

Abstract
Both the induction of SPARC expression and the loss of the p53 tumor suppressor gene are changes that occur early in glioma development. Both SPARC and p53 regulate glioma cell survival by inverse effects on apoptotic signaling. Therefore, during glioma formation, the upregulation of SPARC may cooperate with the loss of p53 to enhance cell survival. This study determined whether the loss of Sparc in astrocytes that are null for p53 would result in reduced cell survival and tumor formation and increased tumor immunogenicity in an in vivo xenograft brain tumor model. In vitro, the loss of Sparc in p53-null astrocytes resulted in an increase in cell proliferation, but a loss of tumorigenicity. At 7 days after intracranial implantation, Sparc-null tumors had decreased tumor cell survival, proliferation, and reduced tumor size. The loss of Sparc promoted microglia/macrophage activation and phagocytosis of tumor cells. Our results indicate that the loss of p53 by deletion/mutation in the early stages of glioma formation may cooperate with the induction of SPARC to potentiate cancer cell survival and escape from immune surveillance.
Keywords: SPARC, Sparc-null, p53, p53-null, TP53, glioma, glioblastoma, macrophage, microglia, immunosuppression, xenograft
INTRODUCTION
Loss of the p53 tumor suppressor gene has been described as an early event in low-grade astrocytoma formation and progression to secondary glioblastoma (GBM) with estimates of 59–77% p53 mutation when studies consider only astrocytomas or secondary GBMs (24–26, 29). Mutations or deletions of p53 were also found in 76.5% of glioma cell lines, often in combination with the loss of other tumor suppressor genes (14). Re-introduction of p53 in glioma cell lines that have mutant or absent p53 results in reduced tumor cell growth both in vitro and in vivo (2, 3, 16). On the other hand, astrocytes cultured from p53 knockout mice, which are wild-type for Sparc, form colonies in soft agar and form tumors in nude mice (6, 7, 42).
Our lab found that SPARC expression is increased in progressive and primary gliomas (grades II–IV) (31), and therefore may also be an early event in glioma formation. We and others have shown that SPARC increases glioma cell survival under conditions of serum-withdrawal (38, 39) or when treated with the chemotherapy drug temozolomide (37). However, despite the positive effect of SPARC on cell survival, we have shown that SPARC suppresses glioma cell proliferation in vitro (30, 40, 41) and glioma tumor growth in vivo (36, 40). Therefore, the early upregulation of SPARC may cooperate with the loss of p53 to bring about glioma formation and development by enhancing cell survival. In this study, we sought to determine whether the loss of Sparc in astrocytes that are null for p53 would result in reduced cell survival and tumor formation in an in vivo xenograft brain tumor model.
Microglia/macrophages (MG/MP) are a major cellular constituent of gliomas. In a study analyzing 11 human glioblastoma tumors, MG/MP content ranged from 8–78% with a mean of 45% (22); and in rodent intracranial glioma models, the MG/MP content ranged from 5–21% (4, 5, 21). However, the scientific literature presents conflicting data with regard to the role of MG/MP in glioma growth and development. Some studies provide evidence that MG/MP promote glioma growth and invasion, while other studies suggest that MG/MP inhibit glioma progression. As SPARC has been shown to suppress the infiltration of macrophages and other immune cells into tumor tissue (1, 28, 32, 34), we also analyzed the MG/MP content of p53-null/Sparc-wt and p53-null/Sparc-null astrocyte xenograft tumors to assess the role of SPARC in glioma-induced immunosuppression in a model of early glioma formation.
We found that the loss of Sparc in p53-null astrocytes results in an increase in cell proliferation; however, there is an inhibition of growth in soft agar. When the astrocytes are grown intracranially, the loss of Sparc results in decreased tumor cell proliferation and a reduced tumor size at 7 days post-implantation and promotes MG/MP activation and the phagocytosis of tumor cells.
METHODS
Mice and breeding
Sparc-null mice on a mixed 129SV/C57BL/6 background were originally obtained from Dr. Chin Howe at the Wistar Institute, Philadelphia, PA. The null mice were backcrossed with B6129SF1/J (Jackson Laboratory), which retained the C57BL/6 background. Heterozygotes were obtained to derive the Sparc-wt (wt) controls and Sparc-nulls (null). Homozygous C57BL/6 TSG-p53 knockout mice (P53N12-M) were obtained from Taconic. The p53-null and Sparc-null mice were mated to generate heterozygotes. Heterozygotes were then mated to generate neonates.
Astrocyte isolation
Using IACUC approved protocols, 1–2 day old neonates were sacrificed and the brains were removed and placed in L15 medium. After the meninges were removed, the medium was aspirated and the brain tissue was minced and disaggregated using trypsin at 37°C for 30 min, vortexing every 5 min. DMEM + 10% FBS was then added and the tissue was centrifuged at 1000 rpm for 5 min. The supernatant was aspirated and the cells were resuspended in fresh DMEM + 10% FBS. The cells were triturated by pipette and then by syringe through a 21 gauge needle. Next, the cells were transferred to T75 flasks pre-coated with 12.5 μg/ml poly-L-lysine, supplemented with DMEM + 10% FBS, and incubated at 37°C. Astrocytes were isolated from cerebral cortex cultures as previously described (23). Briefly, the mixed cultures were grown for 10 days, then agitated by shaking at 100 rpm overnight to remove oligodendrocytes, and pulsed twice with Ara-C (20 μM) to kill dividing cells. The remaining cells were replated and defined as passage 1. Four p53-null/Sparc-null clones were expanded and designated #2, #6, #11, and #30. The Ast11.9 and Ast11.9–2 cell lines are genetically matched p53-null/Sparc-wt controls and were provided by Dr. Oliver Bögler.
Astrocyte p53 genotyping
Southern blot analysis was used for p53 genotyping of astrocytes. DNA was isolated from mouse tails using the DNeasy Blood & Tissue kit (69504, Qiagen). Equal concentrations of DNA from each mouse were digested with BamHI and then electrophoresed on a 1% agarose gel. Gels were then transferred onto Hybond N membrane (GE Healthcare), after denaturing in NaOH /NaCl for 2 × 25 min, followed by neutralization in Tris/NaCl for 2 × 30 min. Transfers were done with 20x SSC buffer overnight. Membranes were rinsed briefly in 2x SCC buffer, dried on filter paper, and UV crosslinked in a Stratagene Crosslinker. Blots were prehybridized for 4 hrs and then hybridized overnight with a P32 random primer labeled murine p53 cDNA as a probe as previously described (6). Knockouts were verified by loss of the wild-type p53 band (~5 kb) and presence of the p53 mutant band (~6.5 kb).
Astrocyte Sparc genotyping
Sparc genotyping was assessed using PCR with 0.2 μM primers including the Sparc forward primer: GAT GAG GGT GGT CTG GCC CAG CCC TAG ATG CCC CTC AC, the Sparc reverse primer: CAC CCA CAC AGC TGG GGG TGA TCC AGA TAA GCC AAG, and the Neomycin reverse primer: GTT GTG CCC AGT CAT AGC CGA ATA GCC TCT CCA CCC AAG. PCR reactions were performed with 100 ng of mouse genomic DNA in a 25 μl reaction including PCR buffer (10 nM Tris-HCL [pH 9.0], 50 mM KCl, 2.5 mM MgCl2, and 0.1% Triton X-100), 0.5 units of Taq DNA polymerase, and 0.2 mM of dNTPs. The reaction was carried out with an initial 1 min at 96°C, followed by 40 cycles of denaturation at 96°C for 45 sec, annealing at 70°C for 45 sec, and extension at 72°C for 6 min, followed by 10 min at 72°C and then a 4°C hold. The PCR product sizes are approximately 300 bp for the wild-type allele and 550 bp for the Sparc-null insertion.
Western blot analysis for SPARC
Protein was isolated using a single detergent lysis buffer as reported (12, 30), and protein concentration was determined using the BCA protein assay (Pierce). DMEM was conditioned by cells for 24 hrs as previously reported (19). Equal concentration of protein or volume of conditioned medium were evaluated by Western blotting as previously described (30) using anti-mouse SPARC antibody (R&D Systems, AF942).
Proliferation assay
For each cell type, 1.5 × 103 cells were seeded into each of 5 wells of a 96-well, white-walled microplate (#3610, Corning, Inc.). Separate plates were made for 0–5 day timepoints. This assay was performed using the FluoReporter Blue Fluorometric dsDNA Quantitation Kit (Molecular Probes) as described previously (40). Data were calculated as a percentage of the 0-day timepoint and combined from 4 separate experiments.
Colony formation in soft agar
Triplicate wells were plated with 1 × 104 or 5 × 104 cells per well in DMEM + 10% FBS with 0.4% low gelling temperature agarose (A9045, Sigma-Aldrich). The cell suspension was overlaid onto a presolidified base layer of DMEM + 10% FBS with 1.0% agarose. Fresh media was added every 2–3 days. After 10 days in culture, live images were captured at 10x and 25x magnification.
Astrocyte Implantation
Cells (5 × 105 cells/5 μl PBS) were implanted into the brains of nude rats as previously described (36) using an IACUC-approved protocol. Tumors were allowed to grow for 7 or 49/50 days. At sacrifice, the rats were anesthetized, perfused with sterile 0.9% saline via cardiac puncture, and then fixed with 10% formalin. Formalin-fixed rat brains were sliced into 2 mm blocks which were routinely processed, paraffin-embedded, and serially sectioned at 5 μm as previously reported (36). Animals/group: for day 7: Ast11.9 has n = 12 and #11 has n = 12; for day 50: Ast11.9 has n = 5 and #11 has n = 8.
Immunohistochemistry
Adjacent serial sections were routinely stained with H&E, immunohistochemically stained for SPARC (1:200 for 45 min; AF942, R&D Systems), the cell proliferation marker Ki-67 (1:250 for 40 min; M7249, Dako), and the macrophage/histiocyte marker CD68 (1:200 for 30 min; MCA341R, Serotec) as previously reported (36). Additional adjacent sections were subjected to the periodic acid Schiff (PAS) reaction with or without diastase (liver tissue was positive control) as previously described (43) to demonstrate phagocytic activity (histiocytes). Images of stained sections were captured on either a Nikon Eclipse E800 microscope with a Nikon DXM1200C digital camera using ImagePro 6.0 Plus (Media Cybernetics) software or a Nikon Eclipse Ni microscope with a Digital Sight DS-U3 digital camera using NIS-Elements AR 4.20 (Nikon) software. Composite figures were prepared using Adobe Photoshop CS6 software.
In Vivo Analysis of Tumor Area
For day 7 tumors, every 10th section of the tumor was stained with H&E. However, as some of the tumors were very small with only a few H&E sections per tumor, tumor volume could not be calculated for all of the tumors, and therefore the tumor area of the largest section through the tumor was imaged at 2x magnification and used to assess tumor size. Nikon NIS-Elements AR 4.20 software was used to outline the tumor and calculate the area in mm2 (n = 8 for Sparc-wt, n = 11 for Sparc-null).
MIB-1 Proliferation Index
One section per tumor, chosen from the sections having the largest tumor cross-section, was stained for Ki-67 with MIB-1 antibody (Dako) and was used to calculate the proliferation index. Using Adobe Photoshop CS5 software, 40x images (1–3 depending on the extent of staining) were overlaid with a grid and total and MIB-1-positive cells were counted. The proliferation index was calculated by dividing the number of MIB-1-positive cells by the total cell number and multiplying by 100 (n = 9 for Sparc-wt, n = 8 for Sparc-null).
CD68 Staining Density Quantitation
The sum density of CD68 staining was measured from one 40x image per tumor of the region with the highest staining density using Nikon NIS-Elements AR 4.20 software. Automated measurement was used to detect the staining with intensity thresholding from 0 (low) to 130 (high). Animals/group: n = 6 for day 7 and n = 5 (Sparc-null) or 8 (Sparc-wt) for day 50.
Statistical analysis
The Wilcoxon rank-sum test was used to compare the probability that Sparc-wt tumors are consistently higher, or lower, than Sparc-null tumors for percentage of MIB-1 stained cells and for tumor area. Probability is estimated as the c-index from a logistic regression model. Confidence intervals were estimated by DeLong’s method. Proliferation rates were estimated using linear regression with generalized estimating equations to account for the correlation (autoregressive-1) within technical replicate. Exponential growth was assumed and a log2 transformation was used. Given that growth is estimated relative to the zero time, the modeled lines were required to pass through the origin.
RESULTS
Confirmation of the loss of p53 and Sparc in knockout mouse astrocytes
Homozygous p53-null and Sparc-null mice were mated to generate heterozygotes which were then mated to generate double-null neonates. In astrocytes isolated from the mice, PCR and Western blot were used to confirm the loss of Sparc (Fig. 1A and B), whereas Southern blot analysis was used to confirm the loss of p53 (Fig. 1C). Four double-knockout (KO) astrocyte cell lines were generated and designated #2, #6, #11, and #30.
Figure 1.

Generation of p53-null/Sparc-wt and p53-null/Sparc-null mouse astrocytes. Homozygous p53-null and Sparc-null mice were mated to generate heterozygotes which were then mated to generate double-null neonates. Four p53-null/Sparc-null astrocyte cell lines were generated and designated #2, #6, #11, and #30. Ast11.9–2 is the control p53-null/Sparc-wt astrocyte cell line. A. PCR and B. Western blot analysis were used to confirm the loss of Sparc. The PCR product sizes are approximately 300 bp for the wt allele and 550 bp for the Sparc-null insertion. The Western blot results show SPARC expression in lysate and conditioned medium (CM). C. Southern blot analysis was used to confirm the loss of p53. Knockouts were verified by loss of the wild-type p53 band (~5 kb) and presence of the p53 mutant band (~6.5 kb).
Loss of Sparc increases p53-null astrocyte proliferation but inhibits growth in soft agar
The growth characteristics of p53-null/Sparc-wt and p53-null/Sparc-null astrocytes were assessed by proliferation assay and growth in soft agar. Analysis of proliferation over 5 days showed that the three p53-null/Sparc-null clones had an increased proliferation rate (15–33%, p<0.01) when compared to p53-null/Sparc-wt cells (Fig. 2A). Daily fold change in cell count is estimated to be 1.87 for Ast11.9 and 2.48, 2.41, and 2.14 for clones #2, #11, and #30, respectively. However, the loss of Sparc inhibited the ability of the two p53-null/Sparc-null clones used (#11 and #30) to form colonies in soft agar (Fig. 2B). Based on the similarity of results for the different Sparc-null clones, the following in vivo studies were performed using clone #11.
Figure 2.
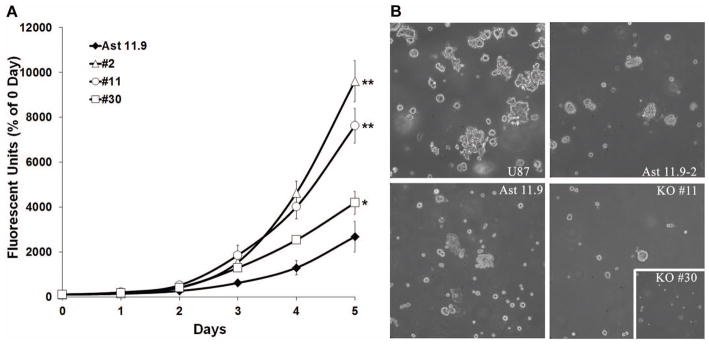
Loss of Sparc increases p53-null astrocyte proliferation but suppresses growth in soft agar. A. p53-null/Sparc-wt (Ast11.9) and p53-null/Sparc-null (#2, #11, and #30) astrocytes were analyzed for proliferation at 0–5 day timepoints by measuring dsDNA content using a fluorescent assay. The assay was repeated 4 times and analyzed for differences in growth rate between the clones. Mean ± SEM per day are presented here. Based on the daily fold change in cell proliferation, there is a significant increase in growth for the Sparc-null clones relative to Ast11.9 (* = p<0.01; ** = p<0.0001). B. Astrocytes were grown in soft agar and imaged after 10 days. Representative images show a reduction in colony-forming ability for the Sparc-null clones #11 and #30 (inset) compared to the Sparc-wt cell lines Ast11.9 and Ast11.9–2 and the glioma cell line U87MG.
Loss of Sparc decreases the in vivo survival and proliferation of p53-null astrocytic tumor cells resulting in a reduced tumor size
To determine the effects of Sparc loss on tumor development in vivo, p53-null/Sparc-wt and p53-null/Sparc-null astrocytes were implanted in the brains of nude rats and intracranial xenografts were assessed at 7 and 50 days post-implantation (Figs. 3–5). At day 50, both Sparc-wt and Sparc-null tumors had few or no tumor cells remaining; however, there was still histological evidence of an immune response (Fig. 4). Therefore, day 7 tumors were assessed for tumor size, proliferation rate, and SPARC expression and representative images from 2 different tumors per genotype are illustrated in Figure 3. The Sparc-null tumors were significantly smaller than the Sparc-wt tumors (p=0.0091, median [range] = 0.709mm2 [0.176–1.099] for Sparc-wt (n=8), and 0.240mm2 [0.079–0.539] for Sparc-null (n=11)) (Fig. 3 & 5A). In addition, the Sparc-null tumors had a significantly lower proliferation index than the Sparc-wt tumors (p=0.0345, median [range] = 8.6% [0–21.8] for Sparc-wt (n=9), and 0.4% [0–1.7] for Sparc-null (n=8)) (Fig. 3 & 5B).
Figure 3.

Loss of Sparc suppresses the in vivo tumor growth and proliferation of p53-null astrocytes. Intracranial xenografts of p53-null/Sparc-wt and p53-null/Sparc-null astrocytes were assessed at 7 days post-implantation. Brains were harvested, formalin-fixed, and paraffin-embedded. Tumor xenograft sections were H&E stained and immunohistochemically stained for mouse SPARC and the proliferation marker Ki-67. Magnifications as indicated. Representative images shown for 2 Sparc-wt and 2 Sparc-null animals out of n = 12 animals/group at day 7.
Figure 5.

Intracranial xenografts were quantitated and analyzed statistically for tumor area (A) MIB-1 proliferation index (B) and CD68 staining density (C). Asterisks denote significantly different from Sparc-wt where * represents p = 0.0091 for tumor area, p = 0.0345 for proliferation index, and p < 0.0001 for CD68 staining density.
Figure 4.

Loss of Sparc promotes macrophage activation and phagocytosis of p53-null astrocytes. Day 7 and day 50 p53-null/Sparc-wt and p53-null/Sparc-null tumor xenograft sections were immunohistochemically stained for CD68 and for periodic acid Schiff (PAS) +/−diastase (D) to assess the presence of microglia/macrophages. Images were taken at 40x magnification with insets at 60x. Representative images from 2 animals out of n = 6 animals/group for day 7 and n = 5 (Sparc-null) or 8 (Sparc-wt) animals/group for day 50.
The day 7 Sparc-wt tumors had a central area of necrosis, nuclear changes evident of tumor cells undergoing apoptosis, and were invasive (Fig. 3). Tumor cells were seen infiltrating along vessels at the brain-tumor interface as well as into adjacent normal brain tissue. In comparison, Sparc-null tumors were smaller in size with apoptotic tumor cells and prominent karyorrhexis throughout the lesions (Fig. 3). These tumors were better circumscribed than Sparc-wt tumors, and no tumor cells were seen invading along vessels at the periphery. A comparison of the number of tumor cells undergoing apoptosis based on nuclear changes in H&E stained sections revealed that there was not a significant difference in the number of apoptotic cells between Sparc-wt and Sparc-null tumors at day 7 (data not shown).
Loss of Sparc promotes macrophage activation and phagocytosis of tumor cells
Staining with CD68 shows that day 7 Sparc-null tumors had a massive infiltration of large phagocytic cells with bubbly cytoplasm. Phagocytic MG/MP were still present at day 50, after most of the tumor cells had been cleared (Fig. 4). The day 7 Sparc-wt tumors had MG/MP scattered throughout the tumor area, however, most of the MG/MP had an activated but not phagocytic appearance. By day 50, there were only a few remaining MG/MP in the area where the tumor cells had previously been located (Fig. 4). The staining density for CD68+ MG/MP was quantitated using Nikon NIS-Elements AR 4.20 software. When a 40x image of the region with the most intense CD68 staining was analyzed, there was a significant increase in staining density in Sparc-null tumors compared to Sparc-wt tumors at both 7 and 50 days post-implantation (p<0.0001, Fig. 5C). This difference in the amount of staining is evident in the images in Fig. 4 and indicates that the MG/MP that are present in Sparc-null tumors are enlarged in size which is consistent with the phagocytic morphology.
Further evidence of a phagocytic process in the Sparc-null tumors is shown by periodic acid Schiff (PAS) staining +/− diastase. Sparc-null tumors had increased PAS staining at both days 7 and 50 compared to Sparc-wt tumors (Fig 4). The PAS stain detects glycogen, glycoproteins, glycolipids, and mucins, which when abundant in lysosomes is a feature characteristic of phagocytic cells. The remaining PAS stain after diastase treatment shows that the glycogen is trapped in lysosomes and not available for metabolic function which is another indication that the cells have become phagocytic.
DISCUSSION
To assess the cooperation of p53 loss/mutation and SPARC upregulation as early events in glioma formation, we compared the in vitro phenotype and intracranial growth of p53-null/Sparc-wt and p53-null/Sparc-null astrocytes. In vitro, we found that the loss of Sparc in p53-null astrocytes resulted in an increase in cell proliferation, but an inhibition of growth in soft agar. When the astrocytes were grown intracranially, the loss of Sparc resulted in decreased tumor cell proliferation and a reduced tumor size at 7 days post-implantation and promoted macrophage activation and phagocytosis of tumor cells.
The in vitro data demonstrate that the loss of Sparc increased the proliferation rate of the p53-null astrocytes. This finding is consistent with our previously published results that the overexpression of SPARC in glioma cells suppresses cell proliferation (30, 40, 41). Despite the increase in proliferation with the loss of Sparc, the cells demonstrated an inability to form colonies in soft agar. These results suggest that the in vitro growth of Sparc-null cells is more anchorage-dependent than that of Sparc-wt cells.
The in vivo intracranial growth of the p53-null/Sparc-wt and p53-null/Sparc-null cells demonstrates that by day 7 there was a reduction in tumor size and cell proliferation for Sparc-null tumors compared to Sparc-wt tumors. This data appears to contradict our previous xenograft results in which SPARC-expressing glioma tumors have a reduced tumor volume and proliferation rate compared to tumors that do not express SPARC (36, 40). However, since our previous xenograft models used p53-wt tumorigenic glioma cells, the different genetic background of the cells, including the loss of p53, may influence the function of SPARC. We know from our previous work that the status of the tumor suppressor protein PTEN in glioma cells can influence the function of SPARC. We have shown that the reconstitution of PTEN in SPARC-expressing glioma cells was able to inhibit SPARC-induced glioma cell migration in vitro and tumor invasion in vivo (40). Thus, the genetic background of the astrocytes, including the presence or absence of tumor suppressor genes, has the ability to influence the function of SPARC.
The glioma literature provides conflicting results on the role of MG/MP in animal models of glioma progression, with some studies showing tumor enhancement and others showing tumor suppression. The current study supports the role of MG/MP in keeping p53-null astrocyte growth under control as evidenced by the lack of tumor development by day 50. The results clearly demonstrate that the loss of p53 is insufficient to fully transform the astrocytes, and therefore the astrocytes may not yet have acquired an immunosuppressive phenotype. However, Sparc provides some degree of immunosuppression by keeping MG/MP from becoming fully activated to a phagocytic state. This is demonstrated by the increase in phagocytic MG/MP observed in Sparc-null tumors. Therefore, the massive infiltration of phagocytic MG/MP in Sparc-null tumors contributes to the reduction in tumor size and cell proliferation in these tumors.
Glioma cells have been shown to express monocyte chemotactic protein-1 (MCP-1) (18), which is a known glioma chemoattractant for MG/MP (27). When glioma cells expressing MCP-1 were grown intracranially, tumors were 3.4 fold larger than control tumors and had a 10-fold increase in microglial cell content (27). Interestingly, exogenous SPARC was shown to inhibit basal and lysophosphatidic acid (LPA)-stimulated MCP-1 production by ovarian cancer cells (33). Therefore, SPARC expression in glioma cells may be involved in regulating MCP-1 expression and the recruitment of macrophages.
Furthermore, some studies provide evidence that MG/MP enhance glioma progression. When MG/MP were ablated locally at the site of tumor injection in a transgenic GL261 glioma model, tumor size was drastically reduced in MG/MP-depleted mice and tumors in depleted mice were almost undetectable by day 42. These results were confirmed when tuftsin, the stimulator of MG/MP, delivered at day 0 significantly increased tumor size and MG/MP numbers in the tumor; whereas conversely, MIF/TKP, an inhibitor of MG/MP, delivered at day 5 inhibited tumor growth and MG/MP infiltration (44).
On the other hand, there are studies suggesting that MG/MP density is inversely proportional to glioma growth. In a transgenic GL261 glioma model in which MG/MP were depleted, tumor volume increased 33% when glioma-associated MG/MP were reduced in number (11). Furthermore, primary MG stimulated with LPS/IFN-γ killed the mouse astrocytoma cell lines SMA-560 and MT539MG after direct co-culture or exposure to microglia supernatant by autophagic death (20).
Our model in this study demonstrates that SPARC may play a role in immunosuppression exerted by gliomas. Interestingly, SPARC can bind to the Stabilin-1 receptor which is expressed by alternatively activated macrophages (17). Stabilin-1 is thought to act as a scavenger receptor for SPARC; however, it is currently unknown whether the endocytosis of SPARC by macrophages alters the intracellular signaling or activation status of the macrophages.
There is an increasing literature suggesting that in tumor models, SPARC has an immunosuppressive function. For example, the administration of melanoma cells with SPARC knockdown in nude mice promoted polymorphonuclear leukocyte (PMN) recruitment and inhibited tumor growth, whereas the in vitro migration and cytotoxic capacity of PMN stimulated by SPARC knockdown cells was suppressed by purified SPARC (1). In animal models of ovarian and bladder cancers in which cancer cells were grown in Sparc-wt and Sparc-null mice, the tumors in Sparc-null mice had significantly higher macrophage infiltration (32, 34). The chemotactic effect of ovarian cancer cells on macrophages was attenuated when the cancer cells expressed SPARC, and co-cultures of ovarian cancer cells with macrophages had a reduced production of the inflammatory mediators IL-6, prostaglandin E2, and 8-isoprostane when the cancer cells expressed SPARC (35). Another study indicating that SPARC regulates macrophage chemotaxis found that pancreatic cancer cells grown in Sparc-null mice had macrophages present throughout the tumor, whereas macrophages were localized to the margin of the tumor in Sparc-wt mice (28). Our current data agree with these studies since the presence of SPARC altered MG/MP activation and delayed tumor cell destruction indicating a role for SPARC in glioma-induced immunosuppression. Further investigation into the interactions between SPARC-expressing glioma cells and glioma-associated MG/MP is warranted.
There is some evidence that SPARC and p53 can regulate one another. The re-expression of p53 in a glioma cell line in which both p53 alleles were lost resulted in a downregulation of SPARC expression in conditioned medium which was not due to altered mRNA levels (15). On the other hand, several studies have found that SPARC expression inversely regulates p53 protein levels. Expression of SPARC in melanoma cells was shown to reduce p53 expression and inhibit p53 induction in response to the cytotoxic drug actinomycin-D (9, 10). In melanoma and cervical cancer cell lines, knockdown of SPARC with siRNA/shRNA resulted in an induction of p53 (8–10, 13). In melanoma cells, this induction of p53 resulted from increased protein stability due to a reduction in total and phospho-MDM2 (9). Therefore, in the non-cancerous cell, p53 may play a role in keeping SPARC levels under control; whereas in the cancer cell with wild-type p53, SPARC may suppress p53 function.
Our results indicate that in glioma cells in which p53 function is lost by deletion/mutation, the induction of SPARC may cooperate with the loss of p53 to potentiate cancer cell survival and escape from immune surveillance. However, since this early model involves only changes in Sparc and p53 and is not sufficient to produce tumors that last and completely escape immune surveillance, future studies should also address the mechanism by which glioma cells can switch from an immunogenic phenotype to an immunosuppressive phenotype. This model will be useful for determining alterations in cytokine expression that regulate MG/MP activation and phagocytosis. Further studies should also determine whether the inhibition of SPARC in developing p53-null gliomas can lead to anti-tumor activity and a reduction in tumor growth when combined with currently used chemotherapeutic agents.
Acknowledgments
We thank Dr. Oliver Bögler for providing the Ast11.9 and Ast11.9-2 cell lines and the p53 cDNA probe for Southern blotting. This work was supported by grants CA86997 and CA138401 from the NCI/NIH to S.A.R. The authors are grateful to the Barbara Jane Levy family for their support.
References
- 1.Alvarez MJ, Prada F, Salvatierra E, Bravo AI, Lutzky VP, Carbone C, Pitossi FJ, Chuluyan HE, Podhajcer OL. Secreted protein acidic and rich in cysteine produced by human melanoma cells modulates polymorphonuclear leukocyte recruitment and antitumor cytotoxic capacity. Cancer research. 2005;65(12):5123–32. doi: 10.1158/0008-5472.CAN-04-1102. [DOI] [PubMed] [Google Scholar]
- 2.Badie B, Drazan KE, Kramar MH, Shaked A, Black KL. Adenovirus-mediated p53 gene delivery inhibits 9L glioma growth in rats. Neurological research. 1995;17(3):209–16. doi: 10.1080/01616412.1995.11740314. [DOI] [PubMed] [Google Scholar]
- 3.Badie B, Kramar MH, Lau R, Boothman DA, Economou JS, Black KL. Adenovirus-mediated p53 gene delivery potentiates the radiation-induced growth inhibition of experimental brain tumors. Journal of neuro-oncology. 1998;37(3):217–22. doi: 10.1023/a:1005924925149. [DOI] [PubMed] [Google Scholar]
- 4.Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46(4):957–61. doi: 10.1097/00006123-200004000-00035. discussion 61–2. [DOI] [PubMed] [Google Scholar]
- 5.Badie B, Schartner JM, Paul J, Bartley BA, Vorpahl J, Preston JK. Dexamethasone-induced abolition of the inflammatory response in an experimental glioma model: a flow cytometry study. Journal of neurosurgery. 2000;93(4):634–9. doi: 10.3171/jns.2000.93.4.0634. [DOI] [PubMed] [Google Scholar]
- 6.Bogler O, Huang HJ, Cavenee WK. Loss of wild-type p53 bestows a growth advantage on primary cortical astrocytes and facilitates their in vitro transformation. Cancer research. 1995;55(13):2746–51. [PubMed] [Google Scholar]
