

Abstract
Intracerebral hemorrhage (ICH) is a subtype of stroke involving formation of hematoma within brain parenchyma, which accounts for 8–15% of all strokes in Western societies and 20–30% among Asian populations, and has a one-year mortality rate greater than 50%. The high mortality and severe morbidity make ICH a major public health problem. Only a few evidence-based targeted treatments are used for ICH management, and interventions focus primarily on supportive care and comorbidity prevention. Even in patients who survive the ictus, extravasated blood (including plasma components) and subsequent intra-hematoma hemolytic products trigger a series of adverse events within the brain parenchyma, leading to secondary brain injury, edema and severe neurological deficits or death. Although the hematoma in humans gradually resolves within months, full restoration of neurologic function can be slow and often incomplete, leaving survivors with devastating neurological deficits.
During past years, peroxisome proliferator-activated receptor gamma (PPARγ) transcription factor and its agonists received recognition as important players in regulating not only glucose and lipid metabolism (which underlies its therapeutic effect in type 2 diabetes mellitus), and more recently, as an instrumental pleiotropic regulator of anti-inflammation, anti-oxidative regulation, and phagocyte-mediated cleanup processes. PPARγ agonists have emerged as potential therapeutic target for stroke. The use of PPARγ as a therapeutic target appears to have particularly strong compatibility toward pathogenic components of ICH. In addition to its direct genomic effect, PPARγ may interact with transcription factor, NF-κB, which may underlie many aspects of the anti-inflammatory effect of PPARγ. Furthermore, PPARγ appears to regulate expression of Nrf2, another transcription factor and master regulator of detoxification and anti-oxidative regulation. Finally, the synergistic co-stimulation of PPARγ and retinoid X receptor, RXR, may play an additional role in the therapeutic modulation of PPARγ function. In this article, we outline the main components of the role of PPARγ in ICH pathogenesis.
Intracerebral Hemorrhage Pathobiology and PPARγ
Intracerebral hemorrhage (ICH) accounts for 8–15% of all strokes in Western societies and 20–30% among Asian populations with a one-year mortality rate greater than 50–60%[1,2,3,4]. Despite advances in the field of stroke and neurocritical care, the 30-day mortality has not changed significantly over the past two decades. The therapeutic interventions that are currently available focus primarily on supportive care and comorbidity management and prevention[5,6,7]. Even in patients who survive the acute ictus (resulting in mass effect and increased intracranial pressure and primary brain injury[8,9]), the extravasated blood and, subsequently, the hemolytic products trigger a series of adverse events within brain parenchyma, causing secondary brain injury, edema and neurological deficits[4,10,11,12,13,14]. Only half of ICH-related deaths occur in the first 2 days after ICH onset[15], strongly pointing at the unique role of secondary brain injury in development of delayed mortality. It is generally accepted that the delayed aspect of ICH injury is multifactorial and, at least in part, is related to hematoma toxicity[16,17,18,19,20], the presence of noxious cellular debris and robust inflammation[11,21,22]. Hemolytic products such as hemoglobin (Hb) and its catabolic by-products (heme and iron), free radical formation (notably through iron involving Fenton-type mechanism), thrombin, metalloproteinases, complement (and other proteases), formation of oxy-modified lipid mediators, and excitotoxicity are generally listed as central components of the delayed damage after ICH[10,23,24,25,26,27]. Although the hematoma in humans gradually resolves within months, restoration of neurological function is slow and most often incomplete, and the neurological deficits can be devastating. Therefore, management of hematoma stability (e.g. preventing re-bleeding) during the acute phase followed by the control of timely clearance of hematoma-deposited blood components (to speed up hematoma resolution) may represent unique targets for the treatment of ICH[28,29,30].
