addr.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Brandes RP, Schroder K. Composition and Functions of Vascular Nicotinamide Adenine Dinucleotide Phosphate Oxidases. Trends Cardiovasc Med. 2008;18:15–19. doi: 10.1016/j.tcm.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 46.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem. 1997;272:13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- 47.Cayatte AJ, Rupin A, Oliver-Krasinski J, Maitland K, Sansilve-stri-Morel P, Boussard MF, Wierzbicki M, Verbeuren TJ, Cohen RA. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets nadph oxidase. Arterioscler Thromb Vasc Biol. 2001;21:1577–1584. doi: 10.1161/hq1001.096723. [DOI] [PubMed] [Google Scholar]
- 48.Jaquet V, Scapozza L, Clark R, Krause KH, Lambeth JD. Small molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11(10):2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 49.Williams HC, Griendling KK. NADPH oxidase inhibitors: new antihypertensive agents? J Cardiovasc Pharmacol. 2007;50:9–16. doi: 10.1097/FJC.0b013e318063e820. [DOI] [PubMed] [Google Scholar]
- 50.Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol Ther. 2008;120:254–291. doi: 10.1016/j.pharmthera.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 51.Cifuentes ME, Pagano PJ. Targeting reactive oxygen species in hypertension. Curr Opin Nephrol Hypertens. 2006;15:179–186. doi: 10.1097/01.mnh.0000214776.19233.68. [DOI] [PubMed] [Google Scholar]
- 52.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 53.Lange S, Heger J, Euler G, Wartenberg M, Piper HM, Sauer H. Platelet-derived growth factor BB stimulates vasculogenesis of embryonic stem cell-derived endothelial cells by calcium-mediated generation of reactive oxygen species. Cardiovasc Res. 2009;81:159–168. doi: 10.1093/cvr/cvn258. [DOI] [PubMed] [Google Scholar]
- 54.Stielow C, Catar RA, Muller G, Wingler K, Scheurer P, Schmidt HH, Morawietz H. Novel Nox inhibitor of oxLDL-induced reactive oxygen species formation in human endothelial cells. Biochem Biophys Res Commun. 2006;344:200–205. doi: 10.1016/j.bbrc.2006.03.114. [DOI] [PubMed] [Google Scholar]
- 55.Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, Neff C, Shah AM, Wingler K, Schmidt HH. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. 2010;56:490–497. doi: 10.1161/HYPERTENSIONAHA.109.149187. [DOI] [PubMed] [Google Scholar]
- 56.Tsai MH, Jiang MJ. Reactive oxygen species are involved in regulating alpha1-adrenoceptor-activated vascular smooth muscle contraction. J Biomed Sci. 2010;17:67. doi: 10.1186/1423-0127-17-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8(9):pii–e1000479. doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milosevic N, Bekhite MM, Sharifpanah F, Ruhe C, Wartenberg M, Sauer H. Redox stimulation of cardiomyogenesis versus inhibition of vasculogenesis upon treatment of mouse embryonic stem cells with thalidomide. Antioxid Redox Signal. 2010;13:1813–1827. doi: 10.1089/ars.2010.3139. [DOI] [PubMed] [Google Scholar]
- 59.Sancho P, Fabregat I. The NADPH oxidase inhibitor VAS2870 impairs cell growth and enhances TGF-beta-induced apoptosis of liver tumor cells. Biochem Pharmacol. 2011;81:917–924. doi: 10.1016/j.bcp.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 60.Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perry BN, Govindarajan B, Bhandarkar SS, Knaus UG, Valo M, Sturk C, Carrillo CO, Sohn A, Cerimele F, Dumont D, Losken A, Williams J, Brown LF, Tan X, Ioffe E, Yancopoulos GD, Arbiser JL. Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J Invest Dermatol. 2006;126:2316–2322. doi: 10.1038/sj.jid.5700413. [DOI] [PubMed] [Google Scholar]
- 62.Berrios RL, Arbiser JL. Novel antiangiogenic agents in dermatology. Arch Biochem Biophys. 2011;508:222–226. doi: 10.1016/j.abb.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhandarkar SS, Jaconi M, Fried LE, Bonner MY, Lefkove B, Govindarajan B, Perry BN, Parhar R, Mackelfresh J, Sohn A, Stouffs M, Knaus U, Yancopoulos G, Reiss Y, Benest AV, Augustin HG, Arbiser JL. Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J Clin Invest. 2009;119:2359–2365. doi: 10.1172/JCI33877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 65.Sedeek M, Callera G, Montezano A, Gutsol A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM, Hebert RL. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2010;299:F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 66.Garrido-Urbani S, Jemelin S, Deffert C, Carnesecchi S, Basset O, Szyndralewiez C, Heitz F, Page P, Montet X, Michalik L, Arbiser J, Ruegg C, Krause KH, Imhof B. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPAR-alpha mediated mechanism. PLoS One. 2011;6:e14665. doi: 10.1371/journal.pone.0014665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 68.Ray R, Murdoch CE, Wang M, Santos CX, Zhang M, Alom-Ruiz S, Anilkumar N, Ouattara A, Cave AC, Walker SJ, Grieve DJ, Charles RL, Eaton P, Brewer AC, Shah AM. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler Thromb Vasc Biol. 2011;31:1368–1376. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 69.Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, Anilkumar N, Yu B, Dong X, Walker SJ, Brandes RP, Shah AM. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci USA. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, Rosen H. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol. 2010;5:981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brinker AM, Ma J, Lipsky PE, Raskin I. Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae) Phytochemistry. 2007;68:732–766. doi: 10.1016/j.phytochem.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 73.Wu F, Han M, Wilson JX. Tripterine prevents endothelial barrier dysfunction by inhibiting endogenous peroxynitrite formation. Br J Pharmacol. 2009;157:1014–1023. doi: 10.1111/j.1476-5381.2009.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y, Perozzo R, Plastre O, Fioraso-Cartier L, Diebold B, Scapozza L, Nauseef WM, Fieschi F, Krause KH, Bedard K. NOX NADPH oxidase isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol. 2011;164(2b):507–520. doi: 10.1111/j.1476-5381.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 76.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatr. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 77.Kim Y, Kim K, Lee H, Han S, Lee YS, Choe J, Kim YM, Hahn JH, Ro JY, Jeoung D. Celastrol binds to ERK and inhibits FcepsilonRI signaling to exert an anti-allergic effect. Eur J Pharmacol. 2009;612:131–142. doi: 10.1016/j.ejphar.2009.03.071. [DOI] [PubMed] [Google Scholar]
- 78.Borbely G, Szabadkai I, Horvath Z, Marko P, Varga Z, Breza N, Baska F, Vantus T, Huszar M, Geiszt M, Hunyady L, Buday L, Orfi L, Keri G. Small-molecule inhibitors of NADPH oxidase 4. J Med Chem. 2010;53:6758–6762. doi: 10.1021/jm1004368. [DOI] [PubMed] [Google Scholar]
- 79.Vendrov AE, Madamanchi NR, Niu XL, Molnar KC, Runge M, Szyndralewiez C, et al. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J Biol Chem. 2010;285:26545–26557. doi: 10.1074/jbc.M110.143917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Briones MA, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, Quinn MT, Salaices M, Touyz RM. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertension. 2011;5:137–153. doi: 10.1016/j.jash.2011.02.001. [DOI] [PubMed] [Google Scholar]