

Abstract
Mouse Hepatitis Virus (MHV) is a single-stranded positive sense RNA virus with the ability to promote acute and chronic diseases in mice. The MHV spike protein (S) is a major virulence determinant which in addition to binding to cellular receptors to mediate cell entry and facilitate virus spread to adjacent cells by cell-cell fusion, also is a molecular mimic of the FcγRII receptor. This molecular mimicry of FcγRII by the MHV S protein is also exhibited by other lineage 2a betacoronaviruses, with the exception of the human coronavirus HCoV-OC43. In this work we undertook a mutational analysis to attempt to identify specific amino acid sequences within the spike glycoprotein crucial for molecular mimicry of FcγRII. Although we were unsuccessful in isolating mutant viruses which were specifically defective in that property, we identified several mutations with interesting phenotypes. Mutation of the cysteine in position 547 to alanine and alanine replacements at residues 581–586 was lethal. Replacing proline 939 with the corresponding HCoV-OC43 residue, leucine, decreased the ability MHV to induce cell-cell fusion, providing experimental support for an earlier proposal that residues 929–944 make up the fusion peptide of the MHV S protein.
Keywords: Mouse hepatitis virus, Coronavirus spike protein, Targeted recombination, Fc receptor, Cell fusion
1. INTRODUCTION
Mouse Hepatitis Virus (MHV) is a member of the family coronaviridae. It includes large single-and positive stranded RNA viruses that induce a spectrum of acute and chronic diseases of the neurological, gastrointestinal and respiratory system in animals and humans, such as the human pathogens SARS-CoV and HCoV-OC43 (1). MHV has been widely utilized as a model for viral pathogenesis, particularly the two most commonly studied strains of MHV, MHV-A59 and MHV-JHM. Infection of C57Bl/6 mice with MHV-A59 causes mild encephalitis, sub-acute demyelination, and acute hepatitis (1, 2). The largely immunologically mediated demyelinating disease in mice serves as an extensively investigated model for demyelinating diseases such as multiple sclerosis (MS) (2, 3).
The major structural proteins of MHV virion include the spike glycoprotein (S) the envelope protein (E) the membrane protein (M) and the nucleocapsid protein (N). Although the multifunctional protein N contributes to the pathogenic outcome of coronavirus infection (4), the MHV S protein plays a major role in the pathogenesis since it recognizes and binds the cellular receptor for the virus, Ceacam1a, mediates cell entry by inducing fusion of viral and cell membranes, subsequently facilitates virus spread to other cells by inducing cell to cell fusion (syncytium formation) at late times in infection when the S protein is abundantly present on the plasma membrane of infected cells (5), and recently it was found to regulate the intracellular transport of the viral genome from the cell surface to the ER (6). S is also a major target of the host immune response, eliciting neutralizing antibodies and CD8+ cytotoxic T cell responses (7).
The MHV S glycoprotein is 1324 amino acids long; the primary 146 kDa translation product is acylated, N-glycosylated, and trimmed subsequently to yield the mature 180 kDa protein. The mature S protein is post-translationally cleaved in most MHV strains by host proteases into two 90kDa subunits: an amino terminal subunit (S1) and a carboxy terminal subunit (S2) (8). S1–S2 cleavage does not destroy virus infectivity; rather it increases the potency of coronaviruses to mediate cell-cell fusion and promotes rapid virus dissemination (8, 9). S1 makes up the unique globular-head structure of the spike and contains the receptor binding activity; the S2 subunit contains two heptad repeats regions that are part of the stalk structure of the spike and are required for membrane fusion, a trans-membrane domain, and a cytoplasmic domain (in the cell) that is in the interior of the virus particle after budding is completed [reviewed in (10)].
In addition, S protein was found to interact with the Fc region of immunoglobulin G (IgG) (11). The MHV S protein behaved as a molecular mimic of the host murine Fcγ receptor (mFcγRII/RIII) in that it bound IgG in an Fc dependent manner, was immunoprecipitated by the 2.4G2 anti-FcγRII/RIII monoclonal antibody, and the Fc binding activity of S protein was expressed on the plasma membrane of infected cells (11, 12). This Fc binding activity is also exhibited by the related type 2a betacoronavirus, bovine coronavirus (BCoV) S protein (13). However, the human coronavirus OC43 (HCoV-OC43), a virus closely related to BCoV, did not exhibit molecular mimicry of FcγRII/RIII in that it was not reactive with the 2.4G2 monoclonal antibody (13).
Analysis of the Fc binding activity of MHV S proteins containing either naturally occurring deletions in the S protein, neutralizing monoclonal antibody escape mutants containing S protein deletions, or recombinant S proteins, suggested that this activity most likely resided in the S1 domain (27, 28), and excluded determinants of Fc binding activity being present in the MHV S1 hyper-variable region (12). To investigate this further we hypothesize that regions containing specific amino acid sequences within the spike glycoprotein of MHV-A59 and other related strains that exhibited FcR binding activity would exhibit sequence similarity with the murine Fc γRII and FcγRIII (mFcγRII and mFcγRIII, respectively) receptors (14), but not with the corresponding amino acids of the HCoV-OC43 S protein that did not exhibit Fc binding activity (13), suggesting to us several regions of interest within the spike glycoprotein as targets for mutagenesis to test this hypothesis. In this study targeted amino acid residues of S protein were replaced with alanine or with the homologous amino acids residues from the S protein of HCoV-OC43. The mutant spike glycoproteins were expressed in murine DBT cells by the vector pCAGGS-S (15), metabolically labeled with 35S and immunoprecipitated by the neutralizing anti-S monoclonal antibodies A2.1 and A2.3 to test that they had folded correctly (16). Subsequently, spike genes encoding proteins with the substituted amino acids were introduced into recombinant isolates of MHV-A59 by targeted recombination (17, 18). Several mutant viruses with interesting phenotypes were recovered. A recombinant virus containing a P939L mutation in the spike protein had a decreased ability to induce cell-cell fusion. A cysteine in position 547 and the amino acids in positions 581–586 within the S1 fragment of spike glycoprotein were essential for virus viability. A C547A mutation greatly decreased the reactivity with the A2.1 and A2.3 monoclonal antibodies suggesting that this cysteine was required for proper folding. Three mutants in which MHV residues were replaced by the corresponding amino acids in the HCoV-OC43 S protein, resulted in viruses that grew less well than the wild type virus. However, none of these regions was found to affect the binding of S to the anti FcγR antibody 2.4G2, implying that the molecular mimicry property is determined not only by sequence similarity to the binding domains of Fc receptor but most probably also the unique secondary and tertiary structure of the protein as well.
2. MATERIALS AND METHODS
2.1 Cells and viruses
DBT and human epithelial kidney cells (293T) were maintained at 37°C and 5% CO2 in Dulbecco’s modified medium (DMEM) supplemented with 10% calf serum (Hyclone). FCWF (Felis catus whole fetus) cells were grown in DMEM supplemented with 10% fetal bovine serum. L2 cells were maintained at 37°C and 3% CO2 in DMEM supplemented with 10% calf serum. The origin and growth of MHV-A59 have been described previously (19, 20). The recombinant A59 (fA59) viruses containing the feline infectious peritonitis virus (FIPV) spike protein ectodomain in place of the MHV-A59 ectodomain (17) was generously provided by Prof. Susan R. Weiss, University of Pennsylvania, and propagated in FCWF cells.
2.2 Plasmid constructions and transfections
MHV (strain A59) S gene was expressed in the pCAGGS-S plasmid vector (kindly provided by Prof. Thomas M. Gallagher) (21). All mutations in the S protein were created by using mutagenic primers and overlap-extension PCR (22), followed by TA cloning of mutagenized amplicons into pGEM-T Easy (Promega) and subsequent sequencing. Cloned amplicons containing the introduced mutation were excised with unique restriction enzymes and replaced the corresponding restriction fragments in S in the pCAGGS-S vector to produce the corresponding plasmids: pCAGGS-S (546–548), pCAGGS-S (546/548), pCAGGS-S (554–556), pCAGGS-S (581–586), pCAGGS-S (562/589), pCAGGS-S (667/687), pCAGGS-S (910/939) and pCAGGS-S (939). All plasmid constructs were sequenced to confirm the presence of the desired mutations (Table 1).
Table 1.
Oligonucleotide primers used to construct the amino acid substitutions in the MHV-A59 spike protein.
| Mutation | Location | Sequence (5′ to 3′)a |
|---|---|---|
| S(546–548) | 25547–25590 | CC TGC CTT GTT AAT GAT gcc gcc gca ATT TTT GCT AAT ATA TTG |
| S(546/548) | 25547–25590 | CC TGC CTT GTT AAT GAT gcc TGC gct ATT TTT GCT AAT ATA TTG |
| S(554–556) | 25577–25611 | TT GCT AAT ATA gcc gca gct GGC ATT AAT AGT GGT |
| S(581–586) | 25664–25709 | GTT ACT GGC ATT TGT gca gca gca gct CTC gcc GGT ATT ACT GGA CAA G |
| S(562) | 25590–25624 | G TTA AAT GGC ATT AAT AGT GGT ctg ACA TGT TCC A |
| S(589) | 25671–25708 | C AAA TAT GAC CTC TAC GGT ATT ctg GGA CAA GGT GTT T |
| S(667) | 25909–25942 | GTT TTT AGC AAT AAT ATT acc CGT GAG GAG AAC C |
| S(687) | 25968–25995 | G GGT TGT GTT GTT AAT GCT tat AAC CGC |
| S(910) | 26622–26657 | GAC CTC CTT TGT GTA CAA TCT TTT aag GGC ATC AAA |
| S(939) | 26729–26759 | CA GCT ATG TTC CCA ctg TGG TCA GCA GCT GC |
The mutated nucleotides are indicated by the lower-case (italic) letters.
For targeted recombination, a fragment of about 3 Kb including the mutation was excised from the corresponding pCAGGS-S expressing vector with SwaI and MluI and transferred to the targeted recombination plasmid pMH54 (5) to produce the corresponding plasmids pMH54 (546–548), pMH54 (546/548), pMH54 (554–556), pMH54 (581–586), pMH54 (562/589), pMH54 (667/687), pMH54 (910/939) and pMH54 (939) respectively.
293T cells were transfected by mixing 4 μg plasmid DNAs with 16 μl of Lipofectamine 2000 (Invitrogen) in 1 ml Opti-MEM (Gibco) and applied to 106 cells for 4 hr and then removed and replaced with complete media. At 24 hr post-tranfection medium was removed from each culture and the cell lysate was prepared as described previously (11) and stored at −80°C.
2.3 Metabolic labeling of cells and immunoprecipitation
Monolayers of DBT cells in 6-well plates were infected with the appropriate viruses at an MOI of 3 plaque forming units per cell at 37°C for 1hr. Transfected 293T cells and infected DBT cells were radiolabeled with 400μCi/ml [35S]-methionine and cysteine for 6–7 hours post transfection or 8 hours post infection until 95–100% of the monolayer was involved in syncytia, respectively. Cytoplasmic extracts of infected and control cells were prepared in 250 μl of lysing buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 1.5 mM MgCl2, 0.5% NP40, 0.2 TIU/ml aprotinin) on ice as described previously (11) and stored at −80°C. Twenty μl of protein G agarose beads (Calbiochem) were incubated with secondary antibody (40 μg of goat anti-mouse IgG or 120 μg of anti-rat IgG, respectively) for 1 hr on ice, than washed twice with PBS and incubated with primary antibodies (A2.1 and A2.3 or 2.4G2, respectively) for an additional hour. Unbound antibodies were washed away with PBS and the protein G beads-antibody complexes were resuspended in MRIP buffer (10 mM phosphate, pH 7.4, 500 mM NaCI, 0.25% NP40, 0.2 TIU/ml aprotinin, 1 mM PMSF) as described before (11). Cell lysates in a volume of 50 μl [for 2.4G2 binding assays 150 μl of the lysates were concentrated into final volume of 50 μl with a Microcon YM-100 centrifugal concentrator (Millipore)] were added to antibody-protein G coated beads and the mixture incubated on ice for 1 hr. The immunocomplexes were collected by centrifugation and washed five times with MRIP buffer. The bound antigens were eluted by heating at 70°C for 5 min in SDS-PAGE sample buffer. The samples were resolved by SDS-PAGE at 10 mA for about 10 hr as described by Laemmli and Favre (23) followed by phosphoimager (GE Healthcare) autoradiography.
2.4 Targeted recombination
Plasmids pMH54, pMH54/S (546–548), pMH54/S (546/548), pMH54/S (581–586), pMH54/S (562/589), pMH54/S (667/687), pMH54/S (910/939) and pMH54/S (939) were digested and linearized with PacI and donor RNAs were transcribed with T7 RNA polymerase as previously described (17, 19). Targeted recombination with MHV-A59 was performed as described previously (17, 19) with a few modifications. Briefly, fMHV corresponding to MHV-A59, in which the sequences encoding the spike protein ectodomain had been replaced by the corresponding feline infectious virus (FIPV) spike ectodomain coding sequence, was used as an acceptor virus. FCWF cells were infected with fMHV and incubated for 6 hr. Cells were nucleofected with the transcribed donor RNAs using program T-020 and the nucleofector V kit (Lonza) and the nucleofected cells were overlaid onto a monolayer of DBT cells. The cultures were incubated up to 72 hr or until cytopathic effect destroyed the monolayer. Recombinant viruses able to enter and replicate in murine cells, and thus which contained the desired MHV-A59 spike ectodomain, were selected by plaque assay on L2 cells. Well-separated plaques containing putative recombinant viruses were picked and underwent a second cycle of plaque purification. The twice plaque cloned viruses were expanded in murine L2 cells, and their recombinant nature was confirmed by RT- PCR and sequencing. Viral titers were determined by plaque assay on monolayers of L2 cells as previously described (24).
2.5 Cell fusion assay
DBT cells were grown in 6-well cluster plates to a density of approximately 106 cells per well, infected with wild type or mutant viruses (3 PFU/cell) and the cells were incubated at 37°C. After 5.5, 7 and 9 hr the nuclei were stained with DAPI and photographed using a fluorescence microscope. The number of nuclei contained in syncytial cells was counted for 10 randomly selected fields for each viral strain.
3. RESULTS
3.1 Alanine substitution of cysteine 547 facilitates partial recognition of the uncleaved S, abolishes the recognition of the cleaved subunit by anti S neutralizing antibodies and prevents recovery of a viable recombinant virus
An alignment of the MHV-A59 and MHV-JHM S protein amino acids sequences to the mFcγRII IgG binding domain revealed several short regions with modest sequence similarity (Fig. 1A), the most robust of which corresponded to residues 546–548 in MHV-A59 (11). We selected this region of the MHV-A59 S protein for our initial studies and created a plasmid, designated pCAGGS-S (546–548), for expressing a mutant S protein in which residues R546/C547/Q548 were changed to alanine. To determine the effect that this mutation had on the overall conformation of the protein we compared its expression and its immunoreactivity with two different conformationally sensitive anti-S monoclonal antibodies, known to neutralize MHV-A59 infectivity and block infection with virus like particles containing the MHV-A59 structural proteins (16, 32) compared to that of wild type S protein expressed in the same vector. Replicate cultures of 293T cells were transfected with pCAGGS-S or pCAGGS-S (546–548) and after 24 hours post transfection the cells were metabolically labeled with 35S (cysteine and methionine) for nine hours, harvested, cytoplasmic extracts prepared and then immunoprecipitated with the anti-spike monoclonal neutralizing antibodies A2.1 and A2.3 (16) or with a polyclonal rabbit anti-spike antibody, B46 (kindly provided by Dr. John Fleming and Dr. K. Holmes, respectively). As shown in Fig. 2A, the S (546–548) mutant protein was barely recognized by both antibodies; signal corresponding to the complete 180 kDa protein was barely discernible compared to the robust signals obtained from the wild type glycoprotein either similarly expressed by transfection, or from cell lysates prepared from MHV-A59 infected DBT cells. Moreover, the cleaved 90 kDa S1 and S2 subunits could not be detected. In contrast the B46 polyclonal antibody strongly immunoprecipitated S (546–548), making it unlikely that the weak reactivity with the A2.1 and A2.3 antibodies was due to decreased stability of the mutant protein relative to wild type S protein (Fig. 2A). Together, these results suggest that alanine substitution mutations at positions 546–548 produced a major change in the conformation of the S protein. We then considered the possibility that this major change in conformation may have largely been due to our mutation of C457, a residue that could play a role in maintaining the overall conformation of the protein through a disulfide bridge. Therefore we made a second construct, containing the R546A and Q548A mutations into pCAGGS-S [pCAGGS-S (546/548)], and transfected it into cells in parallel with the parental pCAGGS-S. After radiolabeling for 9 hours, starting at 24 hours post transfection, lysates were prepared and immunoprecipitated with A2.1, A2.3, or with a polyclonal B46 antibody. As shown in Fig. 2B, both the 90kDa cleaved S1/S2 subunits and the uncleaved full length 180 kDa forms of the S protein were immunoprecipatated by the two monoclonal antibodies as well as the uncleaved protein by the polyclonal B46 antibody (data not shown), suggesting that the R546A/Q548A mutations did not alter the overall conformation of the S protein and allowed it to fold properly.
Fig 1.

FA bioinformatics approach for the identification of targeted mutation regions within spike glycoprotein of MHV/A59. A. Schematic representation of the MHV spike protein structure. The following features are present: RBD – Receptor Binding Domain, PFP – Putative Fusion Peptide, PEP – Putative Fusion Domain, HR – Hydrophobic repeats, TD –Transmembrane Domain, CT – Cytoplasmic Tail. The location of the proline residue at position 939 within the putative fusion peptide is also shown in the schematic. An expansion of the region of weak sequence similarity between MHV S protein and the FcγRII protein is shown below the schematic. Residues of sequence identity are shown in bold underlined lower case text. B. Amino acid sequence comparison of MHV/A59, BCV and HCoV-OC43 spike proteins. Amino acids that were identical in the MHV-A59 and BCV spike proteins but differed from the HCoV-OC43 S protein are in a bold and underlined font.
Fig. 2.

The effect of C547 alanine substitution on the recognition of S protein by neutralizing antibodies A2.1 and A2.3 and on viral recovery. A. 293T cells were transfected with the expression vector pCAGGS-S encoding the native spike protein or the amino acids substituted protein in positions 546–548 of the spike protein [pCAGGS-S (546–548)]. Twenty-four hours after transfection cells were metabolically labeled with [ 35S]-methionine and cysteine for nine hours in 37°C, a lysate was prepared and analyzed by immunoprecipitation with monoclonal neutralizing anti-S antibodies A2.1 and A2.3 or a polyclonal anti-S antibody B46. B. 293T cells were transfected with the expression vector pCAGGS-S or the expression vector encoding the amino acids substitutions in positions 546 and 548 [pCAGGS-S (546/548)], labeled with 35S-methionine and cysteine and immunoprecipitated with anti-S monoclonal or polyclonal antibodies. DBT cells were infected with the recombinant MHV-A59 virus expressing the native spike protein [rA59/S (pMH54)] or the mutated spike protein [rA59/S (546/548)] and metabolically labeled with [35S]-methionine and cysteine for one hour at 9 hours post infection. Cell lysates were immunoprecipitated with the monoclonal neutralizing anti-S antibody A2.3. C. Mean viral titers (left) and plaque diameters (right) of recombinant viruses expressing the native spike protein, rA59/S (pMH54), or the amino acid substituted protein and rA59/S (546/548), respectively. Error bars represent the standard deviations from the mean. S uncleaved denotes the 180 kDa uncleaved spike protein, and S cleaved denotes the 90kDa cleaved S1 and S2 subunits.