The peroxisome proliferator-activated receptors (PPARs), including α, γ, and δ/β are encoded by separate genes and are members of a type II nuclear hormone receptor superfamily of ligand-activated nuclear transcription factors[31,32]. Three different PPARγ transcripts (PPARγ 1, 2 and 3), each a derivative of the PPARγ gene through differential promoter usage[33,34], have been identified. While PPARγ 2 is the isoform primarily expressed in adipose tissue, PPARγ 1 has a broader tissue distribution including presence in the brain[33,35]. The PPARγ regulates target gene expression by binding to conserved DNA sequences termed peroxisome-proliferator response elements (PPREs), as heterodimers with the retinoic acid receptor (RXR)[36,37]. PPARγ functions as a therapeutic target for the treatment of metabolic disorders, e.g., diabetes[32,38,39]. Phosphorylation of serine 112 at the N-terminus of PPARγ2 by MAP kinase and SUMOylation was suggested to regulate its transcriptional activities[40,41]. The ligands for PPARγ include oxidized fatty acids, monounsaturated- and, polyunsaturated- fatty acids such as oleic acid or linoleic acid[42], non-steroidal anti-inflammatory drugs[43], 15deoxy-Δ12,14-Prostaglandin J2 (15d–PGJ2)[44], and a class of compounds, the thiazolidinediones (TZDs)[45]. The PPARγ receptor subtype was originally characterized in adipose tissue as an important regulator of the expression of various key enzymes involved in glucose and lipid metabolism to regulate efficient energy storage[32,38,39]. Through selective activation of PPARγ, the TZDs control insulin sensitivity [44,46]. Two of the TZDs, pioglitazone and rosiglitazone, are approved by the FDA for treatment of diabetes mellitus type 2 (DM2). It is important to stress that these drugs do not change blood insulin levels; rather they make cells more sensitive to its effect.
In response to stroke, it appears that PPARγ mRNA is robustly upregulated in the affected brain tissue, suggesting that the endogenous system is attempting to activate PPARγ pathway via increasing PPARγ transcript [47,48]. While immunohistochemistry confirm that PPARγ protein is increased in the ischemia-affected hemisphere, it seems that the PPARγ DNA binding and PPARγ gene-target expression in this region is not increased, unless animals are treated with PPARγ activator[48]. This may suggest that following brain injury the endogenous activators of PPARγ are not available or in deficit, and that the whole system requires exogenous agonist to activate the PPARγ pathway.
Intracerebral hemorrhage, primarily in the case of large hematomas, could lead to alteration in cerebral perfusion in proximity to the hematoma [49,50]. While, generally, no support exists for direct ischemic penumbra in ICH-affected tissue [50,51], it is likely that even modestly reduced perfusion at the hematoma site in combination with local hypermetabolism [52] (an event demonstrated in the brain in response to intracerebral injection of hemolysates), could lead to restricted cellular injury. PPARγ agonists, by controlling expression of the glucose transporter GLUT-3 [53], could improve glucose utilization and local metabolism and, as such, contribute to cytoprotection after ICH. In addition, the arcuate nucleus, an energy homeostasis and glucose metabolism control center in the brain, contains many neurons that show high expression of PPARγ [54], suggesting a potential role of PPARγ agonists in regulating metabolism by also affecting hypothalamic functions.
Later work on PPARγ noted that PPARγ plays important roles in regulating anti-oxidative processes and inflammation[55]. It is the anti-inflammatory properties of PPARγ ligands that ultimately brought additional attention to the whole class of PPARγ agents [56,57,58,59,60,61,62]. As a transcription factor with pleotropic mechanism of action, in terms of neurological conditions[59], PPARγ was suggested to play important roles in the pathogenesis of Alzheimer’s disease[63,64], Parkinson’s disease and neurodegenerative disorders[65,66], multiple sclerosis[67,68], ischemic stroke[47,69,70,71,72], neurotrauma and spinal cord injury[73,74,75,76], and ICH[77,78]. In studies with tissue culture and other injury models, it became clear that PPARγ is protective not only to neurons[47,72,79], astrocytes[80,81], oligodendrocytes[82], endothelia[83], but also to microglia/macrophages (MMΦ)[78,84,85,86]. Among many potential mechanisms of action, the beneficial effects of PPARγ agonists are proposed to be due to: (i) the suppression of pro-inflammatory mediators, in part by interference with nuclear factor kappa B (NF-κB)[87,88,89], (ii) the upregulation of anti-oxidant enzymes including CuZn-superoxide dismutase (SOD) and catalase[78,90], (iii) the inhibition of excitotoxicity[91,92], and (iv) the promotion of microglia/macrophage-mediated clearance of toxic cellular debris via mechanism involving upregulation of scavenger receptor CD36 expression[78,93,94,95] or (iv) modification of neutrophil phenotype[61].