- 7.Bogler O, Nagane M, Gillis J, Huang HJ, Cavenee WK. Malignant transformation of p53-deficient astrocytes is modulated by environmental cues in vitro. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research. 1999;10(2):73–86. [PubMed] [Google Scholar]
- 8.Chen J, Shi D, Liu X, Fang S, Zhang J, Zhao Y. Targeting SPARC by lentivirus-mediated RNA interference inhibits cervical cancer cell growth and metastasis. BMC cancer. 2012;12:464. doi: 10.1186/1471-2407-12-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fenouille N, Puissant A, Tichet M, Zimniak G, Abbe P, Mallavialle A, Rocchi S, Ortonne JP, Deckert M, Ballotti R, Tartare-Deckert S. SPARC functions as an anti-stress factor by inactivating p53 through Akt-mediated MDM2 phosphorylation to promote melanoma cell survival. Oncogene. 2011;30(49):4887–900. doi: 10.1038/onc.2011.198. [DOI] [PubMed] [Google Scholar]
- 10.Fenouille N, Robert G, Tichet M, Puissant A, Dufies M, Rocchi S, Ortonne JP, Deckert M, Ballotti R, Tartare-Deckert S. The p53/p21Cip1/ Waf1 pathway mediates the effects of SPARC on melanoma cell cycle progression. Pigment cell & melanoma research. 2011;24(1):219–32. doi: 10.1111/j.1755-148X.2010.00790.x. [DOI] [PubMed] [Google Scholar]
- 11.Galarneau H, Villeneuve J, Gowing G, Julien JP, Vallieres L. Increased glioma growth in mice depleted of macrophages. Cancer research. 2007;67(18):8874–81. doi: 10.1158/0008-5472.CAN-07-0177. [DOI] [PubMed] [Google Scholar]
- 12.Golembieski WA, Ge S, Nelson K, Mikkelsen T, Rempel SA. Increased SPARC expression promotes U87 glioblastoma invasion in vitro. International journal of developmental neuroscience : the official journal of the International Society for Developmental Neuroscience. 1999;17(5–6):463–72. doi: 10.1016/s0736-5748(99)00009-x. [DOI] [PubMed] [Google Scholar]
- 13.Horie K, Tsuchihara M, Nakatsura T. Silencing of secreted protein acidic and rich in cysteine inhibits the growth of human melanoma cells with G arrest induction. Cancer science. 2010;101(4):913–9. doi: 10.1111/j.1349-7006.2009.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain pathology. 1999;9(3):469–79. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khwaja FW, Svoboda P, Reed M, Pohl J, Pyrzynska B, Van Meir EG. Proteomic identification of the wt-p53-regulated tumor cell secretome. Oncogene. 2006;25(58):7650–61. doi: 10.1038/sj.onc.1209969. [DOI] [PubMed] [Google Scholar]
- 16.Kock H, Harris MP, Anderson SC, Machemer T, Hancock W, Sutjipto S, Wills KN, Gregory RJ, Shepard HM, Westphal M, Maneval DC. Adenovirus-mediated p53 gene transfer suppresses growth of human glioblastoma cells in vitro and in vivo. International journal of cancer Journal international du cancer. 1996;67(6):808–15. doi: 10.1002/(SICI)1097-0215(19960917)67:6<808::AID-IJC9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 17.Kzhyshkowska J, Workman G, Cardo-Vila M, Arap W, Pasqualini R, Gratchev A, Krusell L, Goerdt S, Sage EH. Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. Journal of immunology. 2006;176(10):5825–32. doi: 10.4049/jimmunol.176.10.5825. [DOI] [PubMed] [Google Scholar]
- 18.Leung SY, Wong MP, Chung LP, Chan AS, Yuen ST. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta neuropathologica. 1997;93(5):518–27. doi: 10.1007/s004010050647. [DOI] [PubMed] [Google Scholar]
- 19.Menon PM, Gutierrez JA, Rempel SA. A study of SPARC and vitronectin localization and expression in pediatric and adult gliomas: high SPARC secretion correlates with decreased migration on vitronectin. International journal of oncology. 2000;17(4):683–93. doi: 10.3892/ijo.17.4.683. [DOI] [PubMed] [Google Scholar]
- 20.Mora R, Abschuetz A, Kees T, Dokic I, Joschko N, Kleber S, Geibig R, Mosconi E, Zentgraf H, Martin-Villalba A, Regnier-Vigouroux A. TNF-alpha- and TRAIL-resistant glioma cells undergo autophagy-dependent cell death induced by activated microglia. Glia. 2009;57(5):561–81. doi: 10.1002/glia.20785. [DOI] [PubMed] [Google Scholar]
- 21.Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. Part 1: Studies of the macrophage content of experimental rat brain tumors of varying immunogenicity. Journal of neurosurgery. 1979;50(3):298–304. doi: 10.3171/jns.1979.50.3.0298. [DOI] [PubMed] [Google Scholar]
- 22.Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. Journal of neurosurgery. 1979;50(3):305–11. doi: 10.3171/jns.1979.50.3.0305. [DOI] [PubMed] [Google Scholar]
- 23.Noble M, Murray K. Purified astrocytes promote the in vitro division of a bipotential glial progenitor cell. The EMBO journal. 1984;3(10):2243–7. doi: 10.1002/j.1460-2075.1984.tb02122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lutolf UM, Kleihues P. Genetic pathways to glioblastoma: a population-based study. Cancer research. 2004;64(19):6892–9. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 25.Ohgaki H, Kleihues P. Genetic profile of astrocytic and oligodendroglial gliomas. Brain tumor pathology. 2011;28(3):177–83. doi: 10.1007/s10014-011-0029-1. [DOI] [PubMed] [Google Scholar]
