

Abstract
To obtain mechanistic insights into the inherent reactivity patterns for copper(I)-O2 adducts, a new cupric-superoxo complex [(DMM-tmpa)CuII(O2˙−)]+ (2) [DMM-tmpa = tris((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)amine] has been synthesized and studied in phenol oxidation-oxygenation reactions. Compound 2 is characterized by UV-vis, resonance Raman and EPR spectroscopies. Its reactions with a series of para-substituted-2,6-di-tert-butylphenols (p-X-DTBP's) afford 2,6-di-tert-butyl-1,4-benzoquinone (DTBQ) in up to 50 % yields. Significant deuterium kinetic isotope effects (KIE's) and a positive correlation of second-order-rate constants (k2's) compared to rate constants for p-X-DTBP's plus cumylperoxyl radical reactions indicate a mechanism involves rate-limiting hydrogen atom transfer (HAT). A weak correlation of (kBT/e) lnk2 vs Eox of p-X-DTBP's indicates the HAT reactions proceed via a partial transfer of charge rather than a complete transfer of charge in the electron-transfer/proton-transfer (ET/PT) pathway. Product analyses, 18O-labeling experiments, and separate reactivity employing the 2,4,6-tri-tert-butylphenoxyl radical provide further mechanistic insights. After initial HAT, a second mole equiv 2 couples to the phenoxyl radical initially formed, giving a CuII-OO-(ArO') intermediate, which proceeds in the case of p-OR-DTBP substrates via a two-electron oxidation reaction involving hydrolysis steps which liberate H2O2 and the corresponding alcohol. By contrast, four-electron oxygenation (O-O cleavage) mainly occurs for p-R-DTBP which gives 18O-labeled DTBQ and elimination of the R group.
Introduction
Investigation of the structure and reactivity of various CuI/O2 adducts (Chart 1), those derived from the reaction of reduced ligand-copper(I) complexes with molecular oxygen (dioxygen) occurs in large part due to their critical roles in metalloenzymes.1 Monocopper complexes A–C or dinuclear species E–G, or their analogs, have been discovered and insights into their electronic structural characteristics and 3-D structures have been obtained.1 Formally high-valent species D has been detected in the gas phase,2 hinted at in coordination chemistry studies3 or discussed computationally.4 Among them, the mononuclear species such as cupric superoxide (A and B), cupric hydroperoxide (C), and copper oxyl (D) species all have been considered as highly reactive intermediates which likely participate in overall reaction sequences occurring in copper enzyme bio-transformations.5
Chart 1.
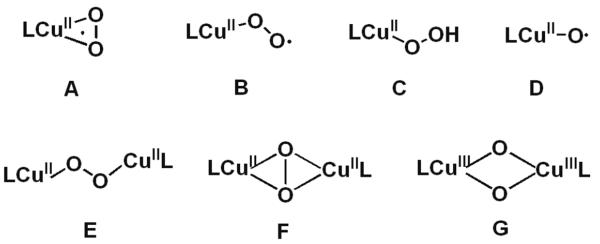
They may hydroxylate the substrate C-H bond in copper enzymes such as peptidylglycine-α-hydroxylating monooxygenase (PHM) and dopamine-β-monooxygenase (DβM)6 or initiate O-H oxidation in copper oxidases such as galactose oxidase (GO)6a,7 or copper amine oxidase (CAO).6a,8 From relatively recent experimental and computational studies, the [CuII(O2˙−)]+ (A or B) moiety has been declared as the reactive intermediate which effects C–H9,10 or O–H7,8 hydrogen-atom transfer (HAT) reactions. Relevant to this is an X-ray crystallographic study on a PHM derivative, which reveals a dioxygen-derived species assigned as an endon bound cupric superoxo species (B) (also discussion below).11 As is relevant to part of the GO catalytic cycle, recent experimental studies support a mechanism wherein O2-binding to this fully reduced copper ion affords a cupric superoxo species (Figure 1) and that this mediates the oxidation of the ligated tyrosine residue resulting in the formation of a cupric hydroperoxide species (C) plus phenoxyl radical.7c Hydrolysis leads to the release of hydrogen peroxide, the observed stoichiometric O2 reduction product in the overall mechanism.
Figure 1.
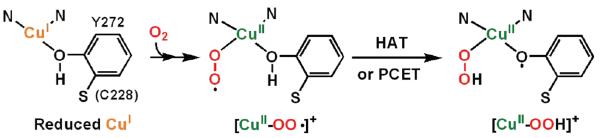
Reduction of molecular oxygen in galactose oxidase (GO). See Supporting Information (Figure S1a) for fuller details.
With this background, one of the research goals that we have recently been emphasizing is on the chemistry of cupric superoxo complexes. It is critical to elucidate chemical/physical properties, spectroscopic characteristics and reactivity toward substrates possessing C-H or O-H bonds. An understanding of such aspects is critical in order to fully elucidate fundamentals involved in oxidative processes and the reduction of dioxygen by copper centers in chemical systems or at the active sites of metalloenzymes.
In this report, we describe the chemistry of a new [CuII(O2˙−)]+ complex and a detailed investigation into its reactions with phenolic substrates. As discussed above, the net hydrogen atom abstraction reaction from a phenol derivative is directly relevant to the enzyme chemistry in GO. Similar reactivity occurs in copper amine oxidase (CAO) where a cupric superoxo species is thought to undergo a proton-coupled electron transfer (PCET) process converting TPQSQ (2,4,5-trihyroxyphenylalanine semiquinone) to TPQIMQ, the one-electron oxidized iminoquinone form (Figure S1b).8 As mentioned, phenols can undergo oxidation by two pathways, both resulting in formation of the neutral phenoxyl radical and transfer of a hydrogen atom to the oxidant, eq 1. Mechanistically, the process can occur by PCET12 or hydrogen atom transfer (HAT) and these are fundamentally important reactions occurring in many biological systems.
| (1) |
Itoh, Fukuzumi and co-workers13 previously investigated phenol (and C-H) oxidation chemistry using binuclear complexes of the type (μ-η2:η2-peroxo)dicopper(II) (F) and/or bis(μ-oxo)dicopper (III) (G). For a series of phenols, kinetic/mechanistic studies demonstrated that these oxidations proceeded via PCET rather than HAT. However, studies on the reactivity of mononuclear CuI/O2 species are scarce and in fact no detailed mechanistic investigations have been described. Only recently have ligand design and synthetic methodologies better allowed for the formation of discrete [CuII(O2˙−)]+ adducts making it possible for the systematic investigation into their inherent chemical and physical properties, along with substrate reactivity.14
Furthermore, the investigation into exogenous phenolic substrate O-H oxidation by [CuII(O2˙−)]+ complexes has been limited to the report of product yields for a few phenol or catechol substrates. Thus, utilizing the tripodal tetradentate ligand NMe2-tmpa (Chart 2), which forms a low-temperature stable end-on bound superoxo copper(II) complex, we found that 2,6-di-tert-butylphenol derivatives (p-X-DTBP; X = OMe, tBu, H) were oxidized to corresponding 2,6-di-tert-butyl-1,4-benzoquinones (DTBQ) (Scheme 1).
Chart 2.
Scheme 1.

We herein describe a detailed study employing a new electron-rich ligand supporting the cupric superoxo complex, [(DMM-tmpa)CuII(O2˙−)]+ (2), (Scheme 1) which is capable of mediating this chemistry for a large series of p-X-DTBP (X = alkoxy/alkyl) substrates. We provide decisive kinetic evidence supporting cupric superoxo complex mediated HAT as the rate-limiting step in phenol O-H bond activation and two- and/or four- electron reduction of molecular oxygen, vide infra. There are three possible reaction pathways in the overall hydrogen-atom transfer from p-X-DTBP to [CuII(O2˙−)]+; (1) electron transfer (ET) followed by proton transfer (PT), (2) PT followed by ET and (3) concerted ET and PT. The former two pathways correspond to PCET processes, whereas the third one, i.e., concerted ET and PT corresponds to a direct HAT process. The mechanistic conclusions are drawn by several corroborating studies employing physical measurements leading to i) kinetic isotope effect (KIE) determination, ii) correlation of the relative reactivity for hydrogen atom abstraction using cumylperoxyl radical (4) as a mechanistic benchmark, iii) comparison of the rate-dependences in the phenol oxidations by 2 and 4 on the one-electron oxidation potentials of the p-X-DTBP's, and iv) activation parameters determined from the reaction kinetics. We also discuss the experimental results from product analysis showing that either overall two-electron oxidation or four-electron oxygenation of the p-X-DTBP's can occur; the results depend on the identity of the substituent X. Detailed pathways leading to the products observed are proposed.
Experimental Section
General Considerations
All materials used were of commercially available reagent quality unless otherwise stated. Acetone was distilled from Drierite under argon atmosphere. Tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MeTHF) inhibitor free was purchased from Sigma-Aldrich and distilled under argon from Sodium/benzophenone prior to use. Pentane was distilled under argon over CaH2. Acetonitrile was purified via passage through a double alumina column solvent purification system from Innovative Technologies, Inc. Di-tert-butyl peroxide was purchased from Nacalai Tesque Co., Ltd and purified by chromatography through alumina which removes traces of the hydroperoxide. Cumene was purchased from Tokyo Kasei Industry Co., Ltd. Air-sensitive compounds were synthesized and transferred under an argon atmosphere using standard Schlenk techniques and stored in an MBraun glovebox filled with N2. [CuI(CH3CN)4]B(C6F5)4 was synthesized as previously reported.15 2,4,6-Tri-tert-butylphenoxyl radical (tBu3ArO•) was synthesized following a procedure reported in the literature16 and characterized with UV-vis absorption band at 384 and 402 nm and sharp EPR signal at g = 2.00. Elemental analyses were performed by Desert Analytics, Tucson, AZ. The 1H-NMR spectrum was measured on a Bruker 300 MHz or a Bruker 400 MHz spectrometer. 2H-NMR was recorded on the broadband coil on a 300 MHz instrument with 2H resonance at 46 MHz. Chemical shifts are reported in ppm downfield against TMS or residual solvent signals unless otherwise specified. Benchtop low temperature UV-vis experiments were carried out on a Cary bio-50 spectrophotometer equipped with a Unisoku USP-203A cryostat using a 1 cm modified Schlenk cuvette. EPR measurements were performed on a Bruker X band EPR 5 mm quartz EPR tubes (Willmad). Electrospray ionization (ESI) mass spectra were acquired using a Finnigan LCQDeca ion-trap mass spectrometer equipped with an electrospray ionization source (Thermo Finnigan, San Jose, CA). Sulfur dioxide (SO2, gas) was prepared by mixing sodium metabisulfite-saturated water and diluted sulfuric acid solution (Figure S2). 2,4,6-Tri-tert-butyl-4-hydroperoxycyclohexa-2,5-dienone was prepared as in the literature,17 and characterized by 1H-NMR spectroscopy.