In this paper, we will focus primarily on the role of PPARγ in ICH. We will discuss the interactions of PPARγ with nuclear factor erythroid 2-related factor (Nrf2; a master regulator of oxidative responses) and NF-κB signaling pathways pertaining to regulation of pro- and anti-inflammatory responses. We will describe a synergistic activation of PPARγ when retinoid X receptor alpha (RXRα) and PPARγ are co-activated to achieve optimal cytoprotection and endogenous cleanup function - the clearance of hematoma-deposited blood components by brain MMΦ after ICH.
PPARγ and Catalase - implication in ICH pathogenesis
The TZDs (e.g. ciglitazone, pioglitazone and rosiglitazone) and cyclopentanone prostaglandins (e.g. 15d–PGJ2) are PPARγ agonists which have been proven to act as potent and safe pro-survival factors for primary neurons subjected to either excitotoxic insult, oxygen-glucose deprivation (OGD) or H2O2-induced oxidative stress. The exact mechanism behind this protective mechanism is not fully known, but one of several potential candidates is a PPARγ-mediated induction of potent anti-oxidative enzymes, such as superoxide dismutase[72,96] and catalase[72,74,97]. Catalase is a well-known gene target for PPARγ[98] and administration of PPARγ agonist, e.g. 15d–PGJ2, after ICH was demonstrated to rapidly induce catalase production in the affected brain[77,78]. This boost in production of anti-oxidative enzymes could be of particular importance for brain cells after ICH, since it was reported that hemoglobin lysis products (a protocol mimicking hematoma environment) reduce tissue levels of free-radical decomposing enzymes[99,100,101]. Catalase is a large homotetrameric protein that is highly abundant in the peroxisome (the membrane-enclosed small organelles that houses various oxidation reactions, in which toxic peroxides are generated as side products), where it serves to protect the cells from the toxic effects of H2O2 by catalyzing its decomposition into O2 and H2O (2H2O2 O2 + 2H2O) without generating free radicals. Interestingly, catalase activity in the brain, as compared with other tissue (e.g. heart, kidney, liver or lung), is relatively low[102]. In response to PPARγ agonist, catalase expression rapidly increased in the ICH-affected brain, demonstrating two temporal peaks with differential spatial distribution. The first peak reflects, primarily, induction of catalase expression in the ICH-affected neurons (as early as 1h after ICH and sustained at higher levels for 6∼24h[77]).The second peak is mainly associated with the catalase induction in MMΦ (appeared 3∼7 days, unpublished data). The rapid catalase production by neurons may likely reflect an adaptive response aimed at improving the H2O2 buffering capacity of neurons and is linked to direct neuroprotection. On the other hand, the upregulation of catalase in MMΦ could facilitate effective phagocytosis-mediated cleanup functions by preventing self-injury to MMΦ. During phagocytosis MMΦ generate high levels of pro-oxidative molecules that, unless neutralized by the MMΦ, may adversely affect phagocytes themselves, as well as other perihematomal brain cells[74,78]. Although the benefits of cytoprotective approaches to reduce damage to neurons, oligodendroglia, astroglia or endothelium have been frequently discussed, the benefits of protecting the phagocytes (MMΦ) from damage at the brain injury site have been seldom addressed. In our ongoing research, subjecting primary microglia to “ICH-like” (hemolytic products plus mild OGD) injury or high (>50 µM) levels of H2O2 in our hands induced significant morphologic and functional damage indicating that these cells can suffer from damage similar to other brain cells. Pre-incubating the microglia with PPARγ activators improves the expression of anti-oxidative enzymes and microglia’s resistance to H2O2 or “ICH-like” injury[78] and could increase resistance to ICH-like damage.
