

Abstract
Several bacterial pathogens decorate their surfaces with sialic acid residues within cell wall components or capsular exopolysaccharides. Sialic acid expression can promote bacterial virulence by blocking complement activation or by engagement of inhibitory sialic-acid binding immunoglobulin-like lectins (Siglecs) on host leukocytes. Expressed at high levels on splenic and lymph node macrophages, sialoadhesin is a unique Siglec with an elongated structure that lacks intracellular signaling motifs. Sialoadhesin allows macrophage to engage certain sialylated pathogens and stimulate inflammatory responses, but the in vivo significance of sialoadhesin in infection has not been shown. We demonstrate that macrophages phagocytose the sialylated pathogen group B Streptococcus (GBS) and increase bactericidal activity via sialoadhesin-sialic acid mediated recognition. Sialoadhesin expression on marginal zone metallophillic macrophages in the spleen trapped circulating GBS and restricted the spread of the GBS to distant organs, reducing mortality. Specific IgM antibody responses to GBS challenge were also impaired in sialoadhesin-deficient mice. Thus sialoadhesin represents a key bridge to orchestrate innate and adaptive immune defenses against invasive sialylated bacterial pathogens.
Keywords: metallophillic macrophage, antibody, siglec, sialic acid
Introduction
The spleen is the largest secondary lymphoid organ in our body, receiving 5% of the cardiac output in blood perfusion, and capable of screening the entire blood volume for pathogens within 20–30 minutes [1]. The crucial role of the spleen in blood filtering is evident in asplenic individuals, who are particularly vulnerable to sepsis with encapsulated bacteria such as Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae [2, 3]. Opening of the arterial blood stream into the marginal sinuses of the spleen reduces blood flow so that pathogens in the systemic circulation can be efficiently phagocytosed by marginal zone macrophages (MZMs) and marginal metallophilic macrophages (MMMs) [4, 5]. Depletion of MZMs and MMMs can result in pathogens escaping to the blood, potentially triggering uncontrolled bacteremia and sepsis [6, 7].
MMMs, but typically not MZMs, express high levels of sialoadhesin (Sn, sialic acid-binding immunoglobulin-like lectin-1 (Siglec-1), CD169) and form an inner ring bordering the marginal zone and the white pulp follicular areas. Among Siglecs, Sn is unique in possessing 17 immunoglobulin-like extracellular domains that may extend its length ∼40 nm beyond the cell surface and recognize sialylated ligands found on many sialylated pathogens, such as sialylated enveloped viruses [8-10], bacteria [11, 12], and protozoa [13]. More recently, Sn has been shown to capture killed sialylated Campylobacter jejuni and able to promote rapid proinflammatory cytokine and type I interferon responses [14]. However, the consequences of Sn-dependent recognition of an invasive sialylated bacteria pathogen on infection outcome have not been addressed.
In this work, we used the sialylated pathogen group B Streptococcus (GBS), a leading cause of human neonatal sepsis and meningitis, as a model for in vitro and in vivo analysis of Sn function. Specifically, we studied whether expression of Sn in MMMs could facilitate splenic trapping of GBS during early infection and/or influence later phase humoral responses, to coordinately combat this invasive blood-borne pathogen.
Materials and Methods
Bacteria and growth condition
GBS of serotypes Ia (A909), Ib (UAB), II (DK23), III (COH1, K79 and D136), and VI (NT-6) are human neonate isolates. COH1ΔNeuA mutant and isogenic COH1 mutants expressing different levels of O-acetylation on the terminal sialic acid were previously described [15, 16]. GBS were grown in Todd-Hewitt broth (Difco, BD Diagnostics) at 37°C without shaking. For functional assays, bacteria were cultivated at 37°C to early log phase and resuspended to OD600 of 0.4 in appropriate buffer to the desired concentrations.
Culture of mouse bone marrow-derived macrophages (MBDMs) and in vitro stimulation with GBS
MBDMs were derived from marrow cells collected from femur and tibia and cultured with conditioned media containing macrophage colony-stimulating factors (M-CSF) for 7 d. MBDMs were then seeded onto 24-well plates at 5 × 105/well with M-CSF (10 ng/ml, R&D Systems) and interferon-α (500 U/ml, PBL IFN Source) to optimize Sn expression. For phagocytosis assays, 2.5 × 106 CFU GBS DK23 was added to 5 × 105 macrophages (MOI = 5). Following 30 min incubation, cells were washed, added medium containing penicillin (5 μg/ml) and gentamicin (100 μg/ml) to kill extracellular bacteria, detached by trypsin, lysed with 0.025% Triton X-100, and plated to enumerate intracellular bacteria. For bacterial killing, 105 GBS DK23 was added to MBDMs at MOI = 0.2 for 1 h, followed by addition of 50 μl of 0.6% Triton X-100 to lyse cells. Recovered GBS were plated for CFU enumeration. For experiments using Sn-neutralizing mAb (clone SER-4, 10 μg/ml), the mAb was added to cells 10 min prior to inoculation with GBS. This neutralizing mAb has been shown to block the sialic acid-mediated interaction between Sn and erythrocytes as well as the interaction between Sn and sialylated meningococcal lipopolysaccharide [11, 17].