The R546A/Q548A mutations were introduced into the plasmid pMH54 and subsequently into a recombinant MHV-A59, rA59/S (546/548), by targeted recombination. A viable recombinant virus expressing these mutations was recovered without difficulty and achieved titers somewhat lower (~1 log) but not statistically different than those achieved by the isogenic control virus rA59 (pMH54) and had a plaque size that was statistically indistinguishable from that of wild type plaques (Fig 2C). Immunoprecipitation of lysates prepared from DBT cells infected with rA59/S (546/548) and labeled for 1 hour from 7–8 h.p.i, with the A2.3 monoclonal antibody confirmed this mutation did not interfere with the overall conformation of the protein (Fig. 2B). Similarly, the S protein carrying the R546A/Q548A mutations was immunoprecipitated by the anti-FcγRII/RIII monoclonal antibody 2.4G.2 (Fig. 3A), making it unlikely that the amino acids in positions 546 and 548 were part of the epitope responsible for the FcγRII molecular mimicry by the S protein. In contrast, multiple attempts to recover a virus containing a C547A mutation in addition to the two alanine replacement mutations in rA59/S (546/548) were unsuccessful and this included an attempt to recover temperature-sensitive viruses by performing targeted recombination at 34°C or 39°C.
Fig. 3.

The effect of alanine residues in positions 554–556 and 546/548 on S protein molecular mimicry of FcγRII. Immunoprecipitation of DBT cells infected with the recombinant viruses rA59/S (546/548) (Panel A) and rA59/S (554–556) (Panels B and C). DBT cells were mock infected, infected with either the isogenic control virus rA59/S(pMH54), or infected with a recombinant virus containing a mutation in S. The cells were incubated until CPE was apparent at 8 hpi and metabolically labeled with [35S]-methionine and cysteine for one hour, after which lysates were prepared and immunoprecipitated with the monoclonal anti-FcγRII antibody 2.4G2 as described in Materials and Methods. S uncleaved denotes the 180 kDa uncleaved spike protein, and S cleaved denotes the 90 kDa cleaved S1 and S2 subunits.
Within the S protein region with the greatest sequence similarity to the FcγRII/RIII proteins (546–556) there was a second trio of amino acids, L554, L 555, and N556, with perfect sequence identity amongst these proteins, A mutant S protein containing alanine replacement mutations at these 3 positions were expressed in pCAGGS-S [pCAGGS-S (554–556)]. Immunoprecipitation with the A2.1 and A2.3 monoclonal antibodies gave results similar to those obtained with pCAGGS-S (546/548), namely the mutant protein was precipitated by these conformational dependent antibodies as efficiently as wild type S protein (data not shown), indicating that the mutant protein was likely able to fold correctly. Sequences encoding the alanine replacement mutations were introduced into plasmid pMH54 by restriction fragment exchange and we subsequently performed targeted recombination to introduce these mutations into MHV-A59. A recombinant virus, designated rA59/S (554–556), containing the desired S protein mutations was successfully recovered. However, unlike the previous recombinant, this virus formed plaques with a mean diameter of 1.17 ± 0.18 mm whereas the plaques formed by the isogenic control virus containing the wild type S sequence had a mean diameter of 2.36 ± 0.20 mm (P<.001). The rA59/S (554–556) virus grew to titers that were equal to those achieved by control virus. Sequencing of the S gene failed to reveal any second site compensatory mutations, and this sequence and plaque phenotype was stable over three passages. Immunoprecipitation of lysates prepared from DBT cells infected with rA59/S (554–556) and labeled for 1 hour with [35S] methionine and cysteine from 7–8 h.p.i, with the A2.1 and A2.3 monoclonal antibody confirmed that this mutation did not interfere with the overall conformation of the protein. Both the non-cleaved and cleaved subunits were recognized by the monoclonal antibodies A2.1 (Fig. 3B) and A2.3 (data not shown). Similarly, immunoprecipitation with the anti-Fcγ receptor antibody 2.4G2 brought down the 180 kDa non-cleaved S protein, making it unlikely that these residues by themselves were involved in molecular mimicry of FcγRII by S protein (Fig. 3C).
3.2 Alanine substitution of the amino acids in positions 581–586 abolishes the recognition of the cleaved subunit by the monoclonal anti S neutralizing antibodies A2.1 and A2.3 and prevents the recovery of a viable recombinant virus
A third region of sequence similarity between the MHV S protein and the murine FcγRII/RIII proteins is located at positions 581–586 (VKYDLY) in the S protein, approximately 30 amino acids C-terminal to the sequences we examined above (Fig. 1A). Thus we replaced five of the amino acids (all but L585) with alanine in the pCAGGS-S expression vector to create the plasmid pCAGGS-S (581–586) and expressed this mutant in 293T cells. Transfected cells were metabolically labeled with 35S-methionine/cysteine and the lysate was immunoprecipitated with the monoclonal anti-spike antibodies A2.1 and A2.3. Although the 180 kDa uncleaved spike glycoprotein was clearly precipitated by both antibodies, the cleaved 90 kDa S1 and S2 subunits were not brought down by either one of the antibodies (Fig. 4). To determine if the spike glycoprotein containing the alanine substitutions in positions 581–586 was fully functional we attempted to recover a recombinant strain containing the S (581–586) mutations utilizing targeted recombination. Three independent attempts to recover a virus expressing the mutation were unsuccessful.
Fig. 4.

The effect of alanine residue substitution in positions 581–586 on the cleavage properties of spike protein and viral recovery. Immunoprecipitation of 293T cells transfected with the expression vector pCAGGS-S or the expression vector encoding the alanine substituted residues in positions 581–586 of the spike protein (pCAGGS-S (581–586)). The cells were metabolically labeled with [35S]-methionine and cysteine for eight hours at 37°C and the lysate was precipitated with the monoclonal anti-S antibodies A2.1 and A2.3 S uncleaved denotes the 180 kDa uncleaved spike protein, and S cleaved denotes the 90kDa cleaved S1 and S2 subunits.
Thus, despite the sequence similarity between the three short regions of MHV/A59 S protein (546–548, 554–556 and 581–586) to the regions of FcγRII sequences that were part of the structural loops that make up the Fc binding site, only two regions of S were found to be functionally essential for the recovery of new virions, the cysteine residue in position 546 and the region that includes the amino acid sequence in positions 581–586. As noted above, mutation of the amino acids at positions 554–556 did not seem to interfere with the ability of the S protein to be immunoprecipitated by the 2.4G2 antibody from viable recovered recombinants (Fig. 3).
3.3 Replacement of the paired amino acids in positions 562/589 and 667/687 with the HCoV-OC43 homologuous residues affects viral replication
Although the spike glycoproteins of several members of the betacoronaviruses are known to possess the property of Fc binding, the human coronavirus OC43 (HCoV-OC43), also a group 2a betacoronavirus, does not exhibit this property, whereas the very closely related bovine coronavirus (BCoV) does bind the Fc domain of rabbit IgG and is recognized by the anti-FcγRII/RIII monoclonal antibody 2.4G2 (13). We hypothesized that a sequence comparison searching for regions that BCoV and MHV S proteins had in common but that differed from the HCoV-OC43 S protein might identify potential sequences involved in the Fc binding activity. This bioinformatics analysis (Fig. 1B) revealed three distinct regions that included specific pairs of amino acids. These amino acid pairs were conserved within the S protein of MHV-A59 and BCoV but not in HCoV-OC43. Two of the regions were located in the S1 domain. To test our hypothesis, we replaced each one of the paired amino acids in MHV-A59 S protein with the homologous residues in the HCoV-OC43 S protein.
The first paired amino acids substitution was the replacement of the threonine residues in positions 562 and 589 of MHV-A59 spike glycoprotein with their homologues in HCoV-OC43 (T562L and T589L). A second paired amino acid substitution was made by the replacement of serine in position 667 and aspartic acid in position 687 with their homologues in HCoV-OC43 (S667T and D687Y). Plasmids driving the expression of these mutant spike proteins containing these paired amino acid substitutions [pCAGGS-S (562/589) and pCAGGS-S (667/687] were transfected into 293T cells. The transfected cells were metabolically labeled for 9 hours, starting at 24 hours after transfection; subsequently lysates were prepared and immunoprecipitated with the monoclonal neutralizing antibodies A2.1 and A2.3 (Fig. 5A). The non-cleaved and cleaved subunits of the S protein were precipitated by both neutralizing antibodies. However, the signal corresponding to the cleaved S1 and S2 subunits expressed by pCAGGS-S (667/687) and immunoprecipitated by the monoclonal antibodies A2.3 and A2.1 was weaker than the intensity of the signal for S1 and S2 obtained with lysates from cells transfected with wild type pCAGGS-S (Fig. 5A). This data is consistent with the S667T and/or D687Y mutations within MHV-A59 spike protein changing the conformation and either decreasing the cleavage efficiency of the spike protein into S1 and S2 subunits or alternatively decreasing the affinity of both A2.1 and A2.3 antibodies to their binding sites in the uncleaved S protein but not the cleaved subunits. The data does not allow us to distinguish between these two possibilities. Viable MHV-A59 recombinant viruses expressing each one of the paired substituted amino acids were successfully recovered and mutant S proteins immunoprecipitated from infected cell lysates by both antibodies A2.1 (data not shown) and A2.3 (Fig. 5A). The titer achieved by the virus containing the mutant rA59/S (562/589) was about 10-fold less than that achieved by wild type (isogenic control) virus and the titer achieved by rA59/S (667/687) was about 100-fold less than wild type virus (Fig. 5B). A comparison of their plaque sizes (Fig. 5B and 5C) was in line with the relative titers achieved by each, with the diameter of plaques formed by rA59/S (562/589) being 70% of those formed by wild type virus and plaques formed by rA59/S (667/687) being 50% the diameter of wild type plaques. Immunoprecipitation reactions of lysates prepared from rA59/S (562/589) (Supplemental Fig. S1), rA59/S (667/687) (data not shown), and isogenic control infected DBT cells with the anti-FcγRII/RIII antibody 2.4G2 showed that these mutations did not affect the recognition of S by this antibody.
Fig. 5.

The effect of the replaced paired residues T562L/T589L and S667T/D687Y on viral replication and reproduction. A. Immunoprecipitation of 293T cells transfected with the expression vector pCAGGS-S or the expression vector encoding the S protein with the paired replaces residues in positions 562/589 and 667/687 of the spike protein (pCAGGS-S (562/589) and pCAGGS-S (667/687), respectively). The cells were metabolically labeled with [35S]-methionine and cysteine for eight hours in 37°C and the lysate was precipitated with monoclonal anti-S antibodies A2.1 and A2.3. S uncleaved denotes the 180 kDa spike protein and S cleaved represents the 90kDa cleaved S1 and S2 subunits. B. Mean viral titers and plaque diameters of the recombinant viruses rA59/S (562/589), rA59/S (667/687) and rA59/S (pMH54) expressing the mutated and the native spike protein, correspondingly. Error bars represent the standard deviation from the mean. C. Plaque morphologies of the recombinant viruses expressing the native and the mutated spike proteins.
3.4 Replacement of proline 939 in the spike protein reduces the fusogenic properties of the virus
A third pair of amino acids that were identical in BCoV and MHV S proteins but differed in the HCoV-OC43 S protein was identified at positions 910 and 939 of the MHV-A59 spike protein, between the S1–S2 cleavage site and the first heptad repeat sequence (Fig. 1). The paired amino acids residues were located in a region highly conserved amongst the 2a lineage of betacoronaviruses and have been hypothesized to be important for the fusion of the coronavirus envelop with the plasma membrane of the host cell (25, 26). The paired amino acids residues asparagine and proline in positions 910 and 939 of the MHV-A59 spike protein were replaced with their homologues from HCOV-OC43 (N910K and P939L, respectively). The spike protein with the paired amino acids substitutions was expressed in 293T cells by the expression vector pCAGGS-S (910/939) and subsequently the uncleaved S protein and the cleaved subunits were precipitated by the neutralizing antibodies A2.1 (Fig. 6A, left panel) and A2.3 (not shown). The substituted amino acids did not affect binding of the N910K/P929L mutant S protein to either monoclonal antibody, nor did it affect S protein cleavage. We next replaced the wild type S sequence with the S (910/939) S sequence by targeted recombination and were successful in recovering a recombinant virus (rA59/S (910/939)) expressing the 180 kDa spike protein with the substituted amino acids and mutant S proteins immunoprecipitated from infected cell lysates with monoclonal antibody A2.3 (Fig. 6A, right panel). Both cleaved and uncleaved S protein were recognized by A2.3, congruent with the results obtained in the transient transfection experiments (Fig 6A, left panel). A one-step growth curve experiment (Fig. 6B) revealed that the rA59/S (910/939) mutant grew with roughly similar kinetics as the wild type isogenic control virus but reached a peak titer that was depressed by approximately 1 log relative to the isogenic control strain. The effect of the mutation on plaque size was much greater, with the rA59/S (910/939) mutant virus forming significantly smaller plaques, approximately 10% the diameter of those produced by the isogenic control strain (Fig. 6C and 6D). Since the amino acid residue in position 939 resided in the hypothesized fusion peptide (25), we made the working assumption that it was likely that the P939L mutation is largely responsible for the phenotype we observed in A59/S (910/939). Thus, a cDNA containing a single amino acid substitution at position 939 (P939L) was constructed and introduced into the MHV-A59 S protein by targeted recombination and a viable mutant [rA59/S (939)] was recovered. The recombinant rA59/S (939) virus grew with slightly slower kinetics than the double mutant virus, vA59/S (910/939) but ultimately reached a peak titer similar to that of the double mutant (Fig 6B). Based on previously published data implicating this region of the S protein as containing the fusion peptide (25), we examined the ability of rA59/S (939) to induce cell fusion. Cells were infected with rA59/S (939) or the isogenic control virus and monolayers were fixed at different times post infection, nuclei were stained with DAPI and visualized by fluorescent microscopy, and photographed. The average number of nuclei contained in multinucleated giant cells per high-powered field was used as a measure of cell fusion. For cultures infected with the rA59/S (939) mutant the average number of nuclei contained in syncytia per field was decreased approximately four-fold compared to cultures infected by the wild type strain and stained at nine hours post infection (Fig. 7).
Fig. 6.

The effect of N910K/P939L mutations on MHV/A59 spike protein expression and virus growth. A. Left Panel. Cells were transfected with the expression vectors pCAGGS-S or pCAGGS-S (910/939) and after 24 hours incubation they were metabolically labeled with [35S]-methionine and cysteine for eight hours at 37°C and cell lysates precipitated with monoclonal anti-S antibody A2.1. Right Panel. Cells were mock infected, infected with either the isogenic control virus rA59/S (pMH54), or a recombinant mutant virus rA59/S (910/939), labeled with [35S]-methionine and cysteine, and lysates prepared and immunopreciptitated with monoclonal antibody A2.3 as described in Material and Methods. B. One-step growth curve of the recombinant viruses rA59/S (939), rA59/S (910/939) and rA59/S (pMH54). Replicate wells of DBT cells in 96-well plates were infected with mutant of wild-type viruses at MOI of 3 and harvested at 0, 4, 8, 12, 16 and 24 h post-infection. Virus titers were determined by plaque assay of triplicate cultures for each time point and the means titers are displayed. Error bars represent the standard errors of the mean. D. Plaque morphologies of the recombinant viruses expressing the native and the mutated spike proteins. Error bars represent the standard deviation of the mean.
Fig. 7.
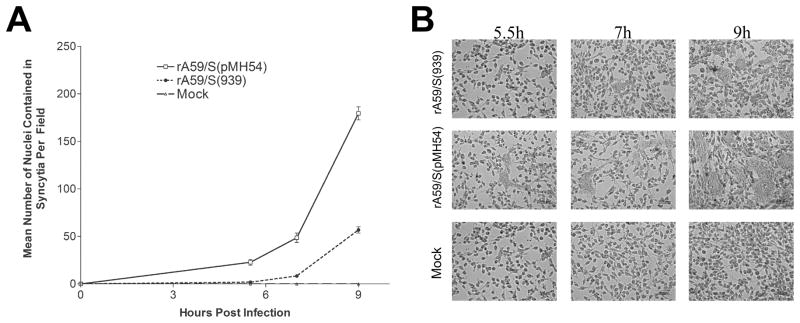
The effect of the P939L substitution mutation in the MHV/A59 spike protein on cell fusion and virus growth. A. DBT cells were infected with recombinant viruses expressing mutated or wild type spike protein, and fixed at 5.5, 7 and 9 h.p.i. Nuclei in the cells were stained with DAPI and the cell fusion score for each virus was determined by counting the number of nuclei in syncytial giant cells per field. Six arbitrary fields were examined for each sample and the mean values plotted. Error bars indicate the standard deviation of three independent experiments. B. Representative images of DBT cells infected with the recombinant viruses after incubation of 5.5, 7 and 9h at 37°C.
As was the case with our prior mutants, the spike proteins containing the 910/939 mutations were immunoprecipitated by the FcγII receptor antibody 2.4G2, as well as the wild type S protein (Supplemental Fig. S1), indicating that the alterations in these sites didn’t interfere with the MHV-A59 spike protein’s ability to mimic of FcγRII.
4. DISCUSSION
In this work we undertook a mutational study to attempt to identify sites within the MHV S protein that were responsible for its molecular mimicry of the murine Fc gamma receptor (mFcγRII and mFcγRIII). Three sequences, at positions 546–548, 554–546, and 581–586, within the spike glycoprotein were candidates to play a role in the recognition of the S protein by the monoclonal anti- mFcγRII /mFcγRIII antibody 2.4G2 because they were conserved in regions of modest amino acid sequence similarity to mFcγRII. These sequences were targeted by alanine replacement mutagenesis. Attempts to determine the reactivity of mutants of the MHV S protein with anti- mFcγRII /mFcγRIII antibody 2.4G2 in transfection experiments were problematical, largely due to difficulties in obtaining sufficient expression levels to obtain robust and reproducible immunoprecipitation reactions of S protein (wild type or mutant) expressed by pCAGG-S with this antibody (data not shown). However, the expression level obtained was sufficiently high for us to perform immunoprecipitation reactions with two conformation- dependent anti-MHV-A59 S monoclonal antibodies (16) to identify mutations that had major effects on S protein folding. S protein carrying the R546A/C547A/Q548A mutation [S (546–548)] was poorly recognized by these conformation-dependent antibodies and the S1 and S2 cleavage products of S were not detected at all, suggesting that this mutation had a major effect on S protein folding. In contrast, S protein carrying alanine substitution mutations in R546 and Q548 but not in C547 was recognized by both of the conformation-dependent antibodies, implicating C547 as being necessary for proper folding of the S protein. This was further supported by our inability to recover a recombinant virus carrying the S (546–548) mutation, whereas the S (546/548) virus was easily recovered by targeted recombination. Some strains of MHV, either viruses recovered in nature, such as MHV-A59, or selected as escape mutants from monoclonal antibody neutralization (27), contain deletions of the S protein relative to the long S proteins found in the MHV-JHM or MHV-S strains. The JHM-X strain contains the longest deletion in the S1 domain amongst these deletion variants and C547 is the first amino acid on the C-terminal side of the deletion (28). Furthermore, an alignment of MHV and mFcγRII shows absolute conservation of this amino acid amongst all strains of MHV and its location in the secondary predicted structure (Fig. 8). We think it likely that C547 participates in a disulfide bridge that is important for proper folding of the S1 domain. In addition, because the deletion in JHM-X is substantial, 153 amino acids, and deletes 15 cysteine residues N-terminal of C547, we think it likely that the partner cysteine to C547 lies somewhere between C547 and the S1/S2 cleavage site at positions 717 and 718. Recovery of recombinant viruses containing the S (546/548) mutation allowed us to determine that alanine replacement of the arginine and glutamine at these two residues did not affect the ability of the S protein to be recognized by the 2.4G2 anti-mFcγRII /mFcγRIII antibody, and thus that this region of sequence similarity was unlikely to be involved in molecular mimicry of FcγRII.
Fig. 8.

Amino acid sequence homology between S protein and mFcγRII regions. A. A scheme showing the homologous regions of S protein and mFcgRII receptor in positions 546–548, 554–556 and 581–586 (lower case letters). B. A comparison between the predicted secondary structures of the binding domains of murine Fcγ receptor type II, mFcγR (Panel A) and the homologous regions of spike protein of MHV/A59 (Panel B) predicted by PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred). The H stands for helix, C for coil and E for strand. The bars for each amino acid represent the confidence of each prediction. The taller the bar is, the higher the confidence. The amino acid sequences of the short regions of sequence similarity in the two structures are underlined similarly.
The mutational studies of the other two regions of sequence similarity between FcγRII and the MHV S protein, at positions 554–556 and 581–586 also failed to identify the epitope that was shared between these two proteins. In the case of the alanine replacement mutations at S residues 554–556 we were able to recover a virus carrying these mutations by targeted recombination and test its reactivity with 2.4G2. For the alanine replacement mutations in S at residues 581–586 this was not possible because we failed to recover a mutant virus by targeted recombination. These mutations altered the pattern of reactivity of mutant S protein with two conformation-dependent anti-A59 S antibodies, indicating that S protein carrying this mutation likely was not folded correctly. Thus, we cannot exclude the possibility that the residues at 581–586 are part of the epitope recognized by 2.4G2.
A comparative bioinformatics approach comparing the sequence from the S proteins of HCoV-OC43, a group 2a beta-coronavirus that does not exhibit molecular mimicry of FcγRII with the sequences of the very closely related BCoV and with the MHV S protein, both of which exhibit this property, identified three potential targets for mutagenesis. Of these three mutants two, vA59/S (562/589) and vA59/S (667/687), contained mutations located in the S1 domain. Both gave rise to viable viruses, although with somewhat impaired replication, but both mutant S proteins were immunoreactive with the 2.4G2 anti-mFcγRII /mFcγRIII antibody, and thus the two regions targeted by our mutations were unlikely to be involved in molecular mimicry of FcγRII. Our third mutant, containing N910K and P939L mutations, was located in the S2 domain. Although this double point mutation did not have alter the molecular mimicry properties of the S protein (immunoreactivity with 2.4G2 anti-mFcγRII /mFcγRIII antibody), the mutation had a marked effect on plaque size, growth kinetics, and rate of cell fusion. These effects were determined to largely reside with the P939L mutation, a residue that is 29 amino acids N-terminal to the first heptad repeat domain (968–1027) within the S2 fragment. The sequences between positions 929 and 944, which contain P939, have been suggested to contain the putative MHV fusion peptide (25). This assignment is controversial in that replacement of several amino acids in the putative fusion peptide did not appear to affect the fusion properties of the S protein, though P939 was not tested in these studies (29). The data in our studies is consistent with the hypothesis that P939 plays a role in the fusion properties of MHV-A59 and strengths the possibility of this region contains the fusion peptide. Alternatively, our data do not rule out the possibility that the P939L mutation either decreases the stability of the S protein or the rate of transport of S protein to the plasma membrane, and thus decreases cell fusion through of those two mechanisms. However, we think it less likely that the P939L mutation has a major effect on S protein stability because this residue is present in an equivalent position in the HCoV-OC43 S protein. HCoV-OC43 also does not induce cell-cell fusion during infection.
These studies ultimately failed to identify mutations in the MHV S protein that abolished molecular mimicry of mFcγRII, as judged by immunoreactivity with the 2.4G2 anti-mFcγRII /mFcγRIII antibody. There are several possibilities for this result, one being that the residues involved with molecular mimicry are not linear but are brought together in space as a result of secondary and tertiary structure in S, as is the case for the Fc binding site formed by gE and gI complex in HSV (30, 31). Other potential explanations include a crucial role for residues making up the Fc binding site in the correct folding of the S protein, making it difficult to identify mutants that fold correctly and have defects in Fc binding activity.
Supplementary Material
The effect of the replaced paired residues T562L/T589L and N910K/P939L on S protein molecular mimicry of FcγRII. Replicate cultures of DBT cells were either mock infected, or infected with the isogenic control virus rA59/S (pMH54), the recombinant virus rA59/S (562/589), or rA/ S (910/939). The cells were incubated until CPE was apparent at 8 hpi and metabolically labeled with [35S]-methionine and cysteine for one hour after which lysates were prepared and immunoprecipitated with the monoclonal anti-FcγRII antibody 2.4G2 as described in Materials and Methods. S uncleaved denotes the 180 kDa uncleaved spike protein.
HIGHLIGHTS.
Identification of functional regions within MHV-A59 spike (S) protein through analysis of sequence similarities with FcγR.
C547 substitution abolishes the recognition of cleaved S by monoclonal antibodies.
Substitution of residues 547 and 581–586 in S prevents the recovery of a viable virus.
Amino acid replacements at positions 562/589 and 667/687 in S affect viral replication.
Replacement of residue 939 in S affects the fusogenic properties of the virus.
Acknowledgments
The authors gratefully acknowledge support from US National Institutes of Health grant AI079857. We also would like to thank Dr. Tom Gallagher for the pCAGGS-S vector, Drs. Susan Weiss and Paul Masters for providing us with the targeted recombination system, Drs. John Fleming and Wendy Gilmore for providing us with anti-MHV-A59 spike protein hybridomas, and Dr. Kathryn Holmes for providing us with a polyclonal anti-MHV-A59 spike protein antibody.
Footnotes
CONFLICT OF INTEREST
There are no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weiss SR, Leibowitz JL. Coronavirus pathogenesis. Adv Virus Res. 2011;81:85–164. doi: 10.1016/B978-0-12-385885-6.00009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiss SR, Leibowitz JL. Pathogenesis of murine coronavirus infections. In: Perlman S, Gallagher T, Snijder EJ, editors. Nidoviruses. ASM Press; Washington, D.C: 2007. pp. 259–278. [Google Scholar]
- 3.Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol. 2000;74:9206–9213. doi: 10.1128/jvi.74.19.9206-9213.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowley TJ, Long SY, Weiss SR. The murine coronavirus nucleocapsid gene is a determinant of virulence. J Virol. 2010;84:1752–1763. doi: 10.1128/JVI.01758-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dveksler GS, Pensiero MN, Cardellichio CB, Williams RK, Jiang G, Holmes KV, Dieffenbach CW. Cloning of the mouse hepatitis virus (MHV) receptor: expression in human and hamster cell lines confers susceptibility to MHV. Journal of Virology. 1991;65:6881–6891. doi: 10.1128/jvi.65.12.6881-6891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu H, Yu D, Zhang X. The spike protein of murine coronavirus regulates viral genome transport from the cell surface to the endoplasmic reticulum during infection. J Virol. 2009;83:10653–10663. doi: 10.1128/JVI.00956-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castro RF, Perlman S. CD8+ T-cell epitopes within the surface glycoprotein of a neurotropic coronavirus and correlation with pathogenicity. J Virol. 1995;69:8127–8131. doi: 10.1128/jvi.69.12.8127-8131.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frana MF, Behnke JN, Sturman LS, Holmes KV. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: host-dependent differences in proteolytic cleavage and cell fusion. J Virol. 1985;56:912–920. doi: 10.1128/jvi.56.3.912-920.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Haan CA, Stadler K, Godeke GJ, Bosch BJ, Rottier PJ. Cleavage inhibition of the murine coronavirus spike protein by a furin-like enzyme affects cell-cell but not virus-cell fusion. J Virol. 2004;78:6048–6054. doi: 10.1128/JVI.78.11.6048-6054.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heald-Sargent T, Gallagher T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses. 4:557–580. doi: 10.3390/v4040557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oleszak EL, Leibowitz JL. Immunoglobulin Fc binding activity is associated with the mouse hepatitis virus E2 peplomer protein. Virology. 1990;176:70–80. doi: 10.1016/0042-6822(90)90231-F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oleszak EL, Perlman S, Leibowitz JL. MHV S peplomer protein expressed by a recombinant vaccinia virus vector exhibits IgG Fc-receptor activity. Virology. 1992;186:122–132. doi: 10.1016/0042-6822(92)90066-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oleszak EL, Kuzmak J, Hogue B, Parr R, Collisson EW, Rodkey LS, Leibowitz JL. Molecular mimicry between Fc receptor and S peplomer protein of mouse hepatitis virus, bovine corona virus, and transmissible gastroenteritis virus. Hybridoma. 1995;14:1–8. doi: 10.1089/hyb.1995.14.1. [DOI] [PubMed] [Google Scholar]
- 14.Tamm A, Kister A, Nolte KU, Gessner JE, Schmidt RE. The IgG binding site of human FcgammaRIIIB receptor involves CC′ and FG loops of the membrane-proximal domain. J Biol Chem. 1996;271:3659–3666. doi: 10.1074/jbc.271.7.3659. [DOI] [PubMed] [Google Scholar]
- 15.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 16.Gilmore W, Fleming JO, Stohlman SA, Weiner LP. Characterization of the structural proteins of the murine coronavirus strain A59 using monoclonal antibodies. Proc Society Exp Biology and Med. 1987;185:177–186. doi: 10.3181/00379727-185-42532. [DOI] [PubMed] [Google Scholar]
- 17.Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol. 2000;74:1393–1406. doi: 10.1128/jvi.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips JJ, Chua M, Seo SH, Weiss SR. Multiple regions of the murine coronavirus spike glycoprotein influence neurovirulence. J Neurovirol. 2001;7:421–431. doi: 10.1080/135502801753170273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leibowitz J, Kaufman G, Liu P. Coronaviruses: propagation, quantification, storage, and construction of recombinant mouse hepatitis virus. Curr Protoc Microbiol. 2011;Chapter 15(Unit 15E):11. doi: 10.1002/9780471729259.mc15e01s21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leibowitz JL, Wilhelmsen KC, Bond CW. The Virus-Specific Intracellular RNA Species of Two Murine Coronaviruses: MHV-A59 and MHV-JHM. Virology. 1981;114:39–51. doi: 10.1016/0042-6822(81)90250-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boscarino JA, Logan HL, Lacny JJ, Gallagher TM. Envelope protein palmitoylations are crucial for murine coronavirus assembly. J Virol. 2008;82:2989–2999. doi: 10.1128/JVI.01906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cormack B. Directed Mutagenesis Using the Polymerase Chain Reaction, Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2001. [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK, Favre M. Maturation Of the Head Of Bacteriophage T4. I. DNA Packaging Events. J Mol Biol. 1973;80:575–599. doi: 10.1016/0022-2836(73)90198-8. [DOI] [PubMed] [Google Scholar]
- 24.Leibowitz JL, DeVries JR, Haspel MV. Genetic analysis of murine hepatitis virus strain JHM. Journal of Virology. 1982;42:1080–1087. doi: 10.1128/jvi.42.3.1080-1087.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chambers P, Pringle CR, Easton AJ. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J Gen Virol. 1990;71 ( Pt 12):3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- 26.de Groot RJ, Luytjes W, Horzinek MC, Van Der Zeijst BAM, Spaan WJM, Lenstra JA. Evidence for a Coiled-coil Structure in the Spike Proteins of Coronaviruses. J Mol Biol. 1987;196:963–966. doi: 10.1016/0022-2836(87)90422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parker SE, Gallagher TM, Buchmeier MJ. Sequence analysis reveals extensive polymorphism and evidence of deletions within the E2 glycoprotein gene of several strains of murine hepatitis virus. Virology. 1989;173:664–673. doi: 10.1016/0042-6822(89)90579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taguchi F, Fleming JO. Comparison of six different murine coronavirus JHM variants by monoclonal antibodies against the E2 glycoprotein. Virology. 1989;169:233–235. doi: 10.1016/0042-6822(89)90061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo Z, Weiss SR. Roles in cell-to-cell fusion of two conserved hydrophobic regions in the murine coronavirus spike protein. Virology. 1998;244:483–494. doi: 10.1006/viro.1998.9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sprague ER, Wang C, Baker D, Bjorkman PJ. Crystal structure of the HSV-1 Fc receptor bound to Fc reveals a mechanism for antibody bipolar bridging. PLoS Biol. 2006;4:e148. doi: 10.1371/journal.pbio.0040148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu-Bin, Murayama T, Ishida K, Furukawa T. Characterization of IgG Fc Receptors Induced by Human Cytomegalovirus. J Gen Virol. 1989;70:893–900. doi: 10.1099/0022-1317-70-4-893. [DOI] [PubMed] [Google Scholar]
- 32.Bos ECW, Luytjes W, Spaan WJM. The function of the spike protein of mouse hepatitis virus of strain A59 can be studied on virus like particles: cleavage is not required for infectivity. Journal of Virology. 1997;71:9427–9433. doi: 10.1128/jvi.71.12.9427-9433.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of the replaced paired residues T562L/T589L and N910K/P939L on S protein molecular mimicry of FcγRII. Replicate cultures of DBT cells were either mock infected, or infected with the isogenic control virus rA59/S (pMH54), the recombinant virus rA59/S (562/589), or rA/ S (910/939). The cells were incubated until CPE was apparent at 8 hpi and metabolically labeled with [35S]-methionine and cysteine for one hour after which lysates were prepared and immunoprecipitated with the monoclonal anti-FcγRII antibody 2.4G2 as described in Materials and Methods. S uncleaved denotes the 180 kDa uncleaved spike protein.