PPARγ and phagocytosis-mediated hematoma resolution
The hematoma size after ICH not only predicts the magnitude of mass effect and direct physical injury, but it also reflects the volume of toxic blood breakdown products, which is the cause of “chemical” secondary damage, deposited in the brain. The larger hematomas may require more time for their resolution (blood clearance) and as such may inflict damage to the surrounding brain tissue (or to impair its repair) for a longer duration of time. Thus, it is not surprising that hematoma size is one of the strongest predictors of poor outcome [103,104]. Based on this assumption several clinical trials targeting surgical hematoma evacuation were initiated [105,106,107,108]. While the overall outcome of these studies is generally neutral, some potentially promising results were seen in patients with superficial lobar hematomas without intraventricular hemorrhage [109,110]. Also, in patients subjected to minimally invasive hematoma evacuation surgery plus rt-PA during hematoma evacuation (MISTIE trial) the procedure was associated with significant reduction in perihematomal edema[107]. These suggest that under circumstances when invasiveness of the surgical approaches are low (e.g. manipulations with superficial aspects of the brain or wash out of blood with the assistance of thrombolytic rt-PA vs. application of pressure suction), the clearance of blood from the brain could be beneficial. While surgical approaches to remove blood clots continue to be evaluated, a new concept of non-surgical approaches to assist blood cleanup through promoting the endogenous microglia/macrophages-mediated phagocytosis are being tested[18]. Normally, depending on the hematoma size, the blood clearance from the brain occurs naturally in 2–4 weeks in rodents[78,111]. Our recent studies indicated that activating PPARγ in microglia/macrophages results in upregulation of expression of CD36, a cell membrane protein, which plays an essential role in phagocytosis-mediated hematoma cleanup after ICH[12].
CD36, a type II scavenger receptor, has been shown to act as a receptor for phosphatidyl serine, thrombospondin and oxidized lipids; in addition, it mediates internalization/phagocytosis of brain apoptotic cells[112,113,114], sickled/asymmetric/dislocated red blood cells (RBC)[78,94,115] and apoptotic neutrophils[116,117,118]. Interestingly, expression of this phagocytosis-aiding protein is under transcriptional control of PPARγ[119], so that its expression could effectively be upregulated pharmacologically by PPARγ agonists and inhibited by selective PPARγ antagonists[120,121,122]. In agreement with this notion, administration of PPARγ activators can efficiently increase expression of CD36 by microglia and improve phagocytosis of RBC, thus promoting hematoma resolution in animal models of ICH[18,78]. This cleanup-aiding function of CD36 and PPARγ was suggested earlier based on findings that deficiency of CD36 in macrophages due to genetic deletion of PPARγ, led to delayed uptake of oxidized low-density lipoprotein (oxLDL) by macrophages and aggravated atherosclerotic lesions[119,123,124]. Thus, CD36 upregulation in MMΦ in response to PPARγ activation may ensure a more efficient interaction between MMΦ and their phagocytosis targets for a timely clearance. This line of research prompted us to launch a clinical trial with pioglitazone in ICH patients[108]. The underlying hypothesis is that pioglitazone through PPARγ activation will assist the enhancement of the endogenous cleanup process and anti-oxidative defense, as well as amelioration of pro-inflammatory responses that altogether will inhibit secondary damage caused by ICH.
PPARγ and two faces of inflammation
After ICH, phagocytosis-mediated clearance of apoptotic or damaged cells and dislocated blood components by MMΦ is believed to play a beneficial role by minimizing the exposure of the brain tissue to this toxic and pro-inflammatory milieu [125,126]. Engulfment of apoptotic cells by MMΦ was proposed to actively suppress production of pro-inflammatory mediators by the phagocyte through promoting release of anti-inflammatory mediators, such as transforming growth factors (TGF-β) and IL-10[127,128,129]. Although clearance of hematoma by MMΦ is necessary to achieve elimination of the hematoma, a source of inflammation, the deleterious molecules generated by MMΦ during phagocytosis could injure the neurovascular component of the brain (e.g. neuron, oligodendrocyte, endothelium), and also the phagocytes themselves[11,130,131]. The main deleterious components of this process include: (i) increased release of pro-inflammatory mediators (e.g. IL-1β, TNFα), (ii) activation of pro-inflammatory transcription factor NF-κB and increased expression of pro-inflammatory enