

Abstract
While the most common dementia is Alzheimer’s disease (AD), a detailed history is needed to rule out rapidly progressive dementias (RPDs). RPDs are less than two years in duration and have a rate of progression faster typical neurodegenerative diseases. Identification of RPDs is important as some are treatable. This review focuses on the spectrum of RPDs, with special emphasis on paraneoplastic disorders and Creutzfeldt-Jakob disease (CJD).
Introduction
While most causes of dementia are neurodegenerative and are characterized by a gradually progressive course, it is increasingly recognized that less common causes may have a more rapid course. In some instances, an apparent rapid course in fact represents the failure of observers to recognize subtle but progressive cognitive impairment that has been present for several years. Commonly, an evaluation in these instances is precipitated by crisis or a specific event (e.g. motor vehicle accident). The most common etiology is Alzheimer’s disease (AD) and a careful history will assist in the diagnosis (Please see previous chapter on AD). It is extremely important to rule out more common etiologies before considering other rare causes.
However, rare sources of dementia that may have rapid progression, moving from asymptomatic to severe stages in the course of weeks to months are increasingly recognized. It is important to be aware of these more rapidly progressive dementias (RPDs) because some are treatable (e.g. autoimmune encephalopathies) while others are not [e.g. Creutzfeldt-Jakob disease (CJD)]. This review focuses on the spectrum of RPDs, with emphasis on those that are potentially treatable. In contrast, CJD is an inevitably fatal, irreversible disease that requires public health reporting.
Rapidly progressive dementias (RPDs)
While no formal diagnostic criteria have been established to define RPDs, most experts use clinical criteria similar to those proposed for CJD. In general, the course of RPD is two years or less in duration and the rate of progression is much faster than observed for more common neurodegenerative diseases 1. Typically the mode of onset is more acute to subacute (i.e. days to weeks), in stark contrast to many neurodegenerative disorders where an exact date of onset is often difficult to identify. Beyond this duration requirement, no unifying features exist that are common to all RPDs. A simple pneumonic for helping organize a framework for possible etiologies of RPD is “VITAMIN C” (V= Vascular, I=Infectious, T= Traumatic, A=Autoimmune, M=Metabolic, I= Idiopathic/Iatrogenic, N=Neoplasm, C=Congenital). These multiple potential etiologies engender a broad differential diagnostic approach (Please see websites listed at the end of the article). At Washington University in St. Louis (WUSTL) we use a systematic, multi-tiered, approach to evaluate RPD (Figure 1). In this review we focus on two primary etiologies of RPD, the autoimmune dementing disorders and CJD. The former represents a treatable form of dementia and the latter is of significant importance from a public health perspective.
Figure 1.
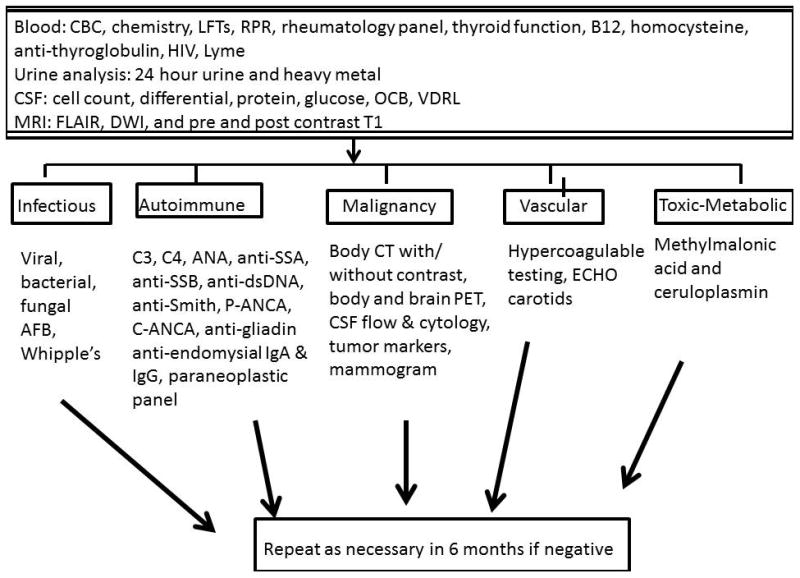
Work-up of a patient with rapidly progressive dementia (RPD).
Autoimmune Dementias
Immune-mediated dementias represent a variety of disorders, ranging from neuropsychiatric lupus to paraneoplastic encephalopathies associated with anti-tumor antibodies to neural antigens 2. These disorders can affect patients of all ages and until recently were considered a relatively rare cause of RPDs. Cognitive impairment in a young individual (< 50 years old) with a history of a dementing disorder should warrant an extensive evaluation for possible autoimmune disorders. Despite being less common, autoimmune etiologies should also be considered in the elderly population when there is a history of RPD. Some clues that may suggest an autoimmune encephalopathy include a history of an acute/subacute onset of symptoms, new onset seizures, sudden psychosis, respiratory impairment, concurrent involvement of other portions of the nervous system (e.g. peripheral neuropathy, myelopathy), or recently diagnosed cancer 3. The history of present illness is the cornerstone of the clinical diagnosis. As detailed a history as possible should be obtained from the patient, when possible, and always from collateral source(s). In the absence of a reliable history or an available collateral source, the physician should have a low threshold for considering an autoimmune encephalopathy as this class of disorders is treatable and cognitive impairment may be reversed.
When considering paraneoplastic etiologies a defining feature of these disorders is that the signs/symptoms cannot be attributed to toxicity of chemotherapy or radiation therapy, infection, coagulopathy or other toxic or metabolic causes. While various mechanisms are likely responsible for the nervous system pathology in paraneoplastic syndromes (beyond the scope of this review), the general pathophysiology is associated with an immune-mediated injury to the nervous system due to a shared antigen, expressed on both tumor cells and within the nervous system 4. The immune system therefore attacks both the cancer cells and the nervous system in its attempt to clear the antigen. The term “paraneoplastic” is commonly applied to any syndrome associated with anti-neuronal antibodies. While many of these disorders occur in the central nervous system (CNS), auto-autoantibodies can also develop in the peripheral nervous system (PNS) 2. This review will focus on disorders of the CNS. Antibodies that are more likely to be associated with an underlying malignancy relative to others are presented in Table 1. Antibody testing often involves analysis of panels of antibodies and any positive result needs to be interpreted within the appropriate clinical context as it may not always be of clinical significance. How to interpret a “positive” result and how the presence of an antibody will be incorporated into the patient’s care are important questions to consider before sending these tests as false positive results are not uncommon 5. The diagnosis of many of the autoimmune etiologies of RPD can be made using a combination of blood tests (see Figure 1), systemic imaging studies to screen for malignancy (including body computerized tomography (CT) and whole body positron emission tomography (PET)), cerebrospinal fluid analysis including glucose, protein, cell count, flow cytometry and cytology, pertinent microbiologic studies and polymerase chain reaction studies (PCRs) for specific viruses, an immunology profile (evaluation for oligoclonal bands and quantitative IgG relative to serum) and biomarkers for neurodegeneration including quantitation of the 14-3-3 antigen and total tau levels, and neuroimaging using magnetic resonance imaging (MRI) to evaluate for abnormalities characteristic of infectious, autoimmune or neurodegenerative etiologies 3. In some instances, repeat evaluations may be required every 6 months to detect malignancies that are suspected but not yet confirmed. It is not uncommon for the paraneoplastic syndrome to predate the detection of an underlying malignancy by months to years 6.
Table 1.
Paraneoplastic disorder etiologies
| Clinical presentation | Most Frequent Tumors | Associated Antibodies |
|---|---|---|
| Limbic encephalitis and Encephalopathy | Small cell lung cancer, testicular cancer, thymoma, teratoma | Anti-Hu (ANNA-1), anti-Yo (PCA-1), anti-Ri (ANNA-2), ANNA-3, anti-Ma1, anti-Ma2, anti-amphiphysin, anti-CRMP5, anti-NMDA (and other neuropil antibodies), anti-VGKC, nicotinic ganglionic AChR autoantibody |
| Cerebellar degeneration | Breast cancer, ovarian cancer, small cell lung cancer, Hodgkin disease | Anti-Yo (PCA-1), anti-Hu (ANNA-1), anti-Ri (ANNA-2), anti mGluR1, anti-VGCC, anti-Ma1, anti-CRMP5(CV2) |
| Opsoclonus-myoclonus | Neuroblastoma, small cell lung cancer, breast | Anti-Ri (ANNA-2), anti-Yo (PCA-1), anti-Hu (ANNA-1), anti-Ma1, anti-Ma2,anti-amphiphysin, anti-CRMP5(CV2) |
| Stiff-person syndrome/PERM | Breast cancer, small cell lung cancer, Hodgkin disease | Anti-amphipysin, anti-GAD, anti-glycine, anti-Ri (ANNA-2) |
| Motor neuron disease | Lymphoproliferative disorders, small cell lung cancer, breast cancer, ovarian cancer | Anti-Hu (ANNA-1), anti-Yo(PCA-1), anti-SGPS, anti-gangliosides GM1, GM2, GD1a and GD1b |
| Peripheral neuropathy | Small cell lung cancer, thymoma, lymphoproliferative disorders | Anti-Hu (ANNA-1), anti-CRMP5(CV2), anti-SGPS, anti-gangliosides GM1, GM2, GD1a and GD1b |
| Neuromyotonia | Thymoma, Hodgkin disease, small cell lung cancer | Anti-VGKC, anti-Hu (ANNA-1) |
| Lambert-Eaton syndrome | Small cell lung cancer | Anti-P/Q VGCC |
CRMP = collapsin response mediator protein; NR, NMDA= N-methyl-D-aspartate; ANNA= antineuronal nuclear antibody; PCA= purkinje cell autoantibody; VGCC= voltage-gated calcium channels; GAD= glutamic acid decarboxylase; TULP1= tubby-like protein 1; PTB= polypyrimidine-tract binding; MAG= myelin-associated glycoprotein; SGPS= sulfated glucuronic acid paragloboside; VGKC= voltage-gated potassium channels; PERM = progressive encephalomyelitis with rigidity and myoclonus; AChR = acetylcholine receptor
Modified from Toothaker and Rubins, Neurologist, 2009.
Treatment for autoimmune dementias varies based on the underlying etiology, the patient’s medical comorbidities and the presence or absence of a possible underlying malignancy 2. Treatment regimens include oral or intravenous steroids, intravenous immunoglobulin, plasma exchange, or other immunomodulatory agents (e.g. rituximab, cyclophosphamide, azathioprine, mycophenylate mofitil, or methotrexate) 5,7. For paraneoplastic disorders, the main priority is to identify and treat the possible underlying malignancy. While some paraneoplastic syndromes may initially respond to immunomodulatory therapies, for the vast majority this response will not persist unless definitive cancer treatment is pursued. With few exceptions, anti-neural antibodies directed against cell surface antigens are often responsive to immunomodulatory therapies, while those directed against intracellular antigens are unlikely to respond to treatment. Examples include antibodies (Ab) directed against the voltage-gated potassium channel complex (VGKC), a cell surface antigen. In fact, the majority of patients with anti-VGKC syndromes do not have an underlying cancer and these patients usually respond very well to immunomodulatory therapies. In contrast, syndromes associated with antibodies against neuronal nuclear antigen 1 (ANNA-1, also known as Hu) are almost always associated with an underlying malignancy and do not respond to treatment unless the underlying cancer is identified and treated 2. Prognosis for paraneoplastic dementias varies and is primarily based on the underlying cancer. For non-paraneoplastic dementias and other autoimmune dementias, the prognosis is often quite good if recognized early. In fact, as the name suggests, steroid-responsive encephalopathy with autoimmune thyroiditis (SREAT, also known as Hashimoto’s encephalopathy) is defined by its marked improvement after administering steroids.
Creutzfeldt-Jakob disease (CJD)
In contrast to autoimmune etiologies, CJD is a fatal RPD without known treatment 8. Most CJD cases are sporadic (sCJD) (85%) in nature but familial (15%) and acquired (≤ 1%) forms exist. The pathogenesis of sCJD remains poorly characterized but is believed to be due to a conformational change in the normal prion protein (PrP to an abnormal form (PrPSc) that is resistant to degradation. The term “prion” was initially coined by Stanley Prusiner in 1982 to describe proteinaceous infectious particles. Prusiner hypothesized once the abnormal PrPSc (for PrP scrapie, a spongiform encephalopathy in sheep) is formed, it acts as a template that subsequently converts normal surrounding PrP to PrPSc. This leads to an exponential growth in PrPSc and subsequent widespread propagation of neuronal degeneration 9. Of note, there is growing evidence that prion-like mechanisms play a role in not only CJD but other, more common, neurodegenerative disorders such as Alzheimer’s disease, amyotrophic lateral sclerosis, and Parkinson’s disease 10.
The resistance of prions to typical sterilization procedures makes exposure to affected brain tissues (particularly dura mater grafts and human growth hormone preparations) a major public health issue, perhaps best exemplified by the marked increase in iatrogenic CJD in Japan, France, the United Kingdom and the United States that started in 1974 and peaked around the year 2000 11. Fortunately, with increased awareness the number of iatrogenic CJD cases (~ 450 in total worldwide) has dramatically decreased since 2000, a public health feat that would not have been possible without the work of physicians to recognize this rare form of dementia. While definitive diagnosis of CJD often requires tissue confirmation, brain biopsy of a patient with suspected CJD is often precluded by for both financial and safety reasons. Many institutions choose to dispose of all instruments used during a biopsy surgery of a suspected CJD patient, a cost prohibitive practice in most circumstances. While strict precautions are used, the theoretical risk of iatrogenic transmission from surgical patients to healthcare personnel due to direct contact with contaminated tissue are also taken into consideration when considering a biopsy 11. Accordingly, more recent criteria for the diagnosis of CJD include less invasive methods.
Currently, the incidence (the number of new cases) of CJD in the United States is estimated to be 1–1.5 per million per year. Rates have not changed over the past two decades despite increased public awareness and a substantial increase in the number of referrals to the National Prion Disease Surveillance Center (NPDSC) 12. The typical age of onset of sCJD is 55–75 years of age (median = 68 years old) with males and females equally affected. The median duration of survival is approximately 4.5 months from time of onset of symptoms to death with around 90% of patients living less than one year 9. We provide more recent guidelines developed at the University of California San Francisco TABLE 2A. It is important to note that cognitive changes may not be the initial presenting symptom but may occur somewhat later in the course. Other common initial signs and symptoms are listed in Table 2B 13. Noninvasive diagnostic tests outlined in the guidelines may help differentiate CJD from other more treatable RPD disorders. Currently the work-up of possible CJD at our institution includes a structural MRI, electroencephalogram (EEG), and cerebrospinal fluid (CSF) and serum analyses (see Figure 1).
Table 2A.
University of California San Francisco Criteria for Probable CJD
| Clinical Symptoms |
|
|
| Rapid cognitive decline with any two of: |
| Myoclonus |
| Pyramidal/extrapyramidal |
| Visual |
| Cerebellar |
| Akinetic mutism |
| Other focal cortical signs |
|
|
| AND Diagnostic Testing |
|
|
| Typical MRI changes on FLAIR and DWI |
| EEG findings of periodic sharp and wave complexes |
|
|
| AND No other condition to explain observed clinical findings |
Modified from Geschwind, Continuum, 2010
Table 2B.
Clinical signs seen in CJD
| Presenting | During* | |
|---|---|---|
| Cognitive | 40% | 100% |
| Cerebellar | 22% | 70% |
| Constitutional | 21% | N/A |
| Behavioral | 20% | N/A |
| Sensory | 9% | N/A |
| Motor (non-cerebellar) | 9% | 62% |
| Visual | 7% | N/A |
Myoclonus = 80%
Modified from Geschwind, Continuum, 2010
Brain MRI has had the greatest impact on improving the accuracy of an antemortem diagnosis of CJD. Characteristic changes on fluid level attenuated inversion recovery (FLAIR) images and diffusion weight imaging (DWI) are extremely helpful for diagnosing CJD. We present characteristic MRI findings from a patient at initial presentation of symptoms (Figure 2). MRI alone has a sensitivity and specificity of 70–95% and 80–100%, respectively 11,13,14. CSF analysis can also assist in the diagnosis. In particular, CSF should be evaluated for elevated levels of 14-3-3 (a non-specific marker of neuronal death) and tau (a non-specific marker of neuronal degeneration). In particular, we have observed a marked elevation in tau in CJD patients (Table 3). Differences exist concerning the sensitivity and specificity of each of these CSF measures for the diagnosis of CJD. Finally, EEG assessment for periodic sharp and wave complexes may also assist in the diagnosis of CJD 14. However, the accuracy of this measurement may vary on when in the time course of the disease it is performed. At best (even at specialty centers like ours) the sensitivity and specificity of each of these tests is approximately 90% 11,13. From our own previous experience with confirmed CJD patients we have seen significant improvement in the diagnosis using a combination of these tests.
Figure 2.
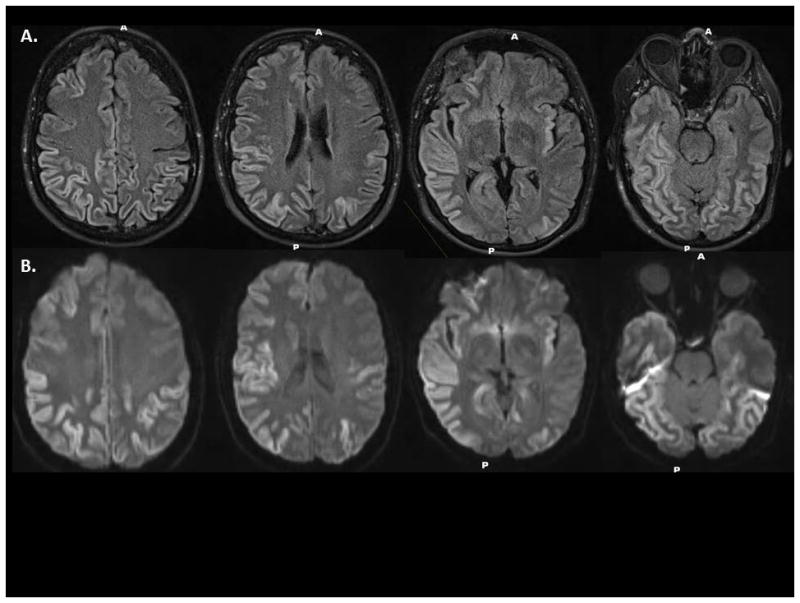
Characteristic magnetic resonance imaging (MRI) findings of a patient with Creutzfeldt-Jakob disease (CJD). Striking diffusion restriction is present in the cortical ribbon on multiple axial slices using (a) fluid level attenuated inversion recovery (FLAIR) and (b) diffusion weight imaging (DWI).
Table 3.
CSF 14-3-3 and tau values for RPD patients evaluated at our institution
| sCJD (n=24) | RPD (n=15) | Sensitivity | Specificity | Odds Ratio (Confidence Interval) | |
|---|---|---|---|---|---|
| CSF 14-3-3 “+” (%) | 68 | 68 | 68 | 71 | 5.4 (1.2 – 23) |
| CSF Tau “+” (%) | 86 | 86 | 86 | 86 | 38 (6 – 262) |
| Mean CSF Tau (pg/ml) | 4651 | 4651 | NA | NA | NA |
| Tau Range (pg/ml) | 440 – 16,131 | 440 – 16,131 | NA | NA | NA |
NA = not applicable
Modified from Wang et al., J Neurol, 2013
Recent advances in using real-time quaking induced conversion (RT-QUIC) testing of CSF may allow for reliable antemortem diagnosis. Researchers have more recently demonstrated that RT-QUIC was capable of detecting fentogram amounts of PrPSC present in CSF of patients with CJD. While this method has great promise it has not been used in large clinical settings 15. Overall, diagnosis of CJD continues to remain very difficult and can be missed. Often the diagnosis of CJD is made at a tertiary referral center such as ours only after an extensive evaluation has been performed.
Unfortunately, no disease modifying therapies exist for CJD. To date, there have been two clinical trials, both of which failed to show any significant benefit with respect to cognitive function or survival in sCJD patients. The PRION-1 study 16,17 was a partially randomized, patient-preference clinical trial using the antimalarial drug quinacrine, a drug selected based on in vitro evidence of prion elimination from a cell culture prion model. An earlier study using flupirtine maleate, a non-opiod analgesic, failed to show any improvements in cognitive function or survival in CJD patients receiving this medication 18.. Currently there are no clinical trials for sCJD planned. However, ongoing interest in developing novel therapies for this this rare disorder exists and treatments may also be relevant for more common neurodegenerative disorders.
While efficacious therapies do not exist for CJD, the role of the clinician remains extremely important in not only identifying possible treatable mimics of CJD but also providing supportive care to not only patients with sCJD but their families. The importance of identifying treatable mimics of CJD has been well illustrated by a large study by Chitravas and colleagues who demonstrated that from 2006 through 2009 the NPDSC received 1106 tissue specimens for diagnostic confirmation of CJD suspected on clinical grounds 19. Three hundred and fifty two (32%) of these specimens showed evidence of an alternative diagnosis and 71 out of these 352 (7% of the total cohort) specimens showed treatable disorders that had gone undiagnosed. The treatable disorders included immune mediated (n=26), neoplastic (n=25), infectious (n=14), metabolic/toxic (n=6). Patterson and colleagues have also noted a similar percent misdiagnosis in regards to cases that were initially identified with other dementias but were subsequently shown at autopsy to be sCJD 20. To this end we concur with the approach of Dr. Michael Geschwind who regards CJD as “the great mimicker of other diseases. It can look like anything (and vice versa).” 11 As emphasized above, a thorough, systematic approach is needed to evaluate an RPD patient in order to rule out possible treatable disorders. We encourage all clinicians, if unsure, to contact our center to ensure that these patients undergo the thorough testing necessary to make this diagnosis. All patients and their families should be encouraged to consider pathologic confirmation of suspected disease. In addition, we provide our contact information as well as the CJD Foundation information for families and patients, and the NPDSC for pathological evaluation at autopsy.
Acknowledgments
Financial Support: Supported by the National Institute of Mental Health (K23MH081786 to B.M.A.) and National Institute of Nursing Research (R01NR012907, R01NR012657, and R01NR014449 to B.M.A).
Footnotes
Conflicts of Interest: Drs. Bucelli and Ances have none to declare.
- National Prion Center- http://www.cjdsurveillance.com/
- CJD Foundation- http://www.cjdfoundation.org/
- Washington University in Saint Louis http://neuro.wustl.edu/research/researchlabs/anceslaboratory/interests/
- University of California San Francisco- http://74.50.49.198/cjd/medical
References
- 1.Williams MM, Storandt M, Roe CM, Morris JC. Progression of Alzheimer’s disease as measured by Clinical Dementia Rating Sum of Boxes scores. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2013 Feb;9(1 Suppl):S39–44. doi: 10.1016/j.jalz.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalmau J, Rosenfeld MR. Update on paraneoplastic neurologic disorders. Community oncology. 2010 May 1;7(5):219–224. doi: 10.1016/s1548-5315(11)70395-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum. 2012 Apr;18(2):366–383. doi: 10.1212/01.CON.0000413664.42798.aa. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbloom MH, Smith S, Akdal G, Geschwind MD. Immunologically mediated dementias. Current neurology and neuroscience reports. 2009 Sep;9(5):359–367. doi: 10.1007/s11910-009-0053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toothaker TB, Rubin M. Paraneoplastic neurological syndromes: a review. The neurologist. 2009 Jan;15(1):21–33. doi: 10.1097/NRL.0b013e3181870aa2. [DOI] [PubMed] [Google Scholar]
- 6.McKeon A, Lennon VA, Pittock SJ. Immunotherapy-responsive dementias and encephalopathies. Continuum. 2010 Apr;16(2 Dementia):80–101. doi: 10.1212/01.CON.0000368213.63964.34. [DOI] [PubMed] [Google Scholar]
- 7.Rosenbloom MH, Atri A. The evaluation of rapidly progressive dementia. The neurologist. 2011 Mar;17(2):67–74. doi: 10.1097/NRL.0b013e31820ba5e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Annals of neurology. 1999 Aug;46(2):224–233. {Perry, 2011 #473} [PubMed] [Google Scholar]
- 9.Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Annals of neurology. 2008 Jul;64(1):97–108. doi: 10.1002/ana.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. The Journal of experimental medicine. 2012 May 7;209(5):889–893. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geschwind MD. Rapidly progressive dementia: prion diseases and other rapid dementias. Continuum. 2010 Apr;16(2 Dementia):31–56. doi: 10.1212/01.CON.0000368211.79211.4c. [DOI] [PubMed] [Google Scholar]
- 12.Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nature neuroscience. 2012 Jul;15(7):936–939. doi: 10.1038/nn.3120. [DOI] [PubMed] [Google Scholar]
- 13.Wang LH, Bucelli RC, Patrick E, et al. Role of magnetic resonance imaging, cerebrospinal fluid, and electroencephalogram in diagnosis of sporadic Creutzfeldt-Jakob disease. Journal of neurology. 2013 Feb;260(2):498–506. doi: 10.1007/s00415-012-6664-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zerr I, Schulz-Schaeffer WJ, Giese A, et al. Current clinical diagnosis in Creutzfeldt-Jakob disease: identification of uncommon variants. Annals of neurology. 2000 Sep;48(3):323–329. [PubMed] [Google Scholar]
- 15.Atarashi R, Satoh K, Sano K, et al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nature medicine. 2011 Feb;17(2):175–178. doi: 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]
- 16.Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet neurology. 2009 Apr;8(4):334–344. doi: 10.1016/S1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geschwind MD. Clinical trials for prion disease: difficult challenges, but hope for the future. Lancet neurology. 2009 Apr;8(4):304–306. doi: 10.1016/S1474-4422(09)70050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology. 2004 Mar 9;62(5):714–718. doi: 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- 19.Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Annals of neurology. 2011 Sep;70(3):437–444. doi: 10.1002/ana.22454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paterson RW, Torres-Chae CC, Kuo AL, et al. Differential Diagnosis of Jakob-Creutzfeldt Disease. Archives of neurology. 2012 Sep 24;:1–5. doi: 10.1001/2013.jamaneurol.79. [DOI] [PMC free article] [PubMed] [Google Scholar]