- 26.Okamoto Y, Di Patre PL, Burkhard C, Horstmann S, Jourde B, Fahey M, Schuler D, Probst-Hensch NM, Yasargil MG, Yonekawa Y, Lutolf UM, Kleihues P, Ohgaki H. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta neuropathologica. 2004;108(1):49–56. doi: 10.1007/s00401-004-0861-z. [DOI] [PubMed] [Google Scholar]
- 27.Platten M, Kretz A, Naumann U, Aulwurm S, Egashira K, Isenmann S, Weller M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Annals of neurology. 2003;54(3):388–92. doi: 10.1002/ana.10679. [DOI] [PubMed] [Google Scholar]
- 28.Puolakkainen PA, Brekken RA, Muneer S, Sage EH. Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell apoptosis. Molecular cancer research : MCR. 2004;2(4):215–24. [PubMed] [Google Scholar]
- 29.Reifenberger J, Ring GU, Gies U, Cobbers L, Oberstrass J, An HX, Niederacher D, Wechsler W, Reifenberger G. Analysis of p53 mutation and epidermal growth factor receptor amplification in recurrent gliomas with malignant progression. Journal of neuropathology and experimental neurology. 1996;55(7):822–31. doi: 10.1097/00005072-199607000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Rempel SA, Golembieski WA, Fisher JL, Maile M, Nakeff A. SPARC modulates cell growth, attachment and migration of U87 glioma cells on brain extracellular matrix proteins. Journal of neuro-oncology. 2001;53(2):149–60. doi: 10.1023/a:1012201300188. [DOI] [PubMed] [Google Scholar]
- 31.Rempel SA, Golembieski WA, Ge S, Lemke N, Elisevich K, Mikkelsen T, Gutierrez JA. SPARC: a signal of astrocytic neoplastic transformation and reactive response in human primary and xenograft gliomas. Journal of neuropathology and experimental neurology. 1998;57(12):1112–21. doi: 10.1097/00005072-199812000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Said N, Frierson HF, Sanchez-Carbayo M, Brekken RA, Theodorescu D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. The Journal of clinical investigation. 2013;123(2):751–66. doi: 10.1172/JCI64782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Said N, Najwer I, Motamed K. Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. The American journal of pathology. 2007;170(3):1054–63. doi: 10.2353/ajpath.2007.060903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Said N, Socha MJ, Olearczyk JJ, Elmarakby AA, Imig JD, Motamed K. Normalization of the ovarian cancer microenvironment by SPARC. Molecular cancer research : MCR. 2007;5(10):1015–30. doi: 10.1158/1541-7786.MCR-07-0001. [DOI] [PubMed] [Google Scholar]
- 35.Said NA, Elmarakby AA, Imig JD, Fulton DJ, Motamed K. SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia. 2008;10(10):1092–104. doi: 10.1593/neo.08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schultz C, Lemke N, Ge S, Golembieski WA, Rempel SA. Secreted protein acidic and rich in cysteine promotes glioma invasion and delays tumor growth in vivo. Cancer research. 2002;62(21):6270–7. [PubMed] [Google Scholar]
- 37.Schultz CR, Golembieski WA, King DA, Brown SL, Brodie C, Rempel SA. Inhibition of HSP27 alone or in combination with pAKT inhibition as therapeutic approaches to target SPARC-induced glioma cell survival. Molecular cancer. 2012;11:20. doi: 10.1186/1476-4598-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Q, Bao S, Maxwell JA, Reese ED, Friedman HS, Bigner DD, Wang XF, Rich JN. Secreted protein acidic, rich in cysteine (SPARC), mediates cellular survival of gliomas through AKT activation. The Journal of biological chemistry. 2004;279(50):52200–9. doi: 10.1074/jbc.M409630200. [DOI] [PubMed] [Google Scholar]
- 39.Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, Rich JN. Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene. 2007;26(28):4084–94. doi: 10.1038/sj.onc.1210181. [DOI] [PubMed] [Google Scholar]
- 40.Thomas SL, Alam R, Lemke N, Schultz LR, Gutierrez JA, Rempel SA. PTEN augments SPARC suppression of proliferation and inhibits SPARC-induced migration by suppressing SHC-RAF-ERK and AKT signaling. Neuro-oncology. 2010;12(9):941–55. doi: 10.1093/neuonc/noq048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vadlamuri SV, Media J, Sankey SS, Nakeff A, Divine G, Rempel SA. SPARC affects glioma cell growth differently when grown on brain ECM proteins in vitro under standard versus reduced-serum stress conditions. Neuro-oncology. 2003;5(4):244–54. doi: 10.1093/neuonc/5.4.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yahanda AM, Bruner JM, Donehower LA, Morrison RS. Astrocytes derived from p53-deficient mice provide a multistep in vitro model for development of malignant gliomas. Molecular and cellular biology. 1995;15(8):4249–59. doi: 10.1128/mcb.15.8.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yunker CK, Golembieski W, Lemke N, Schultz CR, Cazacu S, Brodie C, Rempel SA. SPARC-induced increase in glioma matrix and decrease in vascularity are associated with reduced VEGF expression and secretion. International journal of cancer Journal international du cancer. 2008;122(12):2735–43. doi: 10.1002/ijc.23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhai H, Heppner FL, Tsirka SE. Microglia/macrophages promote glioma progression. Glia. 2011;59(3):472–85. doi: 10.1002/glia.21117. [DOI] [PMC free article] [PubMed] [Google Scholar]