

Abstract
Previously, we demonstrated the sensitivity of RPTEC/TERT1 cells, an immortalized human renal proximal tubule epithelial cell line, to two common environmental carcinogens, cadmium (Cd) and benzo[a]pyrene (B[a]P). Here, we measured BPDE-DNA adducts using a competitive ELISA method after cells were exposed to 0.01, 0.1, and 1 μM B[a]P to determine if these cells, which appear metabolically competent, produce BPDE metabolites that react with DNA. BPDE-DNA adducts were most significantly elevated at 1 μM B[a]P after 18 and 24 hours with 36.34 +/− 9.14 (n = 3) and 59.75 +/− 17.03 (n = 3) adducts/108 nucleotides respectively. For mixture studies, cells were exposed to a non-cytotoxic concentration of Cd, 1 μM, for 24 hours and subsequently exposed to concentrations of B[a]P for 24 hours. Under these conditions, adducts detected at 1 μM B[a]P after 24 hours were significantly reduced, 17.28 +/− 1.30 (n = 3) adducts/108 nucleotides, in comparison to the same concentration at previous time points without Cd pre-treatment. We explored the NRF2 antioxidant pathway and total glutathione levels in cells as possible mechanisms reducing adduct formation under co-exposure. Results showed a significant increase in the expression of NRF2-responsive genes, GCLC, HMOX1, NQO1, after 1 μM Cd × 1 μM B[a]P co-exposure. Additionally, total glutathione levels were significantly increased in cells exposed to 1 μM Cd alone and 1 μM Cd × 1 μM B[a]P. Together, these results suggest that Cd may antagonize the formation of BPDE-DNA adducts in the RPTEC/TERT1 cell line under these conditions. We hypothesize that this occurs through priming of the antioxidant response pathway resulting in an increased capacity to detoxify BPDE prior to BPDE-DNA adduct formation.
Keywords: Mixtures toxicology, BPDE-DNA adducts, renal cancer, RPTEC/TERT1
Introduction
Over 90% of kidney cancers originate in the renal proximal tubule epithelial cells and are classified as renal cell carcinoma (RCC). However, only about 2% of kidney cancer cases can be attributed to a genetic predisposition (Motzer et al., 1996; Polascik et al., 2002). The remaining cases occur in otherwise healthy individuals with no prior familial history (Cancer Facts and Figures 2012, 2012). Substantial evidence of environmental risk factors contributing to the development of RCC suggests that further scrutiny of human mutagens and carcinogens on the cellular and molecular level is warranted (USRDS 2013 Annual Data Report; Chow et al., 2010).
Exposure to polycyclic aromatic hydrocarbons (PAHs) has been associated with an elevated risk of many cancers including skin, lung, bladder, liver, and stomach (Toxic Substances Database, 2011). PAHs are formed as byproducts of incomplete combustion and are ubiquitous in the environment. Major routes of exposure include inhalation and ingestion, which can result from cigarette smoking, consumption of grilled or contaminated foods, and atmospheric pollution associated with the burning of fossil fuels (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. and International Agency for Research on Cancer., 2010). Increased consumption of chargrilled meats has been shown to directly correlate with elevated PAH exposure and risk of RCC (Daniel et al., 2012; Daniel et al., 2011). Additional factors associated with the development of RCC include obesity, smoking, and hypertension (Chow, et al., 2010; Jonasch et al., 2012; Ljungberg et al., 2011).
The ability of cells to repair bulky PAH-DNA adducts may be altered by the presence of environmental contaminants such as cadmium (Cd). Cd has been shown to substitute for zinc ion co-factors in many DNA repair proteins and enzymes specifically responsible for recognizing and repairing DNA adducts (Hartwig and Schwerdtle, 2002). Cd, a heavy metal and known nephrotoxicant, is present in the environment in food, cigarettes, and contaminated water runoff. Human exposure to Cd occurs primarily through inhalation of fine particulates (i.e. tobacco smoke) and consumption of foods such as rice, cereal, and mollusks. (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. and International Agency for Research on Cancer., 2012). Cd accumulates in the liver, kidneys, and bone and is suspected to promote cancers in these organs as well as in the lungs. Cd lacks strong mutagenic properties but may act as a co-carcinogen in the body by inhibiting DNA damage repair processes and increasing oxidative stress in cells (Joseph, 2009; Waisberg et al., 2003).
Because individuals are rarely exposed to single chemical agents or carcinogens in the environment, it is important to study these compounds as humans might encounter them on a daily basis. Exposure to chemical mixtures can result in toxicological outcomes that substantially differ from the expected effects of each compound alone. Toxicants in mixtures may act through similar or distinctly different mechanisms of action. Chemicals and compounds can act antagonistically, additively, synergistically, or one chemical may potentiate the effects of another (Klassen, 2008). Ultimately, these interactions may substantially alter toxicity to different and possibly unexpected degrees. For example, in vitro studies have shown that exposure to binary combinations of PAHs including benzo[a]pyrene (B[a]P) and benzo[b]fluoranthene (B[b]F) results in a significant increase in the formation of DNA adducts than exposure to B[a]P alone. However, exposure to both B[a]P and benzo[k]fluoranthene (B[k]F), a similarly structured PAH, results in a significant reduction in the formation of DNA adducts than exposure to B[a]P alone (Sevastyanova et al., 2007; Staal et al., 2007; Tarantini et al., 2011). The opposing results that occur even after exposure to compounds of the same toxicant class emphasize the need for studies investigating effects elicited after exposure to mixtures of toxicants from similar and different classes.
In order to study the mechanisms of mixture exposure, which may promote RCC, we utilized an immortalized human renal cell line, RPTEC/TERT1. The RPTEC/TERT1 cell line was derived from the renal proximal tubule epithelial cells (RPTEC) of a normal, healthy male donor. These cells were immortalized with the catalytic subunit of the human telomerase reverse transcriptase enzyme (TERT1) (Wieser et al., 2008). Previously, we determined that RPTEC/TERT1 cells exhibit sensitivity and compound-specific responses to B[a]P and Cd treatment (Simon et al., 2014). Our results were consistent with canonical biological responses to both environmental toxicants and demonstrate metabolic competency of the RPTEC/TERT1 cell line. To test our hypothesis that Cd may alter formation of adducts after B[a]P exposure, we have explored concentration-dependent formation of BPDE-DNA adducts through cellular bioactivation of B[a]P. We examined the persistence of those adducts under conditions of pre-treatment with Cd. We intended to determine the effects of Cd on the persistence of BPDE-DNA adducts as a function of time, co-exposure, and oxidative stress.
We hypothesize that exposure to a binary combination of the environmental carcinogens, Cd, a heavy metal, and B[a]P, a representative PAH, acts to alter DNA adduct formation in comparison to levels found after B[a]P exposure alone. As Cd is known to inhibit the recognition and/or repair of PAH-DNA adducts, it is plausible to find persistence of adducts under conditions of co-exposure (Kopera et al., 2004). Alternatively, co-exposure may result in an antagonistic response leading to the formation of fewer DNA adducts through increased detoxification or inhibition of bioactivation. However, our previous work in the RPTEC/TERT1 cell line suggests that the inhibition of bioactivation is unlikely (Simon, et al., 2014). The interaction of chronic, low level exposure to both Cd and PAHs over a lifetime may provide support for environmental contributions to the development of RCC in healthy individuals.
Materials and Methods
Reagents
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless noted otherwise. Cadmium chloride (CdCl2, 202908) was dissolved in fresh complete medium and delivered at 0.1% of the final culture volume to yield the appropriate target concentrations. Benzo[a]pyrene (B[a]P, B1760) was dissolved in dimethyl sulfoxide (DMSO, D8418) and delivered at 0.05% of the final culture volume to yield the appropriate target concentrations. B[a]P preparations and exposures were carried out under low light conditions.
DNA Isolation Reagents
Enzymes used for DNA isolation including RNaseT1, mRNAse A, and proteinase K were purchased from Sigma-Aldrich. Tris-buffered saturated phenol, phenol:chloroform:isoamyl (25:24:1), and 5 PRIME Phase Lock Gel, light, 15mL tubes for DNA isolation were purchased from Fisher Scientific (Pittsburg, PA).
BPDE-DNA Adduct ELISA Reagents
Greiner Bio-One microplates (high-binding, white) were purchased from Fisher Scientific. I-Block casein-based blocking solution and CPD-Star Substrate with Emerald-II Enhancer were purchased from Life Technologies™ (Grand Island, NY). Polyclonal BPDE-DNA antiserum was kindly provided by Dr. Regina Santella. Biotin-labeled goat anti-rabbit secondary antibody (Cat # 111-065-045) was purchased from Jackson ImmunoResearch (West Grove, PA). Streptavidin-alkaline phosphatase conjugate (Cat #21324) was a product of Pierce and purchased from Fisher Scientific. Standard BPDE-DNA adducts were prepared from highly purified calf thymus DNA (Sigma, St. Louis, MO) and benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide(),(anti) from MRIGLOBAL Chemical Carcinogen Repository (Kansas City, MO) according to the procedures described by (Jennette et al., 1977).
Cell Culture
RPTEC/TERT1 cells and culture medium were purchased from Evercyte Laboratories (Vienna, Austria), and grown according to Evercyte’s instructions. Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. RPTEC/TERT1 cells were passaged approximately once or twice per week and subcultured at a 1:2 or 1:3 ratio. Cell culture vessels were purchased from Fisher Scientific and CellTreat® Scientific Products (Shirley, MA) and were tissue culture treated to promote adherent cell growth.
Cell Exposure
Cd was dissolved in fresh complete medium and delivered at 0.1% of final volume to give appropriate dose ranges. B[a]P was dissolved in DMSO and delivered at 0.05% final volume to give appropriate concentration ranges. B[a]P exposures were conducted under low light conditions. Regardless of exposure format, final volume percentage of each chemical was maintained. For co-exposure experiments, 1 μM Cd was used to pre-treat cells for 24 hours before B[a]P exposure. The Cd pre-treatment concentration was determined based on previous characterization of the cell line’s responses to various Cd concentrations. One micromolar Cd was the highest concentration tested that showed no significant cytotoxicity at 24 hours, 48 hours, or 1-week post exposure while demonstrating significantly increased cellular responses at the level of the gene and protein (Simon, et al., 2014).
For DNA isolation, RPTEC/TERT1 cells were treated at confluence in T75cm2 tissue culture treated flasks. After exposure time points, cells were washed twice with cold 1X PBS, collected by centrifugation at 4°C, and stored at −80°C until DNA was isolated.
Gene Expression
RPTEC/TERT1 cells were grown to confluence in 60mm dishes and exposed to Cd or B[a]P as described above. Cells were exposed in triplicate for each concentration and time point examined. Total RNA was isolated from cells after appropriate time points using the QIAshredder (QIAGEN, 79656, Valencia, CA) and RNeasy extraction kit (QIAGEN, 74136) following the manufacturer’s instruction. RNA concentration and purity was assessed using a Thermo Scientific Nanodrop 2000c spectrophotometer. RNA samples were diluted to 0.5μg/μL in nuclease-free water.
Two microliters of each RNA sample were used for cDNA synthesis reactions to deliver 1μg template in a 20μL total reaction volume. cDNA was synthesized using iScriptcDNA synthesis (BioRad, 170–8891, Hercules, CA) protocol as follows: 5 mins at 25°C, 30 mins at 42°C, and 5 mins at 85°C. RNA templates and cDNA were stored at −20°C until use. Gene expression was determined using primer-probe sets from Applied Biosystems® TaqMan® Gene Expression Assays. Actin, beta (ACTB) was used as a reference gene. Primers used are listed in Table 1. The thermal cycling protocol followed the manufacturer’s instructions: 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 95°C for 15s and 60°C for 1min. Reactions were conducted in 20μL volumes with each sample being run in duplicate. All reactions were carried out using a BioRad C1000™ thermal cycler equipped with a CFX96™ Real-Time PCR Detection System.
Table 1.
Primer-probe sets used for RPTEC/TERT1 Gene Expression, Applied Biosystems® TaqMan® Gene Expression Assays
| Gene ID | Gene Function | Gene Location | Assay ID |
|---|---|---|---|
| GCLC | Antioxidant | 6p12 | Hs00155249_m1 |
| HM0X1 | Antioxidant | 22q13.1 | Hs01110250_m1 |
| NQ01 | Quinone Reduction Antioxidant | 16q22.1 | Hs00168547_m1 |
| ACTB | Reference | 7p22.1 | Hs99999903_m1 |
DNA Isolation
Genomic DNA was isolated with a standard phenol chloroform extraction. Briefly, cell pellets were thawed and incubated with 1X TE buffer, RNaseT1, mRNAse A, and SDS for 45 minutes at 37°C. Pellets were incubated with proteinase K for 60 mins at 60°C and then overnight at 37°C. Deproteinized DNA was extracted using 5 PRIME Phase Lock Gel light, 15mL, tubes to increase yield from the aqueous phase. Precipitated DNA was spooled onto a glass pipette, transferred to 70% ethanol, and collected by centrifugation (18,000 rcf for 10 minutes). Ethanol was decanted and DNA was allowed to dry completely before reconstituting in sterile, DNA grade water. DNA concentration and purity was assessed using a Thermo Scientific Nanodrop 2000c spectrophotometer.
BPDE-DNA Adduct ELISA
BPDE-DNA adducts were measured by a competitive ELISA method (Gammon et al., 2002; Mumford et al., 1996; Santella et al., 1988). Briefly, 96-well white microplates were coated by adding 50pg BPDE-substituted DNA in PBS to each microwell. The DNA was sonicated and denatured in a boiling water bath for 5 min before coating. Plates were allowed to dry overnight and washed twelve times the next day with washing buffer (1X PBS/0.05%Tween 20). All subsequent wash steps were also performed twelve times. Plates were treated with I-Block (200 μL/well) for 90 min at 37°C to prevent non-specific binding. Standard curves and samples were prepared by mixing and incubating with the previously characterized polyclonal BPDE-DNA antisera at 1:3,000,000 in I-Block buffer (Mumford, et al., 1996). A 5-point standard curve was used, in triplicate, to give a range of 0.312–10 fM adducts/well. Unknown samples were assessed at 10ug DNA per well in triplicate after sonication and denaturation. The plate was washed after incubation with primary antibody, and a biotin-labeled goat anti-rabbit secondary antibody (1:2,500 in I-Block) was incubated with in each well for 1 hour. After an additional wash, the plate was incubated for one hour with streptavidin-alkaline phosphatase conjugate (1:40,000 in I-Block). After one more wash step, the CPD-Star Substrate with Emerald-II Enhancer was used to produce and amplify signal. Luminescence was read with a Tecan Infinite® 200 PRO multimode reader (Tecan, San Jose, CA).
Adducts were calculated for unknown samples based on percent inhibition of the standard curve and expressed as average number of adducts per 108 nucleotides. Nonspecific background signal detected in vehicle control groups was subtracted.
Total Glutathione Assay
After determined exposure time points, cells were trypsinized, collected, and washed twice in 1× cold PBS. Total glutathione levels in cells were determined using OxiSelect™ Total Glutathione (GSSG/GSH) Assay Kit (Cell Biolabs, Inc., San Diego, CA) according to the manufacturer’s instructions. Cell isolates were diluted at 1:100 for use within the linear range of the assay.
Statistical Analysis
One- and two-way ANOVAs were performed using the GraphPad Prism analytical software, version 6.0 (San Diego, CA). Data total glutathione assays were analyzed using a one-way ANOVA and Dunnett’s multiple comparison tests. Data for gene expression was analyzed using a two-way ANOVA and Tukey’s post hoc test. An alpha of 0.05 was used as the criteria for determining significance.
General linear models were used to test for differences among treatments, treatment groups, and time points for the BPDE-DNA adduct ELISA. Where the initial GLM analysis of variance (GLM-ANOVA) indicated a significant difference, post hoc mean comparisons were conducted using a Tukey correction. Statistical testing was conducted using IBM SPSS Statistics version 19 software (Armonk, NY). An alpha of 0.05 was used as the criteria for determining significance.
Results
BPDE-DNA adducts are formed and detected after exposure to B[a]P but altered after co-exposure to B[a]P and Cd
After 18 hours of exposure to B[a]P alone, BPDE-DNA adducts were detected in RPTEC/TERT1 DNA samples. Although there appeared to be a dose-dependent increase in adduct formation after 18 hours, exposure to 1 μM B[a]P was significantly increased over DMSO vehicle control or lower concentrations, 0.01 and 0.1 μM B[a]P. After 24 hours of exposure to B[a]P alone, adduct formation was most significantly increased at 1 μM B[a]P in comparison to DMSO vehicle control, 0.01 and 0.1 μM B[a]P at both 18 and 24 hours post exposure. Fewer adducts were detected after 24 hours of exposure to 0.1 μM B[a]P in comparison to the same concentration at 18 hours although the difference was not statistically significant (Figure 1, Table 2).
Figure 1.
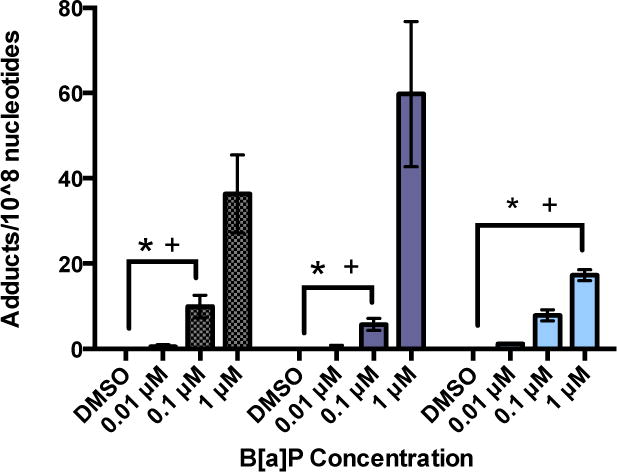
BPDE-DNA adducts are formed and detected in RPTEC/TERT1 cells after exposure to B[a]P but reduced under co-exposure to 1 μM Cd and 1 μM B[a]P. After 18 and 24 hours of treatment with B[a]P a significant increase in the number of adducts was detected at 1 μM B[a]P. Treatment of cells for 24 hours with 1 μM Cd before a 24 hour exposure to concentrations of B[a]P showed a significant decrease in adducts detected at 1 μM B[a]P in comparison to B[a]P alone. Bars represent average adducts/108 nucleotides (n = 3) +/− SEM. * indicates significant difference from 1 μM B[a]P at 18 hours, p < 0.01, + indicates significant difference from 1 μM B[a]P at 24 hours, p < 0.01.
Table 2.
BPDE-DNA adducts formed after B[a]P and Cd exposure in RPTEC/TERT1 cells detected by ELISA
| Exposure Duration | Treatment | Average adducts/108 nucleotides +/− SEM, n = 3 |
|---|---|---|
| 18 hours B[a]P | DMSO | 0.0 +/− 0.15 |
| 0.01 μM B[a]P | 0.58 +/− 0.37 | |
| 0.1 μM B[a]P | 9.92 +/− 2.66 | |
| 1 μM B[a]P | 36.34 +/− 9.14 | |
| 24 hours B[a]P | DMSO | 0.0 +/− 0.21 |
| 0.01 μM B[a]P | 0.0 +/− 0.82 | |
| 0.1 μM B[a]P | 5.72 +/− 1.43 | |
| 1 μM B[a]P | 59.75 +/− 17.03 | |
| 24 hours 1 uM Cd × 24 hours B[a]P | DMSO | 0.0 +/− 0.16 |
| 0.01 μM B[a]P | 1.18 +/- 0.14 | |
| 0.1 μM B[a]P | 7.88 +/− 1.33 | |
| 1 μM B[a]P | 17.28 +/− 1.30 |
In order to assess the ability of Cd to alter adduct formation and persistence, adducts were analyzed under conditions of Cd and B[a]P co-exposure. Cells were exposed to Cd alone for 18 and 24 hours to verify the absence of adducts. There were no BPDE-DNA adducts found above background at either time point after Cd exposure (data not shown). For co-exposure, cells were exposed to a non-cytotoxic concentration of Cd, 1μM, for 24 hours. Cytotoxicity of each compound was based on previous work (Simon, et al., 2014). After 24 hours, cells were exposed to DMSO vehicle control or appropriate concentrations of B[a]P for 24 hours. Adducts detected in groups exposed to lower concentrations of B[a]P remained relatively unchanged between treatment groups. However, cells exposed to 1 μM Cd × 1 μM B[a]P demonstrated significantly reduced levels of adducts in comparison to 1 μM B[a]P alone at either time point (Figure 1, Table 2).
Exposure to Cd increases expression of NRF2 responsive genes
Gene expression changes of the NRF2 responsive genes, glutamate-cysteine ligase, catalytic subunit (GCLC), heme oxygenase 1 (HMOX1), and NAD(P)H dehydrogenase, quinone 1 (NQO1), were examined after exposure to determine if Cd alone or Cd and B[a]P together appeared to induce an antioxidant response that may increase BPDE detoxification and reduce BPDE-DNA adduct formation under co-exposure conditions at 1 μM Cd × 1 μM B[a]P. While GCLC was detected, there was no change among treatment groups after a 24-hour exposure to Cd (Figure 2A). After 24 hours of exposure to 0.1, 1, and 10 μM Cd, there was nearly a 3-fold increase in HMOX1 at 10 μM Cd in comparison to untreated cells and all other concentrations (Figure 2B). Additionally, all concentrations of Cd showed approximately a 2–3-fold increase in NQO1 over that of untreated cells (Figure 2C).
Figure 2.
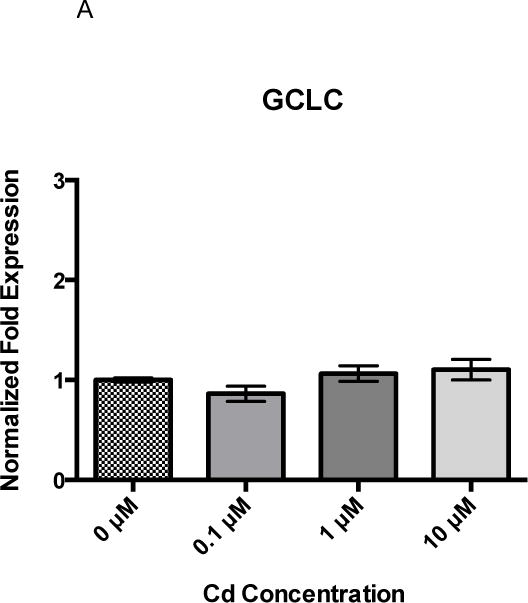
RPTEC/TERT1 cells respond to 24 hour Cd exposure by upregulating HMOX1 and NQO1 but not GCLC. After 24 hours of treatment with Cd at various concentrations, RPTEC/TERT1 cells showed no change in (A) GCLC at any concentration. There was a significant increase in gene expression at the highest concentration, 10 μM Cd, of (B) HMOX1 and (C) NQO1. Bars represent mean fold expression (n = 3) +/− SEM. All genes of interest were normalized to ACTB. 0 μM, where denoted, was set as 1. * indicates significant difference from 0μM Cd, p < 0.01, # indicates significant difference from 10μM Cd, p < 0.01, and + indicates significant difference from 0μM Cd, p < 0.05.
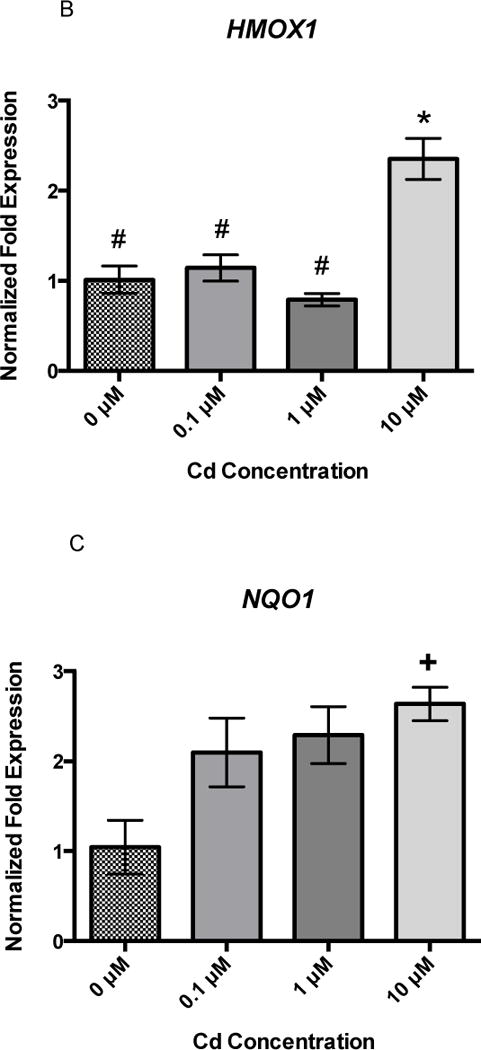
Twenty-four hours of B[a]P exposure did not increase gene expression of GCLC, HMOX1, or NQO1 (Figures 3A, 3B, and 3C). All genes were detected at basal levels by real time PCR. However, co-exposure significantly increased gene expression of all three genes at the highest concentration of 1 μM Cd × 1 μM B[a]P over vehicle control and other co-exposure groups. GCLC gene expression was increased by approximately 2-fold, HMOX1 gene expression was increased by approximately 3-fold, and NQO1 gene expression was increased by approximately 4-fold (Figures 4A, 4B, and 4C). This suggests that co-exposure, under these conditions, triggers a stronger transcriptional antioxidant response than Cd alone.
Figure 3.
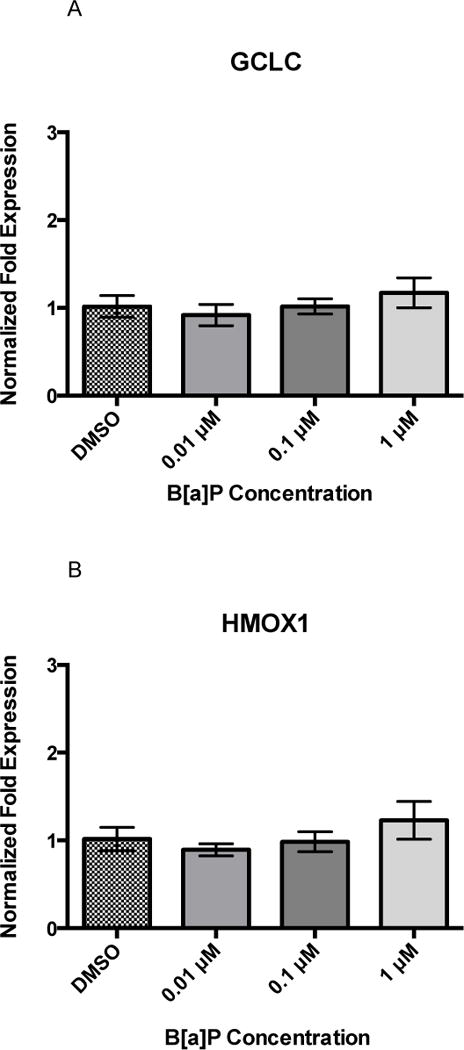
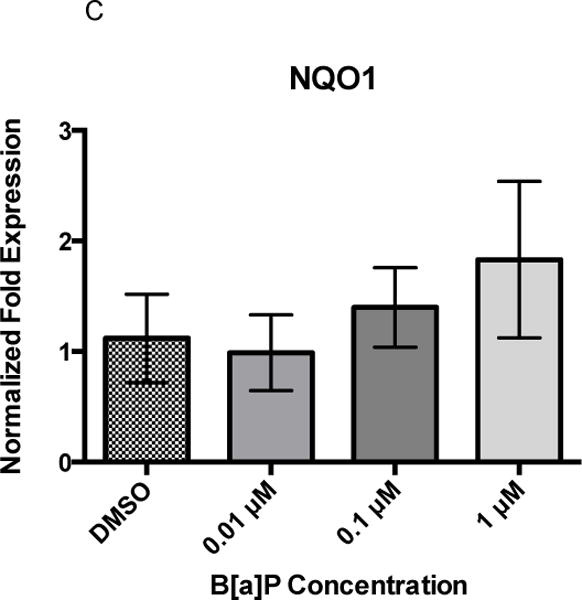
24 hour B[a]P exposure does not induce changes in GCLC, HMOX1, or NQO1. None significantly differ. Bars represent mean fold expression (n = 3) +/− SEM. All genes of interest were normalized to ACTB. DMSO, where denoted, was set as 1.
Figure 4.
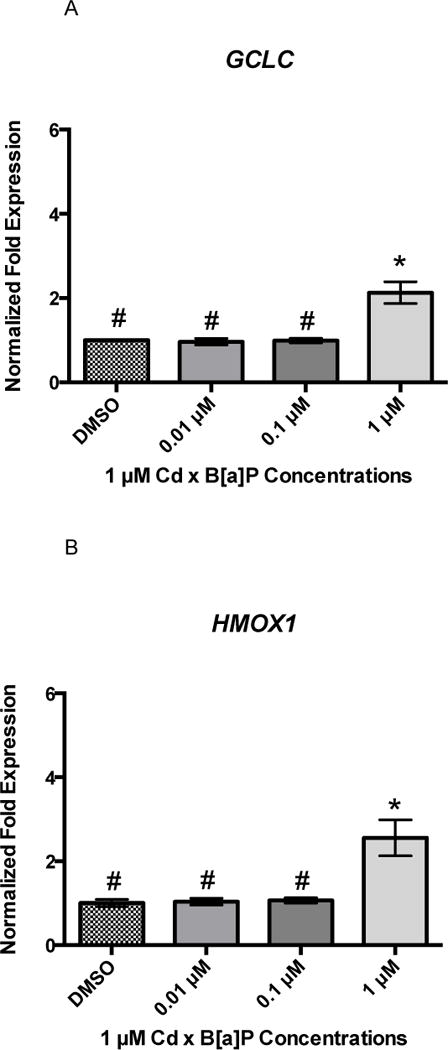
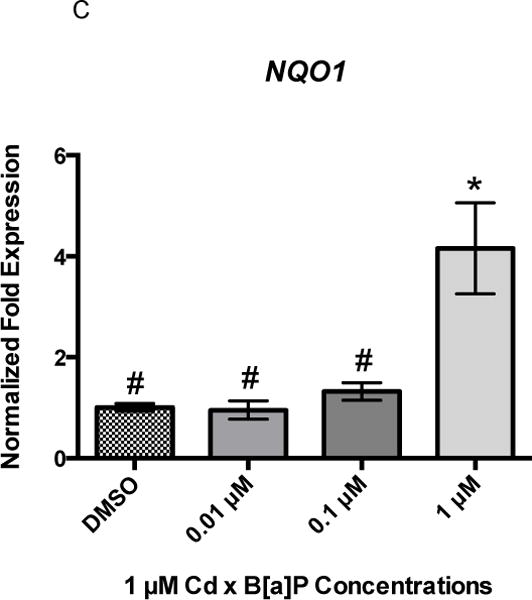
Co-exposure conditions with Cd and B[a]P result in upregulation of GCLC, HMOX1, and NQO1 in RPTEC/TERT1 cells. Cells were exposed to 1 μM Cd for 24 hours followed by a 24 hour exposure to B[a]P at different concentrations. 1 μM Cd was previously determined to be non-cytotoxic to RPTEC/TERT1 cells after 24 hours. RPTEC/TERT1 cells demonstrated significant upregulation of (A) GCLC, (B) HMOX1, and (C) NQO1 after exposure to 1 μM Cd × 1 μM B[a]P. Bars represent mean fold expression (n = 3) +/− SEM. All genes of interest were normalized to ACTB. DMSO, where denoted, was set as 1. * indicates significant difference from DMSO, p < 0.01, and # indicates significant difference from 1μM Cd × 1μM B[a]P, p <0.01.
Cd and Cd × B[a]P exposure increase total glutathione levels in RPTEC/TERT1 cells
Total glutathione was measured after Cd exposure for 24 hours and after co-exposure with B[a]P. Total glutathione levels were approximately double in cells treated with 1 and 10 μM Cd in comparison to untreated groups after 24 hours. Cells treated with 0.1 μM Cd exhibited a slight increase in total glutathione, but this increase was not statistically significant. Total glutathione was also significantly increased in cells pretreated with 1 μM Cd for 24 hours followed by exposure to 0.01, 0.1, and 1 μM B[a]P for 24 hours (Figure 5). This supports our hypothesis that Cd induces a biochemical antioxidant response, and co-exposure to Cd and B[a]P results in a substantial increase in reduced glutathione (GSH) levels possibly greater than those induced by Cd alone.
Figure 5.
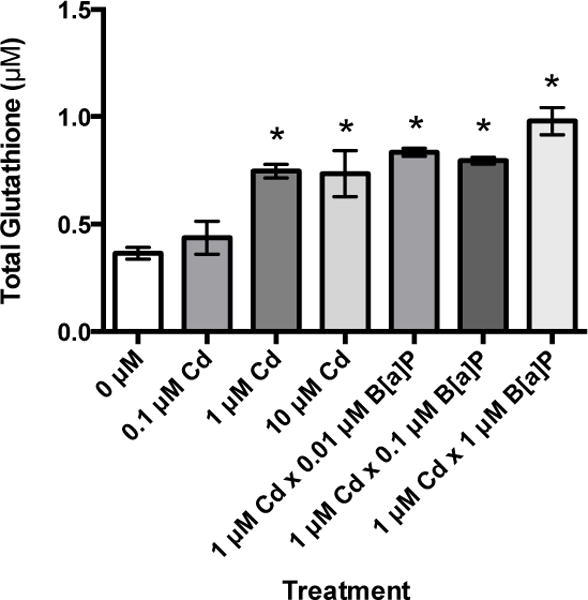
Cd exposure increases total glutathione in RPTEC/TERT1 cells. Total glutathione levels were significantly increased after 24 hours of exposure to 1 and 10 μM Cd. Total glutathione levels were also significantly increased after exposure to 1 μM Cd for 24 hours followed by a 24 hour exposure to B[a]P concentrations. Bars represent mean total glutathione levels (n = 3) +/− SEM. * indicates significant difference from 0 μM Cd, p < 0.05.
Discussion
The multifaceted effects that environmental mixtures have on the human body have been notoriously problematic to resolve. For over a decade, scientists have faced exceedingly difficult challenges in chemical toxicological research when studying chemical mixtures designed to address gaps in our knowledge (Feron and Groten, 2002). However, the importance of pursuing chemical mixture experiments continues to increase with the rise in diseases that have no clear genetic predisposition. Assessments by the American Cancer Society credit heritable mutations in the development of only 5% of all cancers (Cancer Facts and Figures 2012, 2012). Likewise, current evidence suggests that an overwhelming 90% of human disease burden, especially degenerative conditions, can be attributed to environmental factors such as exposure, lifestyle, and diet (Anand et al, 2008; Rappaport, 2011; Rappaport, 2013; Sung et al, 2011). Humans encounter mixtures of chemical compounds daily and throughout their lives; however, relatively little research to date has aimed to address the differential effects that mixtures have on molecular and mechanistic endpoints in comparison to studies focused on individual chemicals and compounds. A recent review on PAH mixtures toxicology illustrates these complexities but also provides strong rationale for approaching these issues (Jarvis et al, 2014). Biological processes and molecular factors that counteract the development of cancer (e.g. increased antioxidant or detoxification capacity) must be studied in the context of exposure to these mixtures. Environmental toxicants that interfere with efficient processing and accurate repair of DNA adducts may increase the mutagenicity of other toxicants by decreasing DNA repair capacity. In this context, such environmental toxicants function as co-carcinogens.
In an effort to characterize the effects of a simple binary mixture on renal proximal tubule cells, we have examined cellular responses of the RPTEC/TERT1 immortalized cell line to B[a]P and Cd, two distinctly different carcinogens. Our previous studies with this cell line have demonstrated its sensitivity to both B[a]P and Cd as well as compound-specific responses (Simon, et al, 2014). Here, we confirm the metabolism of B[a]P to metabolites which form DNA adducts under these conditions. We detected BPDE-DNA adducts at 18 and 24 hours post exposure to B[a]P alone. At 24 hours post exposure, there were fewer adducts detected at intermediate concentrations, 0.01 and 0.1 μM B[a]P, than at 18 hours post exposure. While the numbers of adducts were not significantly reduced, the decrease at these concentrations suggests that there may be some removal or repair of the initial adducts. However, the limited sensitivity of the ELISA method at the lower concentrations of B[a]P tested in these experiments makes these suggestions speculative. At the highest concentration of B[a]P (1 μM) tested alone, we found that significantly greater adduct levels remained at both 18- and 24-hour time points. At this concentration of B[a]P, detoxification and repair mechanisms may have been unable to process the amount of B[a]P metabolites in the examined time period. There may a threshold effect, possibly short-term but in excess of 24 hours, in which bioactivation exceeds detoxification and repair, which generates BPDE-DNA adducts at a rate greater than the rate at which DNA repair processes can remove them. This conclusion is warranted especially if B[a]P treatments alone in this experiment do not induce an antioxidant response, increase detoxification capacity, or increase DNA repair capacity. In contrast, when cells were pre-treated with 1 μM Cd, BPDE-DNA adducts formed after 1 μM B[a]P exposure were significantly reduced. This effect was not observed to this degree at other concentrations of B[a]P after Cd pre-treatment. It is possible that CYP-mediated biotransformation at the lower concentrations of B[a]P was occurring without exceeding detoxification and/or DNA repair capabilities. This would allow cellular mitigation of BPDE-DNA adducts without an increase in induction as supported, in part, by our previous work (Simon, et al., 2014). However, as mentioned previously, the sensitivity of the ELISA method at lower concentrations of B[a]P used in these experiments may not be adequate to distinguish statistically significant differences in BPDE-DNA adduct levels among the treatment and co-treatment groups with adequate precision. We suspect that B[a]P is metabolized at the lower concentrations, but more sensitive analytical methods are necessary to discriminate significant differences in adduct formation and persistence based on treatment regimen. We suggest that future experiments be designed to address such experimental possibilities and statistical power limitations.
The reduction in adducts under co-exposure at the highest concentration of B[a]P may be a function Cd × B[a]P priming the detoxification system through the NRF2 antioxidant pathway. These effects appear to result in increased levels of glutathione and increased inactivation or detoxification of BPDE prior to adduct formation. Future measurements of BPDE-DNA conjugates in conjunction with glutathione levels should be used to confirm this supposition. B[a]P exposure alone, under these conditions, does not appear to induce any such response. While it appears that Cd alone induces a significant antioxidant response resulting in increased levels of glutathione, Cd and B[a]P together at the highest concentrations tested induce an even more robust response. This response to both Cd and B[a]P in our experiments seems to mitigate DNA adduction formation through enhanced detoxification capacity. Our previous work does not support one alternative explanation that Cd inhibits the formation of BDPE through CYP feedback inhibition (Simon, et al., 2014).
The binding of B[a]P to the aryl hydrocarbon receptor increases the expression of xenobiotic response element (XRE) genes and their encoded enzymes, which are responsible for metabolizing B[a]P to reactive intermediates (Shimada and Fujii-Kuriyama, 2004; Xue and Warshawsky, 2005). These reactive intermediates, along with reactive oxygen species (ROS) from heavy metals, can increase the transcription of antioxidant response element (ARE) genes through NRF2 binding (Aleksunes and Manautou, 2007; Nguyen et al., 2009). Activation of this gene battery may be responsible for metabolite detoxification. We suspect that this process, under our experimental co-exposure conditions, reduced level of adducts detected at 1 μM B[a]P following 1 μM Cd pre-treatment. We found the expression of NRF2-targeted genes, GCLC, NQO1, HMOX1, to be significantly increased under experimental conditions coinciding with the most significant reduction in BPDE-DNA adducts under co-exposure. Our results are similar to in vivo studies which have discovered that priming the NRF2 system decreases the levels of adducts formed after B[a]P exposure. Nrf2 knockout mice develop more tumors than wild-type mice when treated with B[a]P alone. When mice are given a Nrf2 activator with B[a]P, wild-type mice develop half as many tumors. However, tumor reduction is not seen in Nrf2 knockout mice given a Nrf2 activator with B[a]P exposure (Aleksunes and Manautou, 2007). In other studies including transformed kidney cell lines from humans and rats, Cd has been shown to induce the NRF2 pathway through an oxidative stress mechanism (Chen and Shaikh, 2009; He et al., 2008; Wilmes et al., 2011). Future studies in the RPTEC/TERT1 cell line should consider the application of a NRF2 inhibitor to mimic in vivo Nrf2 knockout conditions and further verify the responses seen after B[a]P exposure. Several NRF2 activating agents, both natural and synthetic, have been examined as chemoprotectives for chronic disease and overall cell health. Flavonoids, for example, are naturally occurring antioxidants found in cruciferous vegetables, apples, and onions. They have been shown to increase NRF2 mediated expression of NQO1 and GST. Additionally, naturally occurring phytochemicals such as chalcones and coumarins have been shown to act similarly by inducing NRF2 expression of NQO1 and GST to act as anti-inflammatories and antioxidants (Kumar et al., 2014). Similarly, bardoxolone methyl, a synthetic NRF2 activator derived from natural antioxidants, has been successful in increasing kidney function and halting the progression of renal injury in Phase 2 clinical trials in patients with chronic kidney disease (Ruiz et al., 2013).
Our goals were to measure responses in RPTEC/TERT1 cells to defined, non-cytotoxic mixtures of two distinctly different toxicants. While our results suggest an antagonistic, or perhaps a hormetic effect, on the endpoint examined, DNA adduct formation and persistence, further studies are necessary to explore these results and determine effects on other downstream biomarkers. Of particular interest is the mutagenicity of BPDE-DNA adducts under such conditions of co-exposure especially at lower, environmentally relevant concentrations. We hypothesize that increased detoxification capacity is responsible for the reduced levels of BPDE-DNA adducts which may protect cells from these premutagenic lesions. This could be interpreted as a hormetic effect (Calabrese, 2008). It is also possible that, while detoxification capacity is increased, subsequent DNA repair is inhibited by the presence of Cd. Cd inhibition of DNA repair may promote repair mistakes or error-prone translesion synthesis of the remaining adducts leading to a relative increase in mutagenicity under these conditions (for a review, see Hartwig, 2013).
Apparent NRF2 activation and the increased total glutathione levels found in Cd and co-exposure groups are evidence of cellular oxidative stress. Cd and Cd compounds are Group 1 carcinogens and are known to cause cancer in humans (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. and International Agency for Research on Cancer., 2012). DNA damage caused indirectly by Cd, such as oxidative insult and repression of DNA damage repair, must be considered in mutational investigations. Quantifying the levels of PAH-DNA adducts in human studies can serve as a biomarker for exposure as well as provide information on an individual’s DNA repair capacity and mutagenic risk (Gammon, et al., 2002). However, it would be ideal to also measure mutation frequency to confirm mutagenic potential as a function of adduct formation under controlled conditions with in vitro and in vivo models to better represent and understand mechanisms by which mixtures impact humans.
We have conducted these studies in the RPTEC/TERT1 human immortalized cell line because they were derived from a normal, healthy individual, have proven to be metabolically competent, and exhibit canonical responses similar to human kidney cells when exposed to the selected environmental toxicants. However, we acknowledge the difficulties in carrying out robust, controlled experimentation on chemical mixtures. Due to the complicated nature of mixtures toxicology, it remains challenging to extrapolate the results obtained in this or any in vitro or in vivo model to actual human risk. Nevertheless, in vitro models permit higher throughput screening of many compounds and mixtures. As they improve to better model the tissue of interest, in vitro models will prove extremely valuable in mixtures toxicology. Advancements in high throughput screening have allowed scientists to begin to elucidate increasingly complex mechanisms and interactions inherent to mixtures toxicology. As research continues on both environmental and genetic components of disease, the causes of conditions such as cancer are becoming more recognized as environmentally mediated. Thus, it can be hypothesized that the majority of genetic changes resulting in cancer are acquired over a lifetime through one’s interactions with the environment. Therefore, it is critical to better understand the molecular processes that contribute to the initiation of cancer.
Acknowledgments
We would like to thank Dr. Regina M. Santella, Professor of Environmental Health Sciences, Mailman School of Public Health at Columbia University, New York, NY, for training in the BPDE-DNA adduct ELISA method and for generously providing critical reagents used in the assay.
Funding: Funding was provided in part by a generous grant from the Baton Rouge Area Foundation, Baton Rouge, LA. Funding was also provided in part by a grant and cooperative agreement from the NIH/NIEHS 1U19ES20677-01. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS or NIH. Funding and support was also provided by the Tulane Cancer Center and the Louisiana Cancer Research Consortium.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aleksunes LM, Manautou JE. Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicologic pathology. 2007;35:459–73. doi: 10.1080/01926230701311344. [DOI] [PubMed] [Google Scholar]
- Anand P, Kunnumakkara AB, Kunnumakara AB, Sundaram C, Harikumar KB, Tharakan ST, Lai OS, Sung B, Aggarwal BB. Cancer is a preventable disease that requires major lifestyle changes. Pharmaceutical research. 2008;25:2097–2116. doi: 10.1007/s11095-008-9661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese EJ. Hormesis: why it is important to toxicology and toxicologists. Environmental toxicology and chemistry / SETAC. 2008;27:1451–74. doi: 10.1897/07-541. [DOI] [PubMed] [Google Scholar]
- Cancer Facts and Figures 2012. American Cancer Society; Atlanta, Georgia: 2012. Ed.ˆ Eds.) [Google Scholar]
- Chen J, Shaikh ZA. Activation of Nrf2 by cadmium and its role in protection against cadmium-induced apoptosis in rat kidney cells. Toxicol Appl Pharmacol. 2009;241:81–9. doi: 10.1016/j.taap.2009.07.038. [DOI] [PubMed] [Google Scholar]
- Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Nature reviews. Urology. 2010;7:245–57. doi: 10.1038/nrurol.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel CR, Cross AJ, Graubard BI, Park Y, Ward MH, Rothman N, Hollenbeck AR, Chow WH, Sinha R. Large prospective investigation of meat intake, related mutagens, and risk of renal cell carcinoma. Am J Clin Nutr. 2012;95:155–62. doi: 10.3945/ajcn.111.019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel CR, Schwartz KL, Colt JS, Dong LM, Ruterbusch JJ, Purdue MP, Cross AJ, Rothman N, Davis FG, Wacholder S, Graubard BI, Chow WH, Sinha R. Meat-cooking mutagens and risk of renal cell carcinoma. British journal of cancer. 2011;105:1096–104. doi: 10.1038/bjc.2011.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feron VJ, Groten JP. Toxicological evaluation of chemical mixtures. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association. 2002;40:825–39. doi: 10.1016/s0278-6915(02)00021-2. [DOI] [PubMed] [Google Scholar]
- Gammon MD, Santella RM, Neugut AI, Eng SM, Teitelbaum SL, Paykin A, Levin B, Terry MB, Young TL, Wang LW, Wang Q, Britton JA, Wolff MS, Stellman SD, Hatch M, Kabat GC, Senie R, Garbowski G, Maffeo C, Montalvan P, Berkowitz G, Kemeny M, Citron M, Schnabel F, Schuss A, Hajdu S, Vinceguerra V. Environmental toxins and breast cancer on Long Island. I. Polycyclic aromatic hydrocarbon DNA adducts. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2002;11:677–85. [PubMed] [Google Scholar]
- Hartwig A. Metal interaction with redox regulation: an integrating concept in metal carcinogenesis? Free radical biology & medicine. 2013;55:63–72. doi: 10.1016/j.freeradbiomed.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Hartwig A, Schwerdtle T. Interactions by carcinogenic metal compounds with DNA repair processes: toxicological implications. Toxicol Lett. 2002;127:47–54. doi: 10.1016/s0378-4274(01)00482-9. [DOI] [PubMed] [Google Scholar]
- He X, Chen MG, Ma Q. Activation of Nrf2 in defense against cadmium-induced oxidative stress. Chem Res Toxicol. 2008;21:1375–83. doi: 10.1021/tx800019a. [DOI] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans and International Agency for Research on Cancer. A review of human carcinogens. Part C: Arsenic, metals, fibers, and dusts. International Agency for Research on Cancer; 2012. pp. 121–141. Ed.ˆ Eds.) [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans and International Agency for Research on Cancer. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related occupational exposures IARC Press. World Health Organization; Lyon, France Geneva: 2010. [PMC free article] [PubMed] [Google Scholar]
- Jarvis IW, Dreij K, Mattsson A, Jernstrom B, Stenius U. Interactions between polycyclic aromatic hydrocarbons in complex mixtures and implications for cancer risk assessment. Toxicology. 2014;321C:27–39. doi: 10.1016/j.tox.2014.03.012. [DOI] [PubMed] [Google Scholar]
- Jennette KW, Jeffrey AM, Blobstein SH, Beland FA, Harvey RG, Weinstein IB. Nucleoside adducts from the in vitro reaction of benzo[a]pyrene-7,8-dihydrodiol 9,10-oxide or benzo[a]pyrene 4,5-oxide with nucleic acids. Biochemistry. 1977;16:932–8. doi: 10.1021/bi00624a019. [DOI] [PubMed] [Google Scholar]
- Jonasch E, Futreal PA, Davis IJ, Bailey ST, Kim WY, Brugarolas J, Giaccia AJ, Kurban G, Pause A, Frydman J, Zurita AJ, Rini BI, Sharma P, Atkins MB, Walker CL, Rathmell WK. State of the science: an update on renal cell carcinoma. Molecular cancer research : MCR. 2012;10:859–80. doi: 10.1158/1541-7786.MCR-12-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph P. Mechanisms of cadmium carcinogenesis. Toxicol Appl Pharmacol. 2009;238:272–9. doi: 10.1016/j.taap.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Klassen CD. Casarett and Doull’s Toxicology: The Basic Science of Poisons. 7. McGraw Hill; 2008. p. 1310. Ed.ˆ Eds.) [Google Scholar]
- Kopera E, Schwerdtle T, Hartwig A, Ba W. Co(II) and Cd(II) Substitute for Zn(II) in the Zinc Finger Derived from the DNA Repair Protein XPA, Demonstrating a Variety of Potential Mechanisms of Toxicity. Chem Res Toxicol. 2004;17:1452–1458. doi: 10.1021/tx049842s. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kim IS, More SV, Kim BW, Choi DK. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Natural product reports. 2014;31:109–39. doi: 10.1039/c3np70065h. [DOI] [PubMed] [Google Scholar]
- Ljungberg B, Campbell SC, Choi HY, Jacqmin D, Lee JE, Weikert S, Kiemeney LA. The epidemiology of renal cell carcinoma. European urology. 2011;60:615–21. doi: 10.1016/j.eururo.2011.06.049. [DOI] [PubMed] [Google Scholar]
- Motzer RJ, Bander NH, Nanus DM. Renal-cell carcinoma. The New England journal of medicine. 1996;335:865–75. doi: 10.1056/NEJM199609193351207. [DOI] [PubMed] [Google Scholar]
- Mumford JL, Williams K, Wilcosky TC, Everson RB, Young TL, Santella RM. A sensitive color ELISA for detecting polycyclic aromatic hydrocarbon-DNA adducts in human tissues. Mutat Res. 1996;359:171–7. doi: 10.1016/s0165-1161(96)90264-2. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–5. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polascik TJ, Bostwick DG, Cairns P. Molecular genetics and histopathologic features of adult distal nephron tumors. Urology. 2002;60:941–6. doi: 10.1016/s0090-4295(02)01825-3. [DOI] [PubMed] [Google Scholar]
- Rappaport SM. Implications of the exposome for exposure science. Journal of exposure science & environmental epidemiology. 2011;21:5–9. doi: 10.1038/jes.2010.50. [DOI] [PubMed] [Google Scholar]
- Rappaport SM. What is the exposome? Monterey; California: 2013. Ed.ˆ Eds.) [Google Scholar]
- Ruiz S, Pergola PE, Zager RA, Vaziri ND. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney international. 2013;83:1029–41. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santella RM, Weston A, Perera FP, Trivers GT, Harris CC, Young TL, Nguyen D, Lee BM, Poirier MC. Interlaboratory comparison of antisera and immunoassays for benzo[a]pyrene-diol-epoxide-I-modified DNA. Carcinogenesis. 1988;9:1265–9. doi: 10.1093/carcin/9.7.1265. [DOI] [PubMed] [Google Scholar]
- Sevastyanova O, Binkova B, Topinka J, Sram RJ, Kalina I, Popov T, Novakova Z, Farmer PB. In vitro genotoxicity of PAH mixtures and organic extract from urban air particles part II: human cell lines. Mutat Res. 2007;620:123–34. doi: 10.1016/j.mrfmmm.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 2004;95:1–6. doi: 10.1111/j.1349-7006.2004.tb03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon BR, Wilson MJ, Wickliffe JK. The RPTEC/TERT1 cell line models key renal cell responses to the environmental toxicants, benzo[a]pyrene and cadmium. Toxicology Reports. 2014 doi: 10.1016/j.toxrep.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal YC, Hebels DG, van Herwijnen MH, Gottschalk RW, van Schooten FJ, van Delft JH. Binary PAH mixtures cause additive or antagonistic effects on gene expression but synergistic effects on DNA adduct formation. Carcinogenesis. 2007;28:2632–40. doi: 10.1093/carcin/bgm182. [DOI] [PubMed] [Google Scholar]
- Sung B, Prasad S, Yadav VR, Lavasanifar A, Aggarwal BB. Cancer and diet: How are they related? Free radical research. 2011;2011:582869. doi: 10.3109/10715762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantini A, Maitre A, Lefebvre E, Marques M, Rajhi A, Douki T. Polycyclic aromatic hydrocarbons in binary mixtures modulate the efficiency of benzo[a]pyrene to form DNA adducts in human cells. Toxicology. 2011;279:36–44. doi: 10.1016/j.tox.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Toxic Substances Database. Agency for Toxic Substances Disease Registry; Atlanta, GA: 2011. Ed.ˆ Eds.) [Google Scholar]
- National Institutes of Health, editor. United States Renal Data System 2013 Annual Data Report: Atlas of Chronic Kidney Disease and End Stage Renal Diseases in the United States. National Institue of Diabetes and Digestive and Kidney Diseases; Bethesda, MD: 2013. Ed.ˆ Eds.) [Google Scholar]
- Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- Wieser M, Stadler G, Jennings P, Streubel B, Pfaller W, Ambros P, Riedl C, Katinger H, Grillari J, Grillari-Voglauer R. hTERT alone immortalizes epithelial cells of renal proximal tubules without changing their functional characteristics. AJP: Renal Physiology. 2008;295:F1365–F1375. doi: 10.1152/ajprenal.90405.2008. [DOI] [PubMed] [Google Scholar]
- Wilmes A, Crean D, Aydin S, Pfaller W, Jennings P, Leonard MO. Identification and dissection of the Nrf2 mediated oxidative stress pathway in human renal proximal tubule toxicity. Toxicology in vitro : an international journal published in association with BIBRA. 2011;25:613–22. doi: 10.1016/j.tiv.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Xue W, Warshawsky D. Metabolic activation of polycyclic and heterocyclic aromatic hydrocarbons and DNA damage: a review. Toxicol Appl Pharmacol. 2005;206:73–93. doi: 10.1016/j.taap.2004.11.006. [DOI] [PubMed] [Google Scholar]