Flow cytometry analysis of Sn expression
MBDMs were stimulated with IFN-α (500 U/ml), LPS (1 μg/ml, InvivoGen), lipoteichoic acid (1 μg/ml, InvivoGen), GBS capsule (1 μg/ml, purified as described [15]), or heat-killed GBS (108 CFU) for 2 d. Cells were detached by 5 mM EDTA/PBS, stained with FITC-conjugated rat anti-mouse Sn (clone 3D6, Serotec) and analyzed by flow cytometer (FACSCalibur, BD Biosciences).
Siglec-Fc binding assay
Siglec-Fc and FITC-labeled GBS binding studies were modified from a described method [18]. GBS was labeled with 0.1% FITC/PBS for 30 min at 37°C with rotation. 96-well plates were coated with 1 μg/well protein A in 50 mM carbonate buffer (pH 9.5) overnight at 4°C, washed and blocked with 1% BSA/PBS for 1 h at room temperature. 0.5 μg of human and murine Sn-Fc (produced as described [11]) was immobilized to individual wells for ≥ 3 h at room temperature. 107 FITC labeled-GBS was added to each well, and 96-well plate was centrifuged and incubated for 30 min at 37°C. Initial fluorescence intensity was verified, and the residual fluorescence intensity (excitation, 488 nm; emission, 530 nm) was measured using a CytoFluorII fluorescence plate reader after extensive wash to remove unbound bacteria.
Immunofluorescence microscopy based phagocytosis analysis
GBS DK23 was labeled with PKH26 (Sigma) per manufacturer's instruction. Labeled GBS (2.5 × 106 CFU) were added to 5 × 105 macrophages and incubated for 30 min. Macrophages were washed, fixed, permeabilized, blocked with BSA and stained with AlexaFluor 488® phalloidin (Life Technologies) and DAPI for 30 min at room temperature. Images were acquired using the Olympus FV1000 confocal microscope and FV1000 Viewer software.
Intravenous GBS infection and visualization of GBS in the spleen
All studies were approved by the UCSD Committee on the Use and Care of Animals and performed using accepted veterinary standards. Generation of Sn-deficient mice has previously been described [19]. GBS DK23 was labeled with 5-(and-6)-carboxyfluorescein (Invitrogen) per manufacturer's instructions. Mice were intravenously infected with 2 × 108 CFU of labeled GBS via tail vein. Kidney, lung and spleen were collected for CFU enumeration or frozen in OCT solution 1 h after infection. 5-μm spleen cryostat sections were fixed in acetone, and blocked with 1% BSA/PBST and AVIDIN/BIOTIN blocking kit (Vector Laboratories). Sections were stained with rat mAb anti-mouse Sn (clone SER-4) with Alexa Fluor 647-conjugated goat anti-rat IgG (Life Technologies) followed by second step staining with biotinylated rat mAb anti-mouse F4/80 (clone CI:A3-1, Serotec) or B220 (clone RA3-6B2, BD Biosciences) with Alexa Fluor 555-conjugated streptavidin (Life Technologies). Images were from an Olympus FV1000 confocal microscope using FV1000 Viewer software.
GBS intraperitoneal infection mouse model
WT and Sn-deficient mice (10-12 weeks) were infected intraperitoneally with 108 CFU of GBS DK23, and bacterial CFU in the blood monitored 4 h post-infection. To detect anti-GBS antibody responses, mice (10-12 weeks) were first injected with 0.5 mg of anti-Ly-6G mAb (clone 1A8, BioXcell) intraperitoneally to deplete neutrophils 24 h prior to infection with 2 × 107 CFU of GBS DK23. Mouse survival was monitored twice per day for 14 d at which time mice were euthanized and their blood was collected for antibody titer anaysis.
ELISA of anti-GBS antibody isotypes
Fifty microliter of GBS (OD600 = 1.0) in 0.1 M sodium carbonate buffer (pH 9.5) was added into 96-well plates, spun at 2,000 rpm for 10 min, and incubated overnight at 4°C. GBS-coated plates were blocked with 3%BSA/PBST for 1 h at room temperature, and then mouse sera was added in 3-fold serial dilutions and incubated for 2 h at room temperature. Antibody titers were determined with horseradish peroxidase-conjugated immunoglobulin isotype-specific antibodies (Southern Biotechnology) and TMB substrate solution. OD 450 nm was measured with a SpectraMax M3 microplate reader.
ELISA of total antibody isotypes
96-well plates were coated with goat anti-mouse IgG+IgA+IgM (Southern Biotechnology, 0.5 μg/well in 0.1 M sodium carbonate buffer, pH 9.5) at 4°C overnight. Plates were blocked with 3%BSA/PBST for 1 h and sera collected from GBS-infected mice was added (1:5,000 dilution for IgM and IgA,; 1:50,000 dilution for IgG) and incubated for 2 h at room temperature. Antibodies were detected by horseradish peroxidase-conjugated immunoglobulin isotype-specific antibodies and TMB substrate solution. OD 450 nm was measured with a SpectraMax M3 microplate reader.
Results
Sialiadhesin (Sn) binds to sialylated Group B Streptococcus (GBS) and its expression is upregulated by bacterial components and inflammatory stimuli
GBS is a leading cause of neonatal pneumonia and sepsis, and its surface capsular polysaccharide is invariably capped with a terminal α2-3-linked sialic acid (Sia) known to impair phagocytosis and dampen neutrophil bactericidal activities via engaging inhibitory Siglecs and to block complement deposition [18, 20-23]. Sn, a unique Siglec possessing 17 immunoglobulin-like extracellular domains, has been reported to recognize sialylated ligands found on many sialylated pathogens. We sought to determine if Sn could recognize GBS in a Sia-dependent manner as a defense strategy to counteract suppressive signals transduced by inhibitory Siglecs. Seven GBS strains (A909, UAB, DK23, COH1, K79, D136 and NT-6) tested here all bound to human Sn (hSn) and murine Sn (mSn), but the Sia-negative COH1ΔNeuA mutant did not (Fig. 1A).
Figure 1. Sn binds Group B Streptococcus (GBS) and is upregulated by inflammatory stimuli.
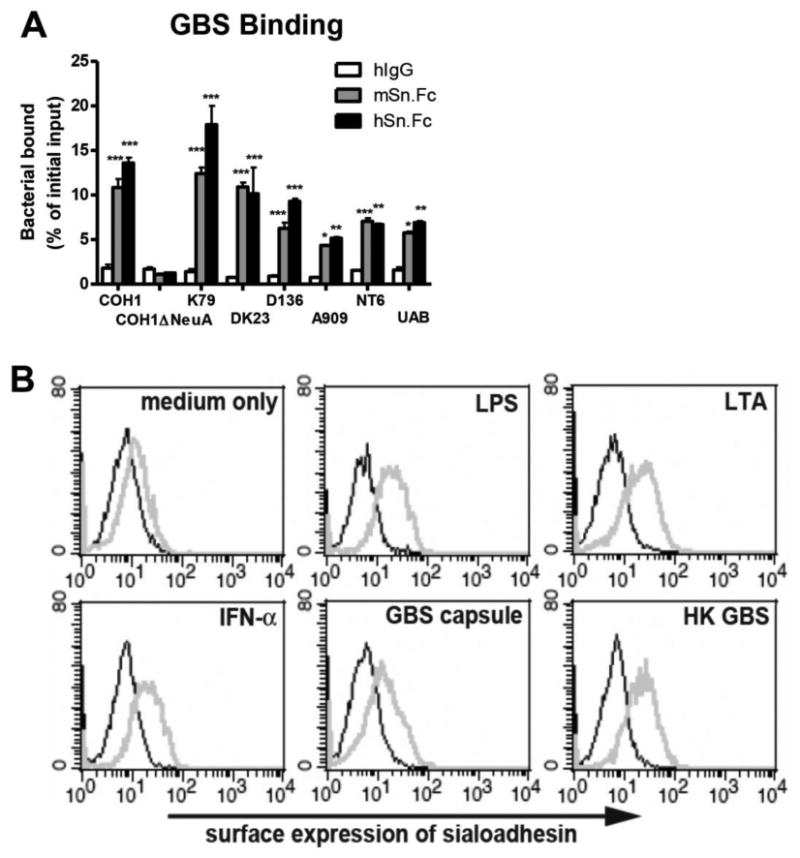
(A) FITC-labeled GBS were added to plates coated with human Sn (hSn), murine Sn (mSn) or control human IgG (hIgG). Binding of bacteria to applied proteins was measured using a fluorescent plate reader after extensive washes. Data shown are means ± sem and the GBS binding difference between control and Sn was determined by two-way ANOVA with Bonferroni posttest. ***P < 0.001; **P < 0.01; *P < 0.05. Experiments have been done with three biological replicates and repeated twice. (B) MBDMs were stimulated with various bacterial components or IFN-α for 2 d, stained with anti-Sn mAb and subjected to FACS analysis to examine Sn expression. Black thin line: isotype control; gray thick line: anti-Sn mAb. Representative data from one of the two reproducible independent experiments was shown.
Expression of Sn on human monocytes is upregulated by IFN-α and agonists for Toll-like receptors [24]. In addition, under infection or pathological conditions, expression of Sn on circulating human monocytes and tissue macrophages can be increased and correlated to disease progression [24-29]. We observed that IFN-a significantly increased Sn expression on mouse bone marrow-derived macrophages (MBDMs), as did lipopolysaccharide (LPS), lipoteichoic acid (LTA), GBS capsular polysaccharide preparation and heat-killed GBS (HK GBS) (Fig. 1B). Upregulation of Sn expression during infection could theoretically influence macrophage phagocytic bactericidal activity to GBS.
Sn is required for efficient uptake and clearance of GBS
To investigate the role of Sn in macrophage phagocytic and bactericidal ability, MBDMs were cultured with IFN-α for 2 d to upregulate Sn expression prior to GBS infection. Compared to Sn-deficient MBDMs, wild type (WT) MBDMs showed greater association with, and internalization of, live PKH26-labeled GBS (Fig. 2A). Corroborating a critical role of Sn in GBS phagocytosis, adding a Sn neutralizing mAb significantly reduced the ability of WT MBDMs to phagocytose GBS to the level of Sn-deficient cells (Fig. 2B). In addition, surviving GBS were also significantly increased in the infected Sn-deficient cells (Fig. 2C). To confirm recognition of GBS capsular Sia by Sn specifically contributes to macrophage phagocytic and bactericidal activity, we studied another GBS serotype III strain, COH1, and isogenic mutants derived from this strain that either lack Sia expression or express differing levels of Sia O-acetylation. O-acetylation is known to interfere with Siglec-mediated Sia binding [15, 16]. WT MBDMs phagocytosed more WT GBS and mutant GBS with reduced Sia O-acetylation than did SN-deficient MBDMs. On the other hand, GBS lacking Sia on their capsule, or GBS with high level of O-acetylation on the capsule Sia, were phagocytosed equally by WT and Sn-deficient MBDMs (Supplementary Fig. 1A and 1B). Non-sialylated GBS and GBS with reduced capsular Sia O-acetylation levels were killed more efficiently by WT BMDMs than Sn-deficient macrophages (Supplementary Fig, 1C). Our findings demonstrate that Sn is critical for macrophage recognition of GBS and contributes to bactericidal activity against the sialylated pathogen.
Figure 2. Sn contributes to phagocytic and bactericidal activity against GBS.
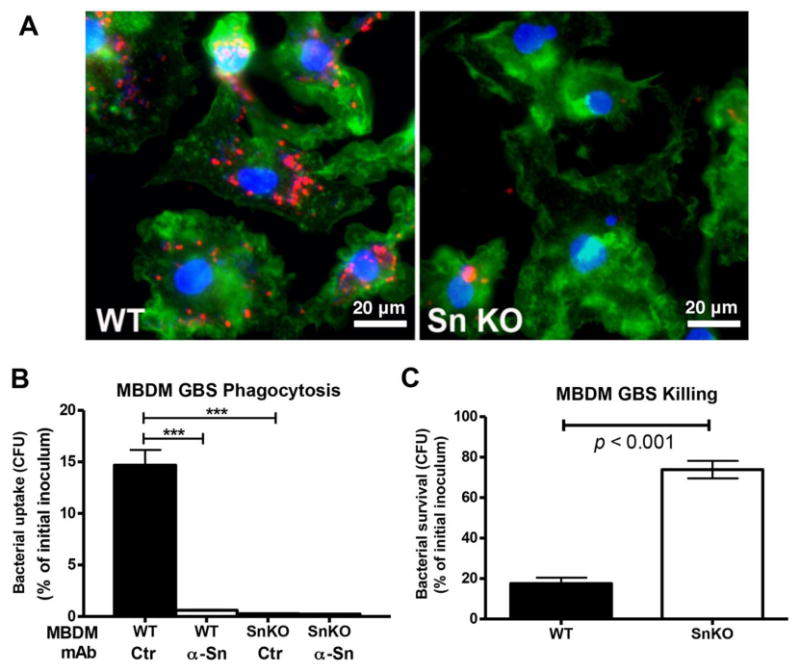
(A) Confocal microscopy images show significantly reduced phagocytosis of GBS in the Sn-deficient (Sn KO) MBDMs. Macrophages were infected with GBS at MOI of 5 for 30 min, and washed extensively before staining cells with actin. Red: GBS; Green: actin; blue, nucleus. (B) Quantitative phagocytosis analysis. MBDMs were infected with GBS in the presence or absence of Sn neutralizing mAb, followed by addition of antibiotics to only recover and enumerate intracellular bacteria CFU. (C) Survival of GBS upon co-incubation with MBDMs. Macrophages were infected with GBS at MOI of 0.2, and surviving GBS was enumerated 1 h post infection by serial plating. Difference between two groups was calculated one-way ANOVA with Tukey's multiple comparison test (B) or by unpaired t test (C). ***P < 0.001. Representative image (A) and data (B and C) were shown from 3 independent experiments.
Efficient capture and control GBS dissemination in vivo by Sn
Splenic MZ macrophages are known to actively sample and trap blood-borne pathogens. Since MMMs express high levels of Sn, we examined whether Sn facilitated MMM recognition of GBS in vivo. 5-(and-6)-carboxyfluorescein-labeled GBS were injected intravenously into mice, and the distribution of GBS was visualized in the spleen co-stained with mAb anti-mouse F4/80 (red pulp macrophage marker) and B220 (B cell marker). Strong co-localization of GBS and Sn+ MMMs was observed in WT animals, and the majority of the GBS were found trapped within the MMMs, with minor spreading into red pulp macrophages, probably due to direct deposit of GBS via small branches of the central artery ending in the red pulp [30] (Fig. 3A, left panel; right panel is for higher magnification). Sn-deficient spleens were still able to capture GBS in the marginal zone, which may reflect a contribution of MZMs or other pattern recognition receptors expressed on the MMMs. Notably, there were significant quantities of GBS identified around the red pulp area, most of them colocalized with the F4/80+ macrophages (Fig 3A, middle panel), while no GBS were observed in the white pulp B cell area of either the WT or Sn-deficient spleens (Fig. 3B). An extended image set with separated channel color is available in Supplementary Fig. 2. In general, more GBS were identified in spleens of Sn-deficient mice compared to WT animals (Fig. 3A and 3B), as confirmed by quantitation of GBS colony-forming unit (CFU) in the spleen (Fig. 3E). Consistent with the greater patrolling and engagement of GBS by Sn+ MMMs in the WT animals, reduced GBS CFUs were recovered from the WT kidney, lung and spleen (Fig. 3C-3E). Our findings indicate that efficient capture of GBS by Sn expressed on MMMs can facilitate GBS clearance locally in the spleen and reduce the magnitude of GBS dissemination to other organs.
Figure 3. Sn facilitates marginal metallophilic macrophage trapping of GBS to limit pathogen dissemination.
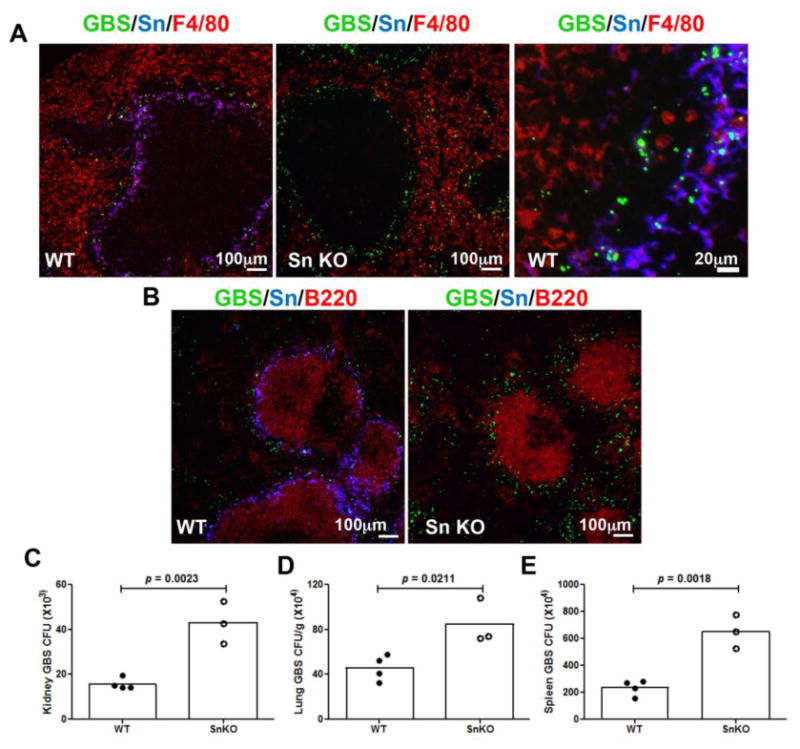
WT and Sn-deficient mice were intravenously injected with 5-(and-6)-carboxyfluorescein labeled GBS and kidney, lung and spleen were collected 1 h post infection. Sections were stained with mAb anti-mouse Sn, F4/80 and B220. Representative images of spleen sections are shown in A and B. Scale bar is 100 μm for all panels except 20 μm for the right panel of A. Comparison of bacterial counts (expressed in CFU) recovered from the kidney (C) lung (D) and spleen (E) of WT mice and Sn-deficient mice 1h post infection. Difference between two groups was calculated by unpaired t test. Representative data (n=3-4 mice each group) were shown from 2 independent experiments.
Sn is important for survival and production of anti-GBS antibodies in neutropenic mice
To date there have been no in vivo studies addressing a role for Sn in controlling active infection. Since expression of Sn is required for macrophages to recognize and clear GBS in vitro and in vivo, we examined whether Sn could impact mouse survival upon GBS challenge. In normal mice, GBS was efficiently removed from the circulation (at least 5 log reduction) within 1 h (Supplementary Fig. 3) after intravenous challenge, Thus, a GBS intraperitoneal challenge model was adapted to provoke systemic infection, as the bacterium can gain access to the blood circulation via this route. WT and Sn-deficient mice were infected intraperitoneally with GBS, and blood CFU was enumerated 4 h later. As shown in Fig. 4A, greater numbers of GBS were recovered from blood of Sn-deficient mice, indicating impaired bacterial clearance. However, when mouse survival was monitored through 7 d post-infection, no differences emerged between the two groups (Fig. 4B). We hypothesized more abundant and rapidly mobilized host neutrophil response may obscure the importance of Sn in this model, since high levels of Sn expression are restricted to MMMs and SCS macrophages in the spleen and lymph nodes, respectively. To study the independent contribution of Sn in control of bacterial infection in the absence of neutrophils, neutrophils were depleted from mice 24 h prior to GBS infection. None of the WT mice died during the 14 d period, but 40% of the Sn-deficient mice died, indicating that absence of Sn rendered mutant mice significantly more susceptible to GBS infection under the neutropenic condition (Fig. 4C).
Figure 4. Sn is important for mouse survival during neutropenia.
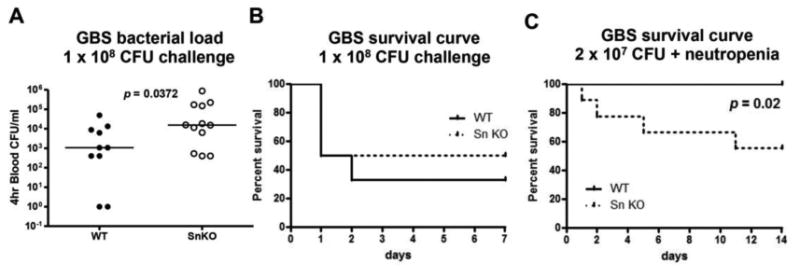
WT and Sn-deficient mice were intraperitoneally infected with GBS, blood was collected 4 h post-infection for CFU enumeration (A) and survival was monitored for 7 d (B). WT and Sn-deficient mice were intraperitoneally challenged with GBS after neutrophil depletion and the survival was recorded for 14 d with n = 10 for WT and n = 9 for Sn-deficient mice (C). Long-rank test was used for statistical analysis for (B) and (C). Representative data were shown from 2 independent experiments.
Sn-deficient mice were previously reported to have an intrinsic reduction in serum IgM amount [19]; therefore, we asked whether serum Ig changes could be identified following GBS challenge. As shown in Fig. 5A, serum IgM and IgG amounts were reduced in the Sn-deficient mice. Since Sn is critical for the phagocytosis of GBS, and SCS Sn+ macrophages can transfer antigens to initiate adaptive immune responses, we then further examined whether anti-GBS humoral immune responses were altered in Sn-deficient mice. We found a significant reduction of the anti-GBS IgM titer in the Sn-deficient mice (Fig. 5B), while similar anti-GBS IgG titers were observed in the two groups (Fig. 5C). Reduced anti-GBS IgMs may further increase the susceptibility of Sn-deficient mice to GBS infection in addition to impairment of immediate innate immune responses, such the macrophage phagocytosis and bactericidal activity.
Figure 5. Sn is important for production of anti-GBS antibodies during neutropenia.
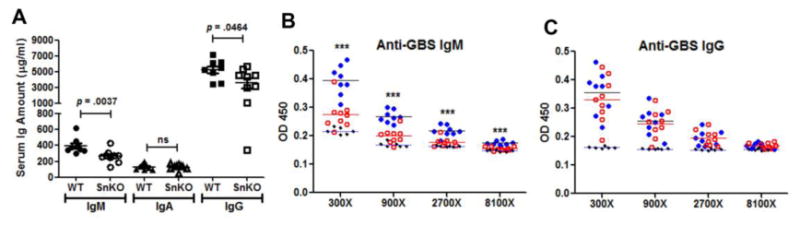
(A) Decreased total IgM and IgG serum levels after GBS challenge in Sn-deficient mice. Total serum immunoglobulins from GBS-infected mice were measured by isotype specific ELISA (n = 8 for WT and n = 9 for Sn-deficient mice). Mann-Whitney test was used for statistical analysis. Anti-GBS IgM (B) and anti-GBS IgG (C) titers were measured from GBS-infected WT (filled blue circle, n = 8), GBS-infected Sn-deficient (open red circle, n = 9), and uninfected WT mice (filed black circle, n = 6). Difference among groups at each dilution was tested by one-way ANOVA with Tukey's multiple comparison test. *** P < 0.001. Representative data were shown from 2 independent experiments.
Discussion
Efficient pathogen elimination is crucial for the host, and will determine the extent and pathological consequences of infection. To combat blood-borne infectious microbes, the spleen is poised to quickly sample and trap circulating pathogens in strategically localized MZMs and MMMs [31]. These rapid innate immune mechanisms help reduce pathogen load until an adaptive immune response, more specific but temporally delayed, is mounted. Here we provide the in vivo demonstration of Sn function in defense against an invasive sialylated bacterial pathogen. This specialized macrophage Siglec not only provides innate immune functions such as phagocytosis and bactericidal activities, but also supports the development of proper humoral responses against the microbe.
Most Siglecs have one or more immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in the cytosolic tail and function as inhibitory receptors. Sialylated pathogens can even exploit ITIM-containing Siglecs to dampen innate immune responses therefore to enhance their survival [18, 32, 33], in addition to reduce complement deposition and activation on the bacterial surface to impair opsonophagocytosis [20, 21]. In contrast, Sn, upregulated upon infection, recognizes the very same Sia epitope and helps preserve a critical innate immune function of these phagocytic cells. As a consequence, Sn has the right properties to mediate critical initial contacts with sialylated pathogens to function directly as a phagocytic receptor or to coordinate with other pattern recognition receptors to mediate efficient phagocytosis. Our data supports this idea showing that applying anti-Sn neutralizing mAb nearly eliminated the phagocytic ability and significantly reduced the bactericidal activity of macrophages, even as they are expected to express numerous other pattern recognition and scavenger receptors (Fig. 2B).
The humoral response provides protective circulating antibodies to neutralize or coat pathogens and facilitate subsequent opsonophagocytosis. The ability of B lymphocytes to interact with an antigen involves antigen-trapping, depositing, and transport systems localized to the major antigen entrance site. Splenic MZ macrophages can facilitate antibody production by transferring antigen captured from the circulation to MZ B cells [4, 34]. In addition, migration of splenic follicular B cells to the outer follicle is induced upon binding of oxysterol produced by MMMs [35-37]. SCS Sn+ macrophages in the LNs transport antigens to invariant natural killer T cells, dendritic cells and B cells, and thus promote the immune responses against invaders [38-40]. Taking all the factors together, we speculate that MMMs are not only required for early antigen trapping and clearance but may also represent a crucial compartment for antigen transferring to B cells and follicular dendritic cells. Although Sn-deficient mice had MMMs located in the right location, they exhibited a small decrease of B220+ cells in the spleen and reduction of serum IgM levels [19]. In response to GBS challenge, Sn-deficient animals showed reduced total serum IgM levels and specific anti-GBS IgM antibodies. Although the production of anti-GBS IgG antibodies was not altered in those Sn-deficient animals, they had reduced total serum IgG levels after GBS challenge. Our findings suggest that expression of Sn on MMMs is required for them to elicit specific humoral responses to counteract invading pathogens.
Whether through specific de novo biosynthesis, scavenging or precursor scavenging, many pathogens including GBS, Campylobacter jejuni, Haemophilus influenzae, Neisseria gonorrhoeae, Neisseria meningitides, Escherichia coli K1 and Pasteurella multocida can display Sias on their cell surface as a method of molecular mimicry, counteracting complement activation and/or engaging inhibitory ITIM-bearing Siglecs on leukocytes. The specialized macrophage Sn receptor has a conserved binding specificity that happens to mirror the type and linkages of Sias expressed by the pathogens mentioned above. Understanding how Sn promotes Sia-dependent phagocytosis and stimulates antibody responses during such infections highlights an additional complexity in the evolutionary arms race between pathogen and host immune defense, wherein microbial glycan expression and its recognition strongly influence outcome.
Supplementary Material
Acknowledgments
This work was supported by the NIH/NHLBI Programs of Excellence in Glycosciences grant P01HL107150 to A.V. and V.N. and by a Wellcome Trust Senior Fellowship WT081882 to P.R.C. We thank technical support from the UCSD Histology Core (Nissi Varki, Director) and Patrick Secrest for mouse husbandry.
Footnotes
Author Contributions: Y.C., A.V. and V.N. designed research. Y.C., J.O., and A.L. performed research. P.R.C. provided vital reagents. All authors analyzed data. Y.C., P.R.C., A.V. and V.N. wrote the paper.
Conflict of Interest: The authors have no financial conflict of interest with this work.
References
- 1.Eichner ER. Splenic function: normal, too much and too little. Am J Med. 1979;66:311–320. doi: 10.1016/0002-9343(79)90554-0. [DOI] [PubMed] [Google Scholar]
- 2.Davidson RN, Wall RA. Prevention and management of infections in patients without a spleen. Clin Microbiol Infect. 2001;7:657–660. doi: 10.1046/j.1198-743x.2001.00355.x. [DOI] [PubMed] [Google Scholar]
- 3.Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23:740–780. doi: 10.1128/CMR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 5.den Haan JM, Kraal G. Innate immune functions of macrophage subpopulations in the spleen. J Innate Immun. 2012;4:437–445. doi: 10.1159/000335216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aichele P, Zinke J, Grode L, Schwendener RA, Kaufmann SH, Seiler P. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol. 2003;171:1148–1155. doi: 10.4049/jimmunol.171.3.1148. [DOI] [PubMed] [Google Scholar]
- 7.Seiler P, Aichele P, Odermatt B, Hengartner H, Zinkernagel RM, Schwendener RA. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur J Immunol. 1997;27:2626–2633. doi: 10.1002/eji.1830271023. [DOI] [PubMed] [Google Scholar]
- 8.Rempel H, Calosing C, Sun B, Pulliam L. Sialoadhesin expressed on IFN-induced monocytes binds HIV-1 and enhances infectivity. PLoS One. 2008;3:e1967. doi: 10.1371/journal.pone.0001967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanderheijden N, Delputte PL, Favoreel HW, Vandekerckhove J, Van Damme J, van Woensel PA, Nauwynck HJ. Involvement of sialoadhesin in entry of porcine reproductive and respiratory syndrome virus into porcine alveolar macrophages. J Virol. 2003;77:8207–8215. doi: 10.1128/JVI.77.15.8207-8215.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou Z, Chastain A, Moir S, Ford J, Trandem K, Martinelli E, Cicala C, Crocker P, Arthos J, Sun PD. Siglecs facilitate HIV-1 infection of macrophages through adhesion with viral sialic acids. PLoS One. 2011;6:e24559. doi: 10.1371/journal.pone.0024559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones C, Virji M, Crocker PR. Recognition of sialylated meningococcal lipopolysaccharide by Siglecs expressed on myeloid cells leads to enhanced bacterial uptake. Mol Microbiol. 2003;49:1213–1225. doi: 10.1046/j.1365-2958.2003.03634.x. [DOI] [PubMed] [Google Scholar]
- 12.Heikema AP, Bergman MP, Richards H, Crocker PR, Gilbert M, Samsom JN, van Wamel WJ, Endtz HP, van Belkum A. Characterization of the specific interaction between sialoadhesin and sialylated Campylobacter jejuni lipooligosaccharides. Infect Immun. 2010;78:3237–3246. doi: 10.1128/IAI.01273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monteiro VG, Lobato CS, Silva AR, Medina DV, de Oliveira MA, Seabra SH, de Souza W, DaMatta RA. Increased association of Trypanosoma cruzi with sialoadhesin positive mouse macrophages. Parasitol Res. 2005;97:380–385. doi: 10.1007/s00436-005-1460-1. [DOI] [PubMed] [Google Scholar]
- 14.Klaas M, Oetke C, Lewis LE, Erwig LP, Heikema AP, Easton A, Willison HJ, Crocker PR. Sialoadhesin promotes rapid proinflammatory and type I IFN responses to a sialylated pathogen, Campylobacter jejuni. J Immunol. 2012;189:2414–2422. doi: 10.4049/jimmunol.1200776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis AL, Nizet V, Varki A. Discovery and characterization of sialic acid O-acetylation in group B Streptococcus. Proc Natl Acad Sci U S A. 2004;101:11123–11128. doi: 10.1073/pnas.0403010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiman S, Dahesh S, Carlin AF, Varki A, Nizet V, Lewis AL. Genetic and biochemical modulation of sialic acid O-acetylation on group B Streptococcus: phenotypic and functional impact. Glycobiology. 2009;19:1204–1213. doi: 10.1093/glycob/cwp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crocker PR, Gordon S. Mouse macrophage hemagglutinin (sheep erythrocyte receptor) with specificity for sialylated glycoconjugates characterized by a monoclonal antibody. J Exp Med. 1989;169:1333–1346. doi: 10.1084/jem.169.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlin AF, Uchiyama S, Chang YC, Lewis AL, Nizet V, Varki A. Molecular mimicry of host sialylated glycans allows a bacterial pathogen to engage neutrophil Siglec-9 and dampen the innate immune response. Blood. 2009;113:3333–3336. doi: 10.1182/blood-2008-11-187302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oetke C, Vinson MC, Jones C, Crocker PR. Sialoadhesin-deficient mice exhibit subtle changes in B- and T-cell populations and reduced immunoglobulin M levels. Mol Cell Biol. 2006;26:1549–1557. doi: 10.1128/MCB.26.4.1549-1557.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubens CE, Wessels MR, Heggen LM, Kasper DL. Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci U S A. 1987;84:7208–7212. doi: 10.1073/pnas.84.20.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wessels MR, Rubens CE, Benedi VJ, Kasper DL. Definition of a bacterial virulence factor: sialylation of the group B streptococcal capsule. Proc Natl Acad Sci U S A. 1989;86:8983–8987. doi: 10.1073/pnas.86.22.8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marques MB, Kasper DL, Pangburn MK, Wessels MR. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect Immun. 1992;60:3986–3993. doi: 10.1128/iai.60.10.3986-3993.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vimr E, Lichtensteiger C. To sialylate, or not to sialylate: that is the question. Trends Microbiol. 2002;10:254–257. doi: 10.1016/s0966-842x(02)02361-2. [DOI] [PubMed] [Google Scholar]
- 24.York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and Toll-like receptor agonists. Arthritis Rheum. 2007;56:1010–1020. doi: 10.1002/art.22382. [DOI] [PubMed] [Google Scholar]
- 25.Bao G, Han Z, Yan Z, Wang Q, Zhou Y, Yao D, Gu M, Chen B, Chen S, Deng A, Zhong R. Increased Siglec-1 expression in monocytes of patients with primary biliary cirrhosis. Immunol Invest. 2010;39:645–660. doi: 10.3109/08820139.2010.485625. [DOI] [PubMed] [Google Scholar]
- 26.Biesen R, Demir C, Barkhudarova F, Grun JR, Steinbrich-Zollner M, Backhaus M, Haupl T, Rudwaleit M, Riemekasten G, Radbruch A, Hiepe F, Burmester GR, Grutzkau A. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis Rheum. 2008;58:1136–1145. doi: 10.1002/art.23404. [DOI] [PubMed] [Google Scholar]
- 27.Cornelissen M, van der Kuyl AC, van den Burg R, Zorgdrager F, van Noesel CJ, Goudsmit J. Gene expression profile of AIDS-related Kaposi's sarcoma. BMC Cancer. 2003;3:7. doi: 10.1186/1471-2407-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Kuyl AC, van den Burg R, Zorgdrager F, Groot F, Berkhout B, Cornelissen M. Sialoadhesin (CD169) expression in CD14+ cells is upregulated early after HIV-1 infection and increases during disease progression. PLoS One. 2007;2:e257. doi: 10.1371/journal.pone.0000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiong YS, Zhou YH, Rong GH, Wu WL, Liang Y, Yang ZX, Geng HL, Zhong RQ. Siglec-1 on monocytes is a potential risk marker for monitoring disease severity in coronary artery disease. Clin Biochem. 2009;42:1057–1063. doi: 10.1016/j.clinbiochem.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 30.Kraal G. Cells in the marginal zone of the spleen. Int Rev Cytol. 1992;132:31–74. doi: 10.1016/s0074-7696(08)62453-5. [DOI] [PubMed] [Google Scholar]
- 31.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, van Rooijen N, Mempel TR, Whelan SP, von Andrian UH. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 32.Cao H, Crocker PR. Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology. 2011;132:18–26. doi: 10.1111/j.1365-2567.2010.03368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khatua B, Bhattacharya K, Mandal C. Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through Siglec-9. J Leukoc Biol. 2012;91:641–655. doi: 10.1189/jlb.0511260. [DOI] [PubMed] [Google Scholar]
- 34.Kraal G, Mebius R. New insights into the cell biology of the marginal zone of the spleen. Int Rev Cytol. 2006;250:175–215. doi: 10.1016/S0074-7696(06)50005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. Int Immunol. 2010;22:413–419. doi: 10.1093/intimm/dxq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, Guerini D, Baumgarten BU, Roggo S, Wen B, Knochenmuss R, Noel S, Gessier F, Kelly LM, Vanek M, Laurent S, Preuss I, Miault C, Christen I, Karuna R, Li W, Koo DI, Suply T, Schmedt C, Peters EC, Falchetto R, Katopodis A, Spanka C, Roy MO, Detheux M, Chen YA, Schultz PG, Cho CY, Seuwen K, Cyster JG, Sailer AW. Oxysterols direct immune cell migration via EBI2. Nature. 2011;475:524–527. doi: 10.1038/nature10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, Yu J, Sutton SW, Qin N, Banie H, Karlsson L, Sun S, Lovenberg TW. Oxysterols direct B-cell migration through EBI2. Nature. 2011;475:519–523. doi: 10.1038/nature10226. [DOI] [PubMed] [Google Scholar]
- 38.Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, Cerundolo V, Batista FD. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol. 2010;11:303–312. doi: 10.1038/ni.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii S, Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Pomares L, Gordon S. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol. 2012;33:66–70. doi: 10.1016/j.it.2011.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.