Ligand Synthesis
DMM-tmpa [tris((4-methoxy-3,5-dimethyl pyridin-2-yl)methyl)amine] ligand utilized in this report was synthesized following a procedure described in the literature.18
2-Phthalimidomethyl-4-methoxy-3,5-dimethylpyridine
TLC on Alumina (1:1 = EtOAc : Hexane), Rf = 0.63;1H-NMR CDCl3 – 8.05 (s, 1H), 7.89–7.86 (dd, 2H, j = 4), 7.72–7.70 (dd, 2H, j = 4), 4.93 (s, 2H), 3.75 (s, 3 H), 2.31 (s, 3H), 2.17 (s, 3H); FAB-MS Calculated MH+ = 297.12392 Found 297.12352
(4-Methoxy-3,5-dimethylpyridin-2-yl)methanamine
TLC on Alumina (95:5 = DCM : MeOH), Rf = 0; 1H-NMR CDCl3 – 8.16 (s, 1 H), 3.87 (s, 2H), 3.71 (s, 3H), 2.20 (s, 3H), 2.16 (s, 3H), 1.98 (s (br), 2 H)
DMM-tmpa
TLC on Alumina (95:5 = DCM : MeOH), Rf = 0.29;1H-NMR CDCl3 - 8.18 (s, 3H), 3.74 (s, 6H), 3.65 (s, 9H), 2.22 (s, 9H), 1.61 (s, 9H); 1H-NMR Acetone-d6 - 8.16 (s, 3H), 3.68 (s, 9H), 3.67 (s, 6H), 2.21 (s, 9H), 1.62 (s, 9H). FAB-MS Calculated MH+ = 465.28657 Found 465.28582
4-Bromo-2,6-di-tert-butyl-2,5-cyclohexadienone
The preparation of 4-bromo-2,6-di-tert-butyl-2,5-cyclohexadienone was accomplished using a modified published procedure.19 1H-NMR CDCl3 – 6.77–6.75 (d, 2H), 5.38–5.36 (t, 1H), 1.24 (s, 9H); 13C-NMR CDCl3 – 185.05, 143.74, 136.60, 77.31
Synthesis of para-Alkoxyl-2,6-di-tert-butylphenols (p-ORDTBP's)
Selected phenols were synthesized from adapted published procedures.20 A 22 mL vial was charged with 810.7 mg of silver triflate and a magnetic stir bar. To this vial, 10 mL of the corresponding alcohol (RO-H) was added. If needed, a minimal amount of dimethylethanol (DME) was used to dissolve the silver salt. To this solution, 900 mg of 4-bromo-2,6-di-tert-butyl-2,5-cyclohexadienone dissolved in 2.5 mL DME was rapidly added resulting in the immediate formation of a precipitate and the solution to turn yellow. After 3 min the solution was poured into a 50 mL solution containing ~1 g of NaSH. The organics were separated by extraction with four times 50 mL portions of pentane. This pentane solution was dried over sodium sulfate and filtered using a 0.45 μm filter. The pentane was removed by vacuum and the phenol was purified by flash chromatography using silica gel eluting with 0~2.5 % gradient of EtOAc : petroleum ether. The phenols were isolated as pale yellow to white solids and the purity was checked by NMR and GC-MS. If necessary, phenols were crystallized from petroleum ether. Phenol purity for kinetic analysis was >95%. The yields of phenols varied according to substituent but were typically 25~70 %.
p-OCH2CH3-DTBP
1H-NMR CDCl3 – 6.758 (s, 2H), 4.757 (s, 1H), 4.02–3.95 (q, 2H, j= 6.9), 1.43 (s, 18H), 1.419–1.372 (t, 3H, j= 6.9); 13C-NMR CDCl3 – 151.83, 147.66, 137.17, 111.262, 63.80, 34.576, 30.22, 15.10; FAB-MS Calculated M+ = 250.19328 Found 250.19312
p-OCD2CD3-DTBP
1H-NMR CDCl3 - 6.758 (s, 2H), 4.757 (s, 1H), 4.02–3.95 (q, 2H, j= 6.9), 1.43 (s, 18H), 1.419–1.372 (t, 3H j= 6.9); 13C-NMR CDCl3 – 151.83, 147.66, 137.17, 111.262, 63.80, 34.576, 30.22, 15.10; FAB-MS Calculated M+ = 255.22466 Found 255.22453
p-OCD3-DTBP
1H-NMR CDCl3 - 6.766 (s, 2H), 4.773 (s, 1H), 1.43 (s, 18H); 2H-NMR CDCl3 - 3.759(s); 13C-NMR CDCl3 – 152.41, 147.68, 110.53, 34.57, 30.18; FAB-MS Calculated M+ = 239.19696 Found 239.19629
p-OCH2CF3-DTBP
1H-NMR CDCl3 - 6.86 (s, 2H), 4.975(s, 1H), 4.40–4.316 (q, 2H, j= 8.4), 1.49 (s, 18H); 13C-NMR CDCl3–150.64, 149.17, 137.49, 129.14–118.082 (q, j = 278) 112.19, 67.71–66.316 (q, j= 35.3) 34.59, 30.121; 19F-NMR 74.61–74.67 (t, j=8.5); FAB-MS Calculated M+ = 304.16501 Found 304.16607
p-OMPP-DTBP (2-methyl-1-phenylpropan-2-yloxy)
1H-NMR CDCl3 - 7.332–7.31 (m, 5H), 6.778 (s, 2H), 4.931 (s, 1H), 2.989 (s, 2H), 1.431 (s, 18H), 1.235 (s, 6H); 13C-NMR CDCl3 – 149.82, 147.18, 138.50, 136.15, 130.77, 127.83, 126.19, 120.88, 79.83, 48.85, 34.27, 30.28, 30.25, 26.23; FAB-MS Calculated M+ = 354.25588 Found 354.25570
Preparation of 2H-O-2,6-di-tert-butyl-4-methoxyphenol
A Schlenk flask containing 1.25 g of p-OMe-DTBP and a stir bar was dissolved in 20 mL of freshly distilled THF. The sample was then chilled to −78 °C and 1.1 equiv of n-butyllithium dissolved in pentane was slowly added to the solution. The solution was allowed to react at low temperature for 30 min, after which 1.4 mL of D2O was added to the solution in ~150 μL portions. The solution was slowly allowed to warm to room temperature and the solvent removed in vacuo yielding a white precipitate. The desired phenol was extracted from the flask by the addition of ~20 mL of freshly distilled pentane and the resulting pentane solution was filtered through a plug of celite. The pentane was removed in vacuo yielding the desired product as a white solid. The sample was stored in the glove box and the 2H content was assessed by the absence of the RO-H proton resonance at ~ 4.8 ppm. 2H content was determined to be >98%.
Redox Potentials of para-Substituted-2,6-di-tert-butylphenols (p-X-DTBP's)
The redox potentials were measured by second harmonic AC voltammetry (SHACV) in CH3CN containing 0.10 M Bu4N+PF6− as a supporting electrolyte using an ALS-630B Electrochemistry Analyzer with a three electrode set up consisting of a platinum disk working electrode, platinum wire counter electrode and an Ag/AgNO3 reference electrode. The voltammograms are plotted against the [Fe(Cp)2]+/0 potential which was measured as an external standard. The E0 values (vs Ag/AgNO3) were converted to those vs the [Fe(Cp)2]+/0 (Fc/Fc+) potential which was measured as an external standard. All electrochemical measurement was carried out at 25 °C under argon atmosphere. Scans were run at 4 mV s−1.
Synthesis of [(DMM-tmpa)CuI(CO)]B(C6F5)4 (1)
This complex was synthesized in a manner very similar to that for the previously reported complex [(NMe2-tmpa)CuI(CO)]B(C6F5)4.21 A 100 mL Schlenk flask containing a stir bar, 100 mg of DMM-tmpa, and 195 mg of [CuI(CH3CN)4]B(C6F5)4 was evacuated and the flask purged with argon on the vacuum line. To this reaction flask, a Claisen adapter fitted with two air-free addition funnels consisting of ~50 mL of THF and ~125 mL of pentane, respectively, was attached. The argon line was replaced with a carbon monoxide (CO) line and the resulting solutions deaerated with briskly flowing CO (g) for 20 min. Approximately 10 mL of the CO-saturated THF solution was used to dissolve the ligand and copper salt. The yellow solution was allowed to stir for ~5 min under a positive pressure of CO. The CO-saturated pentane was added to the THF solution resulting in the solution to turn cloudy and an oil to settle to the bottom of the flask. Excess solvent was decanted off under a CO (g) atmosphere and the resulting oil dried under vacuum yielding a white solid (70 % yield). 1H-NMR Acetone-d6 - 8.39 (s, 3H), 4.13 (s, 6H), 3.82 (s, 9H), 2.29 (s, 9H), 2.24 (s, 9H). Cu-CO stretch 2085 cm−1 in CH3CN, Elemental (Calc. C 50.56, H 2.94, N 4.54; Found C 50.96, H 2.95, N 4.52). Note : The use of the CO-adduct of the copper(I) complex is required in order to prevent disproportionation. The counteranion, B(C6F5)4, is utilized for better solubility of the superoxo-copper(II) complex and products derived from its reactions, at very low temperatures.
Generation of [(DMM-tmpa)CuII(O2˙−)]B(C6F5)4 (2) and Reaction with p-X-DTBP Derivatives
Kinetic Measurements
In the glove box, 0.27 mM solution of [(DMM-tmpa)CuI(CO)]B(C6F5)4 (1) was prepared in a 2.5 mL acetone solvent mixture (10 % MeTHF), and the 1 cm Schlenk cuvette was sealed with a rubber septum. Out of the glove box and at RT, the solution was immediately purged for 15 s with CO (g) using a long syringe needle. The solution was then cooled to the appropriate temperature (−100 °C ~ −85 °C) and dioxygen was gently bubbled through the solution. Clean isosbestic conversion to 2 is obtained within 15 min monitored by UV-vis spectroscopy.21 This green intermediate is stable for hours at −90 °C. Phenol oxidation reactions were initiated by the addition of a stock solution of phenol to the fully generated 2 after three times of Ar/Vacuum purge cycles. Pseudo-first-order rate plots were performed by observing the disappearance of 409 nm band to obtain plots of ln [(A − Af)/(Ai − Af)] vs time(s), which were found to be linear for three or more half-lives. {General Notes: (i) Complex 2 is stable for hour at −90 °C in acetone, as judged by any absorbance loss at λmax = 409 nm. This “lifetime” is well beyond times needed for kinetic studies. (ii) The superoxo complex 2 can also be generated in other solvents such as THF or MeTHF, but it is less stable than in acetone. (iii) If CO (g) is not present in excess in the initial solution of 1 (in 10 % MeTHF/acetone at −90 °C), the binuclear peroxodicopper(II) species [{(DMM-tmpa)CuII}2(μ-1,2-O22−)]2+ readily forms upon addition of O2. (iv) Further, this dicopper species is formed when higher concentrations of 1 (> 1 mM) are employed in the generation of the 2, thus confining the range of concentrations used in all of the studies described. (v) Superoxo complex 2 does slowly decay with warming to above −85 °C, however [{(DMM-tmpa)CuII}2(μ-1,2-O22−)]2+ is not the product.}
Quantification of 2,6-di-tert-butyl-1,4-benzoquinone (DTBQ) with Gas Chromatography-Mass Spectrometry (GCMS)
A Schlenk flask was charged with 10 mL of 0.25 mM acetone solution of [(DMM-tmpa)CuI(CO)]B(C6F5)4 (1) in the glove box, and out of the glove box, the solution was immediately purged with CO (g) at room temperature. The solution was then cooled to the −90°C acetone/liquid nitrogen cooling bath and dioxygen was gently bubbled through the solution to generate [(DMM-tmpa)CuII(O2˙−)]B(C6F5)4 (2). After 2 was fully formed, three times of Ar/Vacuum purge cycles were applied to remove excess dioxygen and a stock solution of substrates (1 ~ 50 equiv) was added to the solution. When the reaction ended, the Schlenk flask was warmed up to room temperature. (Note: we also analyzed solutions quenched at low temperature by addition of SO2, but the results and yields of reactions were not affected). The solvent was removed in vacuo, redissolved in 300 μL of the solvent and transferred in a GC-MS vial with addition of 0.8 μmol of naphthalene as a standard. Then 1 μL was injected into the GC-MS. The area ratio was converted to mole ratio to quantify the yield of DTBQ by using standard curve. All GC-MS experiments were carried out and recorded using a Hewlett-Packard 6890 Series Gas Chromatograph System equipped with 5973N Mass Selective Detector. The GC-MS conditions for the product analysis were: Injector Port Temperature: 250 °C; Column Temperature: Initial Temperature 80 °C; Initial Time, 2 min; Final Temperature 280 °C; Final Time, 2 min; Gradient Rate 10 °C/min; Flow Rate: 14.2 mL/min; Ionization voltage: 1.3 kV.
18O-Labeling Experiments
A Schlenk cuvette was charged with 2.5 mL of 1 mM acetone solution of [(DMM-tmpa)CuI(CO)]B(C6F5)4 (1) in the glove box, and out of the glove box, the solution was immediately purged with CO(g) at room temperature. The solution was then cooled to the −90 °C temperature and 18O2 was gently bubbled through the solution to generate [(DMM-tmpa)CuII(O2˙−)]B(C6F5)4 (2) monitored by UV-vis spectroscopy. 18O2 (Icon 6393) was prepared in 100 mL of Hamilton gastight syringe equipped with a three-way valve and needle outlet. After 2 was fully formed, three times of Ar/Vacuum purge cycles were applied to remove excess dioxygen and a stock solution of substrates was added to the solution. When the reaction ended, the Schlenk cuvette was taken out to be warmed up at room temperature. The solvent was removed in vacuo, redissolved in 300 μL of the solvent and transferred in a GC-MS vial. Then 1 μL was injected into the GC-MS.
OR Product Analysis using 2H-NMR
Reactions were performed using a 0.75 mM solution of [(DMM-tmpa)CuI(CO)]B(C6F5)4 (1) in acetone. The [(DMM-tmpa)CuII(O2˙−)]B(C6F5)4 (2) was generated in a similar fashion described above at −95 °C. After oxygenation a solution containing 5 equiv of p-OR-DTBP (OR = OCD3 or OCD2CD3) and d6-benzene (as internal reference) was added to the NMR tube. The sample was loaded to the spectrophotometer at low temperature and the spectrum recorded. After which, the sample was removed from the instrument allowed to be warmed to room temperature and the spectrum of the solution recorded.
Oxidation of Phenols by the Cumylperoxyl Radical (4)
Kinetic measurements were performed on a JEOL X-band spectrometer (JES-ME-LX) at 183 K. Typically, photoirradiation of an oxygen-saturated acetone solution containing di-tert-butyl peroxide (1.0 M) and cumene (1.0 M) with a 1000 W Mercury lamp resulted in formation of cumylperoxyl radical (g = 2.0156) which could be detected at low temperatures. The g values were calibrated by using a Mn2+ marker. Upon cutting off the light, the decay of the EPR intensity was recorded with time. The decay rate was accelerated by the presence of p-X-DTBP's (1.0 × 10−2 M). Rates of hydrogen transfer from p-X-DTBP's to PhCMe2OO• were monitored by measuring the decay of EPR signal of PhCMe2OO• in the presence of various concentrations of p-X-DTBP's in acetone at 183 K. Pseudo-first-order rate constants were determined by a least-squares curve fit using a personal computer. The first-order plots of ln (I - I∞) vs time (I and I∞ are the EPR intensity at time t and the final intensity, respectively) were linear for three or more half-lives with the correlation coefficient, ρ > 0.99. In each case, it was confirmed that the rate constants derived from at least five independent measurements agreed within an experimental error of ±5 %.
Hydrogen Peroxide Quantification
Detection of H2O2 as a product has been performed with CH3CN-saturated NaI solution as in a recent report.22 2.5 mL of a 0.6 mM solution of [(DMM-tmpa)CuII(O2˙−)]B(C6F5)4 (2) was generated in a Schlenk cuvette as a typical way. After the reaction with p-X-DTBP was ended, the solution was taken out and warmed to room temperature. 70 μL of solution was added into 2.0 mL of a CH3CN-saturated NaI solution at room temperature in the darkness. The UV-vis spectrum of this solution displayed the formation of triiodide (I3−) at 362 nm and the yield was calculated by comparing with a standard H2O2 solution of known concentrations. 2,4,6-Tri-tert-butyl-4-hydroperoxycyclohexa-2,5-dienone was also found to oxidize the iodide ion.
Attempt to Detect Formaldehyde
The detection of the formaldehyde was accomplished spectrophotometrically via an aqueous-based Nash assay.23 The Nash reagent was prepared by dissolving 7.5 g ammonium acetate, 100 μL of 2,4-pentadione and 150 μL acetic anhydride in 50 mL water. A 8 mg of [(DMM-tmpa)CuI(CO)]B(C6F5)4 (1) was dissolved in 5 mL acetone and transferred to a Schlenk flask, capped with a rubber septum and bubbled with CO(g) for ~ 30 seconds. This reaction vesicle was put in a −95 °C acetone bath. The formation of [(DMM-tmpa)CuII(O2˙−)]B(C6F5)4 (2) was achieved by O2 displacement for ~10 min at which time 7.4 mg of para-methoxy-2,6-di-tert-butylphenol dissolved in 200 μL acetone was added to the solution. The reaction was allowed to react at −95 °C for 45 min at which time the reaction was quenched at low temperature with SO2 (g). The solution was allowed to be warmed up to room temperature and a 250 μL aliquot of the reaction solution was added to a vial charged with 2 mL of the Nash reagent cocktail. The reaction mixture was capped, sealed and heated to 60 °C for 90 min at which time the absorbance at 413 nm was recorded.
Attempt to Detect Isobutylene
The attempt for detecting isobutylene(gas) as one of the products was performed by using GC-MS. An isobutylene-saturated acetone solution was injected as an authentic standard and it could be well detected by GC-MS. However, when the reaction solution was tested, there was no evidence of isobutylene. We attribute this to the low concentrations (maximum 1 mM) used for reaction.24
Resonance Raman Spectroscopy
Resonance Raman spectra were recorded on a Princeton Instruments ST-135 back-illuminated CCD detector and on a Spex 1877 CP triple monochromator with 1200, 1800, and 2400 grooves/mm holographic spectrograph gratings. Excitation was provided by a Coherent I90C-K Kr+ ion laser (λex = 407nm). The spectral resolution was <2 cm−1. Spectra were recorded at 5 mW power at the sample, and the samples were cooled to 77 K in a quartz liquid nitrogen finger Dewar (Wilmad). Baseline spectra were collected using ground, activated charcoal. Isotopic substitution was achieved by oxygenating with 18O2 (Icon, Summit, NJ).
Results and Discussion
Spectroscopic Characterization of [(DMM-tmpa)CuII(O2˙−)]+ (2)
There exist three structurally characterized cupricsuperoxo complexes. One is a synthetic complex from Fujisawa-Kitajima with side-on η2-bound O2˙− fragment. It is a ground state singlet species (S = 0) and from resonance Raman (rR) spectroscopy νO-O = 1043 cm−1.25 There is also the already mentioned case of the PHM protein X-ray structure, considered to have the CuII(O2˙−) formulation and end-on bound O2 fragment.11 The physical properties of this species within the protein are still lacking. Closely related to our own case here, 2, is the now very well known complex from Sundermeyer and Schindler,26 [(TMG3tren)CuII(O2˙−)]+ (TMG3tren = (1,1,1-tris[2-[N2-(1,1,3,3-tetramethyl guanidino)] ethyl]amine), with analogous tripodal tetradentate ligand, and end-on superoxo binding (∠ Cu-O-O = 123°) (X-ray structure, Figure 2) with νO-O = 1120 cm−1 (Δ18O2 = −63 cm−1). The electronic structure of this molecule has been delineated by Solomon and coworkers27 and the molecule possesses a triplet ground state with ferromagnetically coupled spins on both the Cu(II) ion and superoxo fragment. In fact, the finding of a triplet ground state for cupric-superoxo complexes possessing tripodal tetradentate ligands is general.28
Figure 2.

Structural representation of [(TMG3tren)CuII(O2˙−)]+.26
Based on the physical properties of [(DMM-tmpa)CuII(O2˙−)]+ (2), as described here, it has a physical and electronic structure very similar to that of [(TMG3tren)CuII(O2˙−)]+. Complex 2 was generated in acetone and/or MeTHF at −90 °C via displacement of CO(g) by bubbling O2 gas through a solution of [(DMM-tmpa)CuI(CO)]+ (1); a further discussion of why/how this procedure is employed, with references, is given in the Experimental Section. Thus, 2 has been presently characterized by UV-vis and resonance Raman (rR) spectroscopies. The absorption spectrum of this green colored complex is shown in Figure 3a and three primary absorption bands are observed: 409, 587 and 743 nm with ε = 4250, 1100 and 1030 M−1 cm−1, respectively. This complex is EPR silent (Figure S3). Complex 2 is stable enough (t1/2 > ~3 h; – 90 °C) to investigate its reactivity toward external substrates. Compared to the parent tmpa ligand (without any pyridyl substituents) CuII-superoxo complex, which can only be observed as a fleeting intermediate at −128 °C in MeTHF,29 the electron donating groups on the DMM-tmpa ligand (as also for NMe2-tmpa; Chart 2),21 provide significant electron density to the copper center resulting in stabilization of [(ligand)CuII(O2˙−)]+ species.
Figure 3.

(a) Absorption spectra of 1 (0.27 mM) and 2 after addition of O2 (g) in acetone (10 % MeTHF) at 183 K. (b) rR spectra of 2 (0.7 mM) measured in MeTHF (λex = 407 nm). Red, 16O2; blue, 18O2.
Resonance Raman (rR) spectra of [(DMM-tmpa)CuII(O2˙−)]+ (2) (λex = 407 nm) reveal two dioxygen isotope sensitive vibrations (Figure 3b). An O-O vibration is observed at 1121 cm−1 which shifts to 1058 cm−1 (Δ18O2 = −63 cm−1) upon 18O2 substitution. An additional isotope sensitive vibration attributed to the Cu-O stretch occurs at 474 cm−1 (Δ18O2 = −18 cm−1). These parameters closely match those for [(TMG3tren)CuII(O2˙−)]+ and other cupric-superoxo complexes with tripodal tetradentate N4 ligands.21,27,30
Reactivity of [(DMM-tmpa)CuII(O2˙−)]+ (2) towards para-Substituted-2,6-di-tert-butylphenols
As mentioned in the Introduction, the broader perspective for why the present study was undertaken is our general knowledge of [CuII(O2˙−)]+ reactivity is very limited. There are a few highly interesting examples of C–H oxidation reaction (vide infra), and as also indicated, we provided initial descriptions of phenol oxygenation (giving benzoquinones) with both [(NMe2-tmpa)CuII(O2˙−)]+ (Chart 2)21 and [(TMG3tren)CuII(O2˙−)]+ (Figure 2).32 At first, we planned to employ either or both of these complexes for detailed phenol oxidation/oxygenation reactivity studies, but found them to have a very narrow range of substrates that could be oxidized (in terms of O–H BDE) and even where oxidation occurred the reactions were exceptionally slow (Figure S4), thus not amenable to kinetic investigations, as compared to what we find here for reactions of complex 2. A very simple analysis and conclusion would be that the superoxo moiety in [(NMe2-tmpa)CuII(O2˙−)]+, with its very electron releasing pyridyl p-NMe2 substituents, is less electrophilic than the superoxo complex in 2, with its pyridyl p-OMe substituents. As to how this translates to reactivity of phenols with varying O-H BDE, we can judge from the BDE values of substrates in Table 1, the BDE of the Cu-OOH complex must be larger than 79.6 kcal mol−1, but smaller than 82.7 kcal mol−1. Initial survey of reaction of 2 with a variety of phenols indicated the kinetic studies and product analyses were viable for a good range of phenols, both p-alkoxy-2,6-di-tert-butylphenols (p-OR-DTBP's) and p-alkyl-2,6-di-tert-butylphenols (p-R-DTBP's) (Table 1).
Table 1.
Phenol BDE's, Redox Potentials (Eox), Second-Order Rate Constants (183 K) forp-X-DTBP Phenol Oxidations by 2 and 4 and Reaction Yields (See Figure S10 for kinetics details)
| substituents (X) | BDEa kcal mol−1 | Eox, V(vs Fc/Fc+)c | k2 (2) M−1 s−1 | k2c (4)d M−1 s−1 | DTBQ Yield | |
|---|---|---|---|---|---|---|
| OR | OCH2CH3 | - | 0.532 | 24 | 714 | - |
| OCH3 | 79.6 | 0.526 | 23 | 520 | 49% | |
| OCD3 | - | 0.496 | 21 | 513 | - | |
| OCH3, -OD | - | 0.585 | 2.1 | 58 | - | |
| OMPPb | - | 0.614 | 0.84 | 350 | 44% | |
| OCH2CF3 | - | 0.805 | 0.81 | 329 | - | |
| R | CH3 | 80.1 | 0.81 | 0.042 | 185 | - |
| CH2CH3 | 80.0 | 0.875 | 0.027 | 160 | - | |
| sec-butyl | - | 0.884 | 0.023 | 152 | - | |
| CH3, -OD | 0.896 | 0.010 | - | - | ||
| tert-butyl | 82.3 | 0.927 | 0.008 | 106 | 38% | |
| H | 82.7 | 1.074 | NR | - | - | |
Bond Dissociation Energy in DMSO.31
OMPP = 2-methyl-l-phenylpropan-2-yloxy.
These were determined from SHACV measurements, see Experimental Section. The experimental error is ± 0.01 V.
The experimental error is ± 5%.
Kinetic and Thermodynamic Studies
Demonstration of hydrogen atom transfer chemistry
Kinetic isotope labeling studies on the oxidation of the two phenol substrates p-OMe-DTBP and p-Me-DTBP by [(DMM-tmpa)CuII(O2˙−)]+ (2) were performed under pseudo-first-order conditions at 183 K by following the disappearance of the 409 nm band as shown in Figure 4a.33 In both cases, the decay behavior observed fits to a first-order kinetics and yields an observed rate constant (kobs) which was linear with respect to the substrate concentration as shown in Figure 4b. A second-order-rate constant (k2) of 23 M−1 s−1 was obtained for the oxidation of proteo p-OMe-DTBP. Using substrate which had been subjected to deuterium (2H-O) exchange (see Experimental Section), a significant deceleration of the reaction rate was observed, yielding a second-order-rate constant of 2.1 M−1 s−1. Thus, a primary kinetic isotope effect (KIE) of 11 was obtained. Using p-Me-DTBP, a lower but significant KIE of 4.2 was observed based on the determination of k2 = 4.2 × 10−2 M−1 s−1 and 1.0 × 10−2 M−1 s−1 obtained for the oxidation of the proteo and deuterium labeled phenols, respectively. Thus, it appears that the oxidations of both the p-alkoxy-DTBP and p-alkyl-DTBP by 2 occur via a rate limiting O-H activation event.
Figure 4.

(a) UV-vis spectral changes observed by addition of p-Et-DTBP (20 mM) to 2 (0.27 mM) in acetone (10 % MeTHF) at 183 K. kobs = 5.73 × 10−4 s−1. (b and c) Plots of kobs's against the concentrations of substrates (circles) and deuterated 2H-O substrates (squares) for p-OMe-DTBP (b) and p-Me-DTBP (c) to determine second-order-rate constants and KIE's.
Comparison of KIEs to other metal-superoxo mediated oxidations
There have been no prior reports of O–H KIEs by cupric superoxo complexes. However, a primary KIE of 10.6 and 10.9 are reported for the C–H activation of hippuric acid and dopamine by PHM and DBM, respectively,34 thought to be effected by a protein cupric superoxide moiety.6b,10 In a bioinspired synthetic system, a value of 12.1 was reported for the oxidation of BNAH (1-benzyl-1,4-dihydronicotinamide) for the PV-tmpa [bis(pyrid-2-ylmethyl) ([6-(pivalamido)pyrid-2-yl]methyl)amine] cupric superoxo complex.30 A lower magnitude of 4.1 was reported for the intramolecular benzylic C–H oxidation of a phenethyl ligand arm in a chelated copper(II)-superoxo complex reported by Itoh and coworkers.35 In addition, this value (KIE = 12) is approximately equal to what is observed for the oxidation of a water-soluble tri-substituted phenol by a chromium superoxo described by Bakac and coworkers.36 All of these results are consistent with the substrate activation via homolytic O–H bond cleavage.
Cumylperoxyl radical (4) plus phenol substrate HAT reactions for comparison
To provide further evidence that hydrogen atom transfer (HAT) is the rate-determining step (r.d.s.) in these reactions, we studied the same substrates with cumylperoxyl radical (4); the latter is known to effect “pure” HAT chemistry with phenols (and N,N-dimethylanilines).13,37 This hydrogen atom acceptor reacts with phenols to yield cumene hydroperoxide and phenoxyl radical, here, we wished to compare the behavior of 4 with phenols to that of the reactions of [(DMM-tmpa)CuII(O2˙−)]+ (2) and phenols (Scheme 2). Thus, to understand the relationship between second-order-rate constants for 2 (k2) and 4 (k2C) (Table 1), the reaction of the whole series of substrates with 2 and 4 has been explored monitoring the chemistry with the use of UV-vis and EPR spectroscopies, respectively (see Experimental Section). A large KIE value (KIE = 9.0) was observed for hydrogen atom transfer from p-OMe-DTBP to 4 as shown in Figure S5. The k2 values display a linear correlation with k2C, which further indicates HAT is involved in rate-limiting step of phenol oxidations by 2 (Figure 5). If the r.d.s. was to be pure HAT, as in case of 4, the slope would be one. The larger slope observed (= 4.85) from the plot obtained indicates that some degree of contribution of partial transfer of charge is also involved in the r.d.s. for 2 plus phenol reactions.
Scheme 2.

Figure 5.

Correlation between log k2 of 2 and log k2C of 4 with p-X-DTBP's, respectively. Slope is 4.85.
Rate constants (k2) correlation to phenol redox potentials ?
We also considered possible relationships between the second-order-rate constants for the reaction of [(DMM-tmpa)CuII(O2˙−)]+ (2) with phenols (k2) and substrate redox potentials. In fact, k2 values for the reactions increase with decreasing the redox potentials, i.e., with increasing driving force of electron transfer from phenols to 2. The plot of (kBT/e) lnk2 vsEox exhibits a linear correlation with a negative slope = −0.29 as shown in Figure 6. By contrast, for the case of cumylperoxyl radical (4), the plot of (kBT/e) lnk2C vsEox exhibits a much weaker dependence on the Eox values and the slope is only −0.05. Although the bond dissociation energy (BDE) of p-X-DTBP is known to decrease by electron-donating substituents,38 the difference in BDE between OCH3 and tert-butyl group is only 2.7 kcal mol−1, which is equivalent to 0.12 eV, whereas the difference in Eox between OCH3 and tert-butyl group is 0.40 eV (Table 1). Based on thermochemical cycles,37 the difference in BDE (ΔBDE) is given by eq 2,
| (2) |
where ΔpKa is the difference in the pKa values of p-X-DTBṖ+ and entropy changes are neglected. The pKa value becomes smaller with increasing in the Eox value and the constant BDE value (eq 2) indicates that an increase in the Eox value is partially cancelled by a decrease in the pKa value. Such cancellation may be the reason why the BDE value is much less sensitive to electron-donating substituents in Table 1 as compared to the Eox value.
Figure 6.

Plots of (kBT/e) lnk2 for the reactions of p-X-DTBP's with 2 (squares) and 4 (circles) against the one-electron oxidation potentials (Eox) of substrates. The slopes are −0.29 and −0.05, respectively.
Mechanistic considerations
There are three possible reaction pathways in the apparent hydrogen-atom transfer from p-X-DTBP to [CuII(O2˙−)]+; (1) electron transfer (ET) followed by proton transfer (PT), (2) PT followed by ET and (3) concerted ET and PT (Scheme 3). The observation of large KIEs in Figure 4 indicates that PT should be involved in the rate-determining step. In such a case, PT should be the rate-determining step following the ET equilibrium, which is endergonic, in the ET/PT pathway (1).39 In this case the observed second-order-rate constant (k2) is given by eq 3,
| (3) |
where kp is the rate constant of proton transfer from p-X-DTBṖ+ to [CuII(O22−)]0 and Ket is the ET equilibrium constant between p-X-DTBP and [CuII(O2˙−)]+ (Ket << 1). Because the Brønsted slope of kp is between 0 and 0.5 for an exergonic proton-transfer reaction,40 and the slope of the plot of (kBT/e) lnKetvsEox is −1.0, the predicted slope of a plot of (kBT/e) lnk2 vsEox is between −0.5 and −1.0.
Scheme 3.

In the case of the PT/ET pathway (2), PT should also be the rate-determining step followed by fast ET. In this case the slope of the plot of (kBT/e) lnk2 vsEox is positive, because an increase in the Eox value with electron-withdrawing substituents results in an decrease in the pK'a value (more acidic) when the proton transfer from p-X-DTBP to [CuII(O2˙−)]+ becomes thermodynamically more favorable. The observed negative slope (−0.29) clearly rules out the PT/ET pathway. Although the ET/PT pathway affords the negative slope of a plot of (kBT/e) lnk2 vsEox is between −0.5 and −1.0, the observed slope (−0.29) is less negative than −0.5. In the case of the concerted ET/PT pathway (one-step hydrogen atom transfer), the slope is −0.05 as observed for hydrogen atom transfer from p-X-DTBP to 4. The smaller slope than expected for transfer of a full unit of charge has been reported to result from only partial transfer of charge in hydrogen-atom transfer reactions from hydrogen donors to the triplet excited state of benzophenone.41 Thus, it is most likely that hydrogen transfer from p-X-DTBP proceeds via a partial transfer of charge rather than an ET/PT pathway in which a full unit of charge is transferred. Thus, more electron deficient ligands may be required in order to increase the hydrogen-transfer reactivity of [CuII(O2˙−)]+ by facilitating charge transfer from p-X-DTBP to [CuII(O2˙−)]+.
Activation parameters and comparisons
The rates of the [(DMM-tmpa)CuII(O2˙−)]+ (2) + p-X-DTBP's reactions were of course temperature dependent. However, we could only examine a narrow temperature range, as colder (than 173 K) conditions involved solution freezing, while self decomposition of 2 was too extensive when warming above 188 K. Also, the slow self-decomposition precluded the generation of good quality data except for the fastest reaction, that being with the p-OMe-DTBP substrate. Never-the-less, an Eyring plot developed from data between 173 K and 188 K for this substrate yielded activation parameters as follows: ΔH‡ = 3.6 ± 0.6 kcal mol−1 and ΔS‡ = −32 ± 3 cal mol−1 K−1 (Figure S6). The large negative activation entropy suggests that the r.d.s. involves a well ordered transition state.
Although no comparable activation parameters for p-X-DTBP oxidation by cupric superoxide or other Cu-oxygen complexes have been reported, there are a few values measured for oxidation of phenol derivatives by various other metal ion complexes, including CuIII, RuVI, MnV, and RuIII (Table 2).42 Excepting the case of reaction of a RuIII-pterin complex which was concluded to involve PCET, all other cases are stated as involving HAT in the rate-limiting step. In particular, the HAT reaction of a manganese(V)-oxo corrolazine complex plus 2,4-di-tert-butylphenol (2,4-DTBP) affording MnIV(OH) plus 2,4-di-tert-butylphenoxyl radical, gives both ΔH‡ and ΔS‡ values close to those obtained from our reaction of 2 and p-OMe-DTBP.
Table 2.
Kinetic Parameters in Various Metal Complexes plus Phenol Oxidation Reactions.
| Reactants | ΔH‡, kcal mol−1 | ΔS‡, cal mol−1 K−1 | |
|---|---|---|---|
| CuII-O2˙−a | p-OMe-DTBP | 3.6 ± 0.6 | −32 ± 3 |
| Cu(III)42a | 2,4-DTBP | 8.3 ± 1.1 | −27 ± 3 |
| Ru(VI)42b | Phenol | 11.3 ± 0.8 | −14 ± 2 |
| Mn(V)42c | 2,4-DTBP | 6.3 ± 1.4 | −35.6 ± 2.3 |
| Ru(III)42d | p-tBu-DTBP | 1.6 ± 0.2 | −36 ± 2 |
This work
KIE temperature dependence?
We also investigated the temperature dependence of the KIE values for the reaction with deuterated p-OMe-DTBP (Table 3, Figure S6). While the temperature range is again limited, the results suggest there is no temperature dependence on the magnitude of the primary KIEs. Thus, the Arrhenius pre-exponential factor for the phenol O-H and O-D isotopically labeled analogues remain essentially constant as a function of temperature. This behavior is consistent with a substrate oxidation mechanism involving classical rate limiting H–atom abstraction.43
Table 3.
Temperature Dependence on the KIEs of the HAT Reactions from p-OMe-DTBP to [(DMM-tmpa)CuII(O2˙−)]+ (2)
| kH, M− s−1 | kD, M−1 s−1 | kH/kD | |
|---|---|---|---|
| 173 K | 11 | 1.0 | 11 |
| 178 K | 14 | 1.2 | 12 |
| 183 K | 23 | 2.1 | 11 |
| 188 K | 26 | 2.7 | 10 |
Summary of the kinetic-thermodynamic studies
As detailed above, our in-depth studies are consistent with conclusion that the cupric superoxo complex [(DMM-tmpa)CuII(O2˙−)]+ (2), as a kind of prototype for the initial adducts formed in chemical or biological CuI/O2 interactions, reacts with p-OMe-DTBP (and the other phenols) via a HAT oxidative mechanism. Likewise, as mentioned above, rate-limiting HAT occurs for the galactose oxidase (GO) active-site special Tyr, but PCET in copper amine oxidase (CAO) has been suggested by Klinman7c and Roth.8 In relevant C-H bond activation by [CuII(O2˙−)]+ species, Klinman and Blackburn reported C-H bond cleavage in N-benzoylglycine by PHM having large intrinsic isotope effects.34a They also demonstrated hydrogen tunneling as occurring in PHM with a lack of a temperature dependence of an intrinsic isotope effect on the C-H bond cleavage of the substrate hippuric acid.44 As also mentioned, Itoh and co-workers reported intramolecular C-H hydroxylation (HAT) by a cupric superoxo species supported by tridentate ligand possessing a distorted tetrahedral geometry.35a
Product Analyses and Further Mechanistic Insights
Phenol oxidations lead to benzoquinone products
As described above in detail, the initial products generated after rate-limiting hydrogen atom transfer occurs are presumed to be a [(DMM-tmpa)CuII(OOH)]+ (3) and a para-X-2,6-di-tert-butylphenoxyl radical (Scheme 2). Such a phenoxyl radical would possess distinctive λmax = 384 and 402 nm absorptions and exhibit a sharp g ~ 2.0 EPR spectroscopically detectable signal. However, neither of these spectroscopic signals could be observed. Thus, qualitative and quantitative product analyses were carried out allowing us to understand and build up the proposed overall mechanism and stoichiometry of the reactions observed. In fact, for all phenolic substrates, benzoquinones are produced. As mentioned above, reactions of 2 with p-OR-DTBP and p-R-DTBP proceed via different pathways, two-electron oxidation and four-electron oxygenation, respectively, and these further aspects of the overall chemistry are now detailed.
Reaction of [(DMM-tmpa)CuII(O2˙−)]+ (2) with p-alkoxy-2,6-di-tert-butylphenols (p-OR-DTBP's)
All synthetic reactions were carried out by mixing excess phenol substrate with 2, at −90 °C. The cryogenic solutions were interrogated by UV-vis, NMR and EPR spectroscopies. As described for the initial kinetics of the system (vide supra), 2 rapidly disappears. Low-temperature UV-vis (new d-d bands) and EPR (EPR silent to EPR active) spectroscopies reveal that a new mononuclear CuII complexes has formed, but NMR spectroscopy suggests that no quinone has yet formed and only starting extra phenol is present. However, warming of these initial reaction solutions to RT followed by NMR and GC-MS analyses indicates that up to 50 % yields of 2,6-di-tert-butyl-1,4-benzoquinones (DTBQ's) form (Scheme 1). An 18O labeling experiment (see Experimental Section) where 2 was generated using 18O2 gas revealed that no 18O was incorporated into the DTBQ. This is explained below; only the overall two-electron oxidation of the substrate phenol takes place while hydrogen peroxide and alcohol products are also formed. The fate of the initial para-OR fragment, after warming, was identified by 2H(D)-labeling experiments using p-OCD3/OCD2CD3-DTBP substrates (Figure S9). In each case, the product detected was the corresponding alcohol (methanol and ethanol), not the oxidized form (formaldehyde and acetaldehyde). The inorganic product is a mononuclear copper(II) complex [(DMM-tmpa)CuII-(OH(H))]+/2+, and H2O2 is produced (~50 % yield), as determined by EPR/UV-vis spectroscopy (see Figures S7, S8 & S13) and iodometric titration, respectively.45
Proposed pathway for [CuII(O2˙−)]+ (2) reaction with p-OR-DTBP substrates (Scheme 4)
Scheme 4.
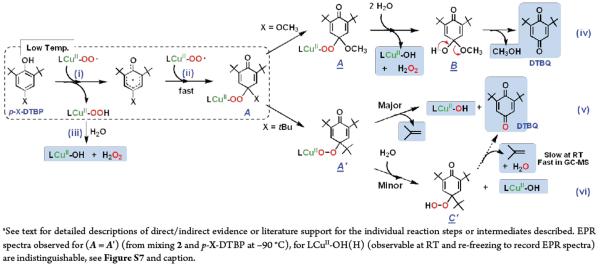
Proposed Pathways for [CuII(O2˙−)]+ Reactivity with p-OMe-DTBP or p-tBu-DTBP Substrates Leading to Observed Products.a
Initially, a hydrogen atom is transferred from the phenol to [CuII(O2˙−)]+ as the (low-temperature) r.d.s., where [CuII(OOH)]+ and the corresponding phenoxyl radical form (Scheme 4, step i). The [CuII(OOH)]+ intermediate would `hydrolyze' giving H2O2 and [CuII(OH)]+ (Scheme 4, step iii), (upon warming up to RT). Strong support for this claim comes from our independent generation of authentic complex [CuII(OOH)]+ (3), and demonstration that it releases H2O2 (which was directly identified) and forms the hydroxide complex, [CuII(OH)]+. This was identified as the Cu product by matching EPR and UV-vis spectroscopic data with authentically generated compounds (see Supporting Information). The formation of the eventual benzoquinone and other products is explained by (a) a fast radical-radical coupling reaction of the newly generated ArO• moiety and a second equiv [CuII(O2˙−)]+, affording a peroxy CuII-OO-(ArO') species A (Scheme 4, step ii), (b) then this reacts with solvent water to give B, which eliminates the alcohol (RO-H) derived from the p-OR-DTBP substrate, step iv. {Note: if this hydrolysis step involved CuII–OOH rather than CuII-OO–(ArO') bond cleavage, then the corresponding hydroperoxo intermediate C (HOO-(ArO')) (Scheme 5) forms, rather than our postulated B with HO-(ArO') formulation. However, C would be incapable of producing methoxide/methanol plus benzoquinone, our observed products. If C or CuII-peroxo derivative A were to form, CH2=O would be produced (Scheme 5), but none was observed (Experimental Section)}. Kinetic aspects for quinone formation were not investigated in detail, as this step occurs later, with warming of the reaction solutions (Also, see Experimental Section).
Scheme 5.

Reaction of [(DMM-tmpa)CuII(O2˙−)]+ (2) with p-alkyl-2,6-di-tert-butylphenols (p-R-DTBP's)
For the case where the para-substituent is an alkyl group like tert-butyl, the reaction of p-R-DTBP plus [(DMM-tmpa)CuII(O2˙−)]+ (2) proceeds differently, via an overall four-electron substrate oxygenation. The initial reaction, steps i and ii proceed analogously, producing the CuII-OO–(ArO') species A' (Scheme 4). This however, can undergo a tert-butyl substituent oxidation via elimination of isobutylene, then directly giving the DTBQ and Cu(II) products observed (Scheme 4, steps v).46 Isobutylene or tBuOH (= isobutylene + H2O) elimination from tert-butyl substituted phenols under oxidative conditions is well precedented.47 We, however, were not able to detect isobutylene as a product.24,48
As can be seen from Scheme 4 (step v), the product DTBQ in the [(DMM-tmpa)CuII(O2˙−)]+ (2) plus p-R-DTBP substrate reaction, should have one of its O-atoms derived from molecular oxygen (derived from 2). Indeed, this appears to be the case. The reaction of p-tBu-DTBP with 2 provides for a quantitative yield of DTBQ (based on the stoichiometry of two molecules of 2 per one mole of phenol substrate). For 18O-labeled 2 (derived from 1 plus 18O2), reaction with p-tBu-DTBP affords a 40 % of 18O-incorporated DTBQ (GC-MS). The smaller isotope incorporation yield than expected may result from the 18O-16O exchange reaction with water, because carbonyl compounds are known to undergo oxygen exchange reactions with water, catalyzed by acids or bases.49
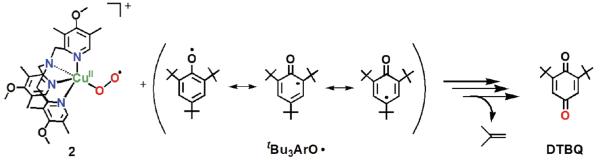
[CuII(O2˙−)]+ (2) coupling with 2,4,6-tri-tert-butylphenoxyl radical
A set of experiments that provides further and strong support for our mechanistic picture of the chemistry comes from reacting [(DMM-tmpa)CuII(O2˙−)]+ (2) with isolated tBu3ArO• (Scheme 6). The addition of one equiv tBu3ArO• to 2 was monitored by UV-vis and EPR spectroscopies. A fast reaction occurs;50 the intermediate 2 quickly disappears (UV-vis monitoring) and the Cu(II) EPR spectrum previously observed for the [(DMM-tmpa)CuII(OH(H))]+/2+ product appears. The yield of DTBQ was 98 % and a 70~80 % 18O-incorporation into the DTBQ product was observed. Thus, the 2 + tBu3ArO• occurs with a 1:1 stoichiometry (Scheme 4, step ii), and this then supports the [CuII(O2˙−)]+ : phenol = 2 : 1 overall stoichiometry. In contrast to the case of p-OR-DTBP, the oxygen incorporated into the oxidized product (DTBQ) obtained from the reaction of p-R-DTBP with 2 originates from 2 via O-O bond cleavage followed by the elimination of the oxidized R group (Scheme 4, step v).
Scheme 6.
A minor reaction pathway contributing to p-R-DTBP oxidation
If the reaction of [(DMM-tmpa)CuII(O2˙−)]+ (2) with p-R-DTBP (or that of 2 + tBu3ArO•) occurred only via step v (Scheme 4), the product solution should not oxidize iodide since no H2O2 would have been generated. However, we observed 20~30 % of triiodide absorption band formation when testing for peroxide. We explain this by invoking the “minor” step vi, where hydrolysis of A' occurs giving 2,4,6-tri-tert-butyl-4-hydroperoxycyclohexa-2,5-dienone (C'), which (i) does itself oxidize iodide ion (see Experimental Section), and (ii) slowly converts to DTBQ (at RT) but (iii) fully converts to DTBQ in the GC-MS experiment.
Precedent and comparison with cobalt and nickel superoxide phenol oxidations
In the overall process involving reaction of the p-X-DTBP, steps i and ii (top line; Scheme 4), a second mole-equiv of [(DMM-tmpa)CuII(OH)]+ and H2O2 are produced, all consistent with the identity and yields of products obtained, and thus the reaction stoichiometry. Nishinaga and coworkers, in classical studies of the reactions of [CoIII(O2˙−)]+ (formed from CoII and O2), also observed this same reaction stoichiometry, involving two [CoIII(O2˙−)]+ moieties with one phenol substrate. Further, Nishinaga's work provides precedent for the proposed (Scheme 4) reaction of [(DMM-tmpa)CuII(O2˙−)]+ (2) with the initially generated phenoxyl radical intermediate;51 Nishinaga51b also obtained a cobalt peroxy species CoIII-OO-(ArO'), and its X-ray structure for a para-tBu phenol substrate (Scheme 7). Further, in order to explain the products observed in very recent investigations by Driess and co-workers,52 involving phenol reactions with a new [NiII(O2˙−)]+ complex, an intermediate analogous to A' was proposed to form from p-R-DTBP's (R = H, Me, tBu) substrates.
Scheme 7.

A prior computational study of [CuII(O2˙−)]+ reaction with 2,6-di-tert-butylphenol (p-H-DTBP) leading to a 2,6-di-tert-butyl-1,4-benzoquinone (DTBQ)
In 2009, based on DFT calculations, Güell et al.53 proposed a mechanism (Scheme 8) of p-HDTBP oxidation by a cupric superoxide complex close analogue of 2, [(NMe2-tmpa)CuII(O2˙−)]+ (see Introduction), which differs considerably from what we have found and proposed in the present study (Scheme 4). They suggest HAT occurs at the first step (as we observe), but then it is suggested that the LCuII-OOH species formed undergoes Cu–OOH homolytic cleavage and the hydroperoxyl radical thus formed attacks the phenoxyl radical, overall leading to copper(I) and hydroperoxy organic. This C-O bond formation process is described as the r.d.s. Then, the resulting copper(I) facilitates O–O homolytic cleavage forming hydroxyl radical plus a copper(II)-O-ArO' complex. The last step involves HAT (or PCET) from the ring to hydroxyl radical leading to the formation of copper(I), a water molecule and DTBQ. Thus, their overall chemistry is formally a catalytic reaction (regenerating copper(I), to react again with more O2 to give the cupric superoxo species), with different stoichiometry than we observed and a different r.d.s. as mentioned. However, we should point out that the substrate used in their calculations was p-H-DTBP, a perhaps rather special case compared to the series of substrates we examined, Table 1. In fact, we find that p-H-DTBP is unreactive toward [(DMM-tmpa)CuII(O2˙−)]+ (2).
Scheme 8.

Conclusion
In this report, we have obtained detailed mechanistic insights into the oxidation of a wide series of para-substituted-2,6-di-tert-butyl-phenols (p-X-DTBP's) by a newly synthesized cupric superoxo complex supported by an electron-rich ligand, [(DMM-tmpa)CuII(O2˙−)]+ (2). With detailed kinetic investigations, we proved that hydrogen atom abstraction is the first step and the rate-determining-step for the oxidation of p-X-DTBP's. The key observations supporting this were the finding of a large deuterium kinetic isotope effect, a good correlation with reactivity of the cumylperoxyl radical toward the same substrates, comparison of the rate-dependences in the phenol oxidations on the one-electron oxidation potentials of the p-X-DTBP substrates, and the observed activation parameters for the reaction. The hydrogen atom transfer from p-X-DTBP's proceeds via a partial transfer of charge rather than a complete transfer of charge in the ET/PT pathway. The qualitative and quantitative product analyses and reactivity study carried out with the 2,4,6-tri-tert-butylphenoxyl radical (tBu3ArO•) reacting with 2, allowed us to build the case for the proposed overall stoichiometry of reaction and furthermore, the detailed mechanism. Two moles of copper(II) superoxo species are required to oxidize para-substituted-2,6-di-tert-butylphenols to 2,6-di-tert-butyl-1,4-benzoquinones. It is found that the reaction of 2 toward para-alkoxy-2,6-di-tert-butylphenol proceeds via two-electron oxidation with hydrolysis while reaction with para-alkyl-2,6-di-tert-butylphenol proceeds via a process involving four-electron oxygenation chemistry.
This study contributes significantly to our understanding of the fundamental chemistry and oxidative capabilities of initial copper(I)-dioxygen adducts, i.e., cupric-superoxide complexes. As described in the Introduction, CuII(O2˙−) species are implicated in reactions with both C–H and O–H containing substrates, and the latter biomimetic chemistry was the focus of attention here. The supporting ligand is well known to modulate the structural, electronic structure/bonding, as well as the reactivity nature of copper complexes, here with O2 derived fragments bound to the copper ion. Thus, future efforts will include the generation of CuII(O2˙−) complexes with rather differing supporting ligands, to explore the range of reactivity possible for such primary copper-dioxygen adducts.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the NIH (E.I.S., DK31450; K.D.K., GM28962), JSPS (S.F., 20108010; K.O., 23750014) and JST (S.F., ALCA) for financial support of this research.
Footnotes
Supporting Information. Reaction schemes for copper proteins, synthetic details, EPR spectra, additional kinetic data and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; (b) Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (c) Hatcher LQ, Karlin KD. J. Biol. Inorg. Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]; (d) Peterson RL, Kim S, Karlin KD. Comprehensive Inorganic Chemistry II (Second Edition) Elsevier; Oxford: Amsterdam: 2013. pp. 149–177. [Google Scholar]
- (2).(a) Schröder D, Holthausen MC, Schwarz H. J. Phys. Chem. B. 2004;108:14407–14416. [Google Scholar]; (b) Dietl N, van der Linde C, Schlangen M, Beyer MK, Schwarz H. Angew. Chem., Int. Ed. 2011;50:4966–4969. doi: 10.1002/anie.201100606. [DOI] [PubMed] [Google Scholar]
- (3).(a) Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. J. Am. Chem. Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, Tolman WB. J. Am. Chem. Soc. 2011;133:17602–17605. doi: 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. J. Am. Chem. Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. [DOI] [PubMed] [Google Scholar]; (d) Maiti D, Narducci Sarjeant AA, Karlin KD. Inorg. Chem. 2008;47:8736–8747. doi: 10.1021/ic800617m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Nakao Y, Hirao K, Taketsugu T. J. Chem. Phys. 2001;114:7935–7940. [Google Scholar]; (b) Decker A, Solomon EI. Curr. Opin. Chem. Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]; (c) Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Inorg. Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- (5).Itoh S. Curr. Opin. Chem. Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- (6).(a) Klinman JP. Chem. Rev. 1996;96:2541–2561. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]; (b) Klinman JP. J. Biol. Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- (7).(a) McGuirl MA, Dooley DM. Curr. Opin. Chem. Biol. 1999;3:138–144. doi: 10.1016/S1367-5931(99)80025-8. [DOI] [PubMed] [Google Scholar]; (b) Rokhsana D, M. SE, Brown DE, Dooley DM. Amine Oxidase and Galactose Oxidase. In: Itoh S, Karlin KD, editors. Copper-Oxygen Chemistry. Vol. 4. John Wiley & Sons, Inc.; Hoboken: 2011. pp. 53–106. [Google Scholar]; (c) Humphreys KJ, Mirica LM, Wang Y, Klinman JP. J. Am. Chem. Soc. 2009;131:4657–4663. doi: 10.1021/ja807963e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu Y, Mukherjee A, Nahumi N, Ozbil M, Brown D, Angeles-Boza AM, Dooley DM, Prabhakar R, Roth JP. J. Phys. Chem. B. 2012;117:218–229. doi: 10.1021/jp3121484. [DOI] [PubMed] [Google Scholar]
- (9).Evans JP, Ahn K, Klinman JP. J. Biol. Chem. 2003;278:49691–49698. doi: 10.1074/jbc.M300797200. [DOI] [PubMed] [Google Scholar]
- (10).(a) Chen P, Solomon EI. J. Am Chem. Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]; (b) Chen P, Bell J, Eipper BA, Solomon EI. Biochemistry. 2004;43:5735–5747. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- (11).Prigge ST, Eipper B, Mains R, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- (12).Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Osako T, Ohkubo K, Taki M, Tachi Y, Fukuzumi S, Itoh S. J. Am Chem. Soc. 2003;125:11027–11033. doi: 10.1021/ja029380+. [DOI] [PubMed] [Google Scholar]
- (14).During the review of this present manuscript, Itoh and co-workers reported on the “redox reactivity” of a [CuII(O2˙−)]+ complex first described in 2009.35 The complex with tridentate N3 ligand does not oxidize phenols, but only reacts with them in an acid-base fashion.
- (15).Liang H-C, Kim E, Incarvito CD, Rheingold AL, Karlin KD. Inorg, Chem. 2002;41:2209–2212. doi: 10.1021/ic010816g. [DOI] [PubMed] [Google Scholar]
- (16).Manner VW, Markle TF, Freudenthal JH, Roth JP, Mayer JM. Chem. Commun. 2008:256–258. doi: 10.1039/b712872j. [DOI] [PubMed] [Google Scholar]
- (17).Nishinaga A, Shimizu T, Matsuura T. J. Org. Chem. 1979;44:2983–2988. [Google Scholar]
- (18).Zhang CX, Kaderli S, Costas M, Kim E.-i., Neuhold Y-M, Karlin KD, Zuberbühler AD. Inorg. Chem. 2003;42:1807–1824. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- (19).Fyfe CA, Vanveen L. J. Am. Chem. Soc. 1977;99:3366–3371. [Google Scholar]
- (20).Omura K. J. Org. Chem. 1996;61:7156–7161. doi: 10.1021/jo9609945. [DOI] [PubMed] [Google Scholar]
- (21).Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- (22).Kim S, Saracini C, Siegler MA, Drichko N, Karlin KD. Inorg. Chem. 2012;51:12603–12605. doi: 10.1021/ic302071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).(a) Nash T. Biochem. J. 1953;55:416–421. doi: 10.1042/bj0550416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thesis: Zhang, C. X. Ph.D. The Johns Hopkins University; Baltimore MD: 2001. [Google Scholar]
- (24).If higher concentrations of [(DMM-tmpa)CuII(O2˙#−)]+ (2) are employed, 2 reacts with [(DMM-tmpa)CuI(CO)]+ (1) forming dicopper(II) species, see Introduction (structure type E) and the Experimental Section concerning the synthesis of 1 and the generation of 2.
- (25).(a) Fujisawa K, Tanaka M, Morooka Y, Kitajima N. J. Am. Chem. Soc. 1994;116:12079–12080. [Google Scholar]; (b) Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. J. Am Chem. Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- (26).Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Angew. Chem. Int. Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- (27).Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, Sundermeyer J, Schindler S, Solomon EI. Inorg. Chem. 2010;49:9450–9459. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ginsbach JW, Peterson RL, Cowley RE, Karlin KD, Solomon EI. Inorg. Chem. 2013;52:12872–12874. doi: 10.1021/ic402357u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lucas HR, Meyer GJ, Karlin KD. J. Am. Chem. Soc. 2010;132:12927–12940. doi: 10.1021/ja104107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. J. Am. Chem. Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bordwell FG, Zhang X-M. J. Phys. Org. Chem. 1995;8:529–535. [Google Scholar]
- (32).Maiti D, Lee D-H, Gaoutchenova K, Würtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew. Chem., Int. Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- (33).It is only for the p-alkyl-DTBP cases that it was convenient to take a large number of full spectra, as in Fig. 3a, to follow the kinetics. For all other cases, reactions which were much faster, only the 409 nm absorption value changes were monitored, to deduce pseudo first-order rate constants.
- (34).(a) Francisco WA, Merkler DJ, Blackburn NJ, Klinman JP. Biochemistry. 1998;37:8244–8252. doi: 10.1021/bi973004y. [DOI] [PubMed] [Google Scholar]; (b) Miller SM, Klinman JP. Biochemistry. 1983;22:3091–3096. doi: 10.1021/bi00282a011. [DOI] [PubMed] [Google Scholar]
- (35).(a) Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. J. Am. Chem. Soc. 2009;131:2788–2789. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]; (b) Kunishita A, Ertem MZ, Okubo Y, Tano T, Sugimoto H, Ohkubo K, Fujieda N, Fukuzumi S, Cramer CJ, Itoh S. Inorg. Chem. 2012;51:9465–9480. doi: 10.1021/ic301272h. [DOI] [PubMed] [Google Scholar]
- (36).Nemes A, Bakac A. Inorg. Chem. 2001;40:746–749. doi: 10.1021/ic001063l. [DOI] [PubMed] [Google Scholar]
- (37).Fukuzumi S, Shimoosako K, Suenobu T, Watanabe Y. J. Am. Chem. Soc. 2003;125:9074–9082. doi: 10.1021/ja035156o. [DOI] [PubMed] [Google Scholar]
- (38).Pratt DA, Dilabio GA, Mulder P, Ingold KU. Acc. Chem. Res. 2004;37:334–340. doi: 10.1021/ar010010k. [DOI] [PubMed] [Google Scholar]
- (39).If the ET step is exergonic, this should occur first, followed by rate-determining PT. In such a case, phenol radical cation derivatives should be observed, but they were not. If an exergonic ET were the rate-determining step, followed by rapid PT, no KIE would be observed in contrast to our experimental observations. We further note that [CuII(O2˙−)]+ complex 2 is unreactive to the strong reductant decamethylferrocene.
- (40).(a) Matsumoto T, Ohkubo K, Honda K, Yazawa A, Furutachi H, Fujinami S, Fukuzumi S, Suzuki M. J. Am. Chem. Soc. 2009;131:9258–9267. doi: 10.1021/ja809822c. [DOI] [PubMed] [Google Scholar]; (b) Marcus RA. J. Phys. Chem. 1966;72:891. [Google Scholar]
- (41).(a) Guttenplan JB, Cohen SG. J. Am. Chem. Soc. 1972;94:4040–4042. [Google Scholar]; (b) Wagner PJ, Lam HMH. J. Am. Chem. Soc. 1980;102:4167–4172. [Google Scholar]
- (42).(a) Lockwood MA, Blubaugh TJ, Collier AM, Lovell S, Mayer JM. Angew. Chem. Int. Ed. 1999;38:225–227. [Google Scholar]; (b) Yiu DTY, Lee MFW, Lam WWY, Lau T-C. Inorg. Chem. 2003;42:1225–1232. doi: 10.1021/ic026184v. [DOI] [PubMed] [Google Scholar]; (c) Lansky DE, Goldberg DP. Inorg. Chem. 2006;45:5119–5125. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]; (d) Miyazaki S, Kojima T, Mayer JM, Fukuzumi S. J. Am. Chem. Soc. 2009;131:11615–11624. doi: 10.1021/ja904386r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).(a) Kohen A, Klinman JP. Acc. Chem. Res. 1998;31:397–404. [Google Scholar]; (b) Kwart H. Acc. Chem. Res. 1982;15:401–408. [Google Scholar]
- (44).Francisco WA, Knapp MJ, Blackburn NJ, Klinman JP. J. Am. Chem. Soc. 2002;124:8194–8195. doi: 10.1021/ja025758s. [DOI] [PubMed] [Google Scholar]
- (45).(a) The smaller yield of H2O2 than expected from Scheme 4 may result from partial decomposition during the reaction. (b) The DMM-tmpa ligand in Cu(II) reaction products remains intact (based on direct TLC and MS analyses); no ligand oxidation occurred.
- (46).In fact, we were not able to detect isobutylene as a product, see the Experimental Section for details and explanation.
- (47).(a) Wali A, Das J, Pillai SM, Ravindranathan M. Green Chem. 2002;4:587–591. [Google Scholar]; (b) Mandal S, Macikenas D, Protasiewicz JD, Sayre LM. J. Org. Chem. 2000;65:4804–4809. doi: 10.1021/jo991625m. [DOI] [PubMed] [Google Scholar]; (c) Nishinaga A, Itahara T, Shimizu T, Matsuura T. J. Am. Chem. Soc. 1978;100:1820–1825. [Google Scholar]; (d) Peters A, Trumm C, Reinmuth M, Emeljanenko D, Kaifer E, Himmel H-J. Eur. J. Inorg. Chem. 20092009:3791–3800. [Google Scholar]; (e) Gupta R, Mukherjee R. Tet. Lett. 2000;41:7763–7767. [Google Scholar]; (f) Cvetkovic M, Batten SR, Moubaraki B, Murray KS, Spiccia L. Inorg. Chim. Acta. 2001;324:131–140. [Google Scholar]; (g) Gupta M, Upadhyay SK, Sridhar MA, Mathur P. Inorg. Chim. Acta. 2006;359:4360–4366. [Google Scholar]; (h) Singla M, Mathur P, Gupta M, Hundal MS. Transition Met Chem. 2008;33:175–182. [Google Scholar]; (i) Mukherjee C, Weyhermuller T, Bothe E, Chaudhuri P. C. R. Chimie. 2007;10:313–325. [Google Scholar]; (j) Singh AP, Gupta R. Eur. J. Inorg. Chem. 2010:4546–4554. [Google Scholar]; (k) Bakshi R, Mathur P. Inorg. Chim. Acta. 2010;363:3477–3488. [Google Scholar]; (l) Ahuja G, Mathur P. Inorg. Chem. Commun. 2012;17:42–48. [Google Scholar]
- (48).For the substrate p-Me-DTBP, which was studied for its kinetics (Table 1), there is a question as to what the final organic product could be. The mechanism applied to the p-tBu-DTBP substrate which would eliminate iso-butylene, cannot apply to p-Me-DTBP, Methanol or a Cu(II)-OMe species might be formed, as suggested by one reviewer, but a detailed analysis was not performed.
- (49).(a) Damuel D, Silver BL. Adv. Phys. Org. Chem. 1965;3:123–186. [Google Scholar]; (b) Kawanishi Y, Suzuki Y, Miyazawa A. Chem. Eng. J. 2011;167:531–535. [Google Scholar]
- (50).Upon benchtop mixing of 2 and tBu3ArO•, (less then one minute, with 1:1 ratios of reagents), the reaction is already over by the time the first UV-vis spectrum could be recorded, as indicated by the complete loss of the UV-vis signals ascribed to 2 (Figure 3) and an expected 402 nm peak for tBu3ArO•. It is outside the scope of this study to attempt stopped-flow (triple mixing) experiments which would be required.
- (51).(a) Nishinaga A, Tomita H, Matsuura T. Tet. Lett. 1980;21:3407–3408. [Google Scholar]; (b) Nishinaga A, Tomita H, Nishizawa K, Matsuura T, Ooi S, Hirotsu K. J. Chem. Soc. Dalton Trans. 1981:1504–1514. [Google Scholar]
- (52).Company A, Yao S, Ray K, Driess M. Chem.-Eur. J. 2010;16:9669–9675. doi: 10.1002/chem.201001138. [DOI] [PubMed] [Google Scholar]
- (53).Güell M, Luis J, Solà M, Siegbahn P. J. Biol. Inorg. Chem. 2009;14:229–242. doi: 10.1007/s00775-008-0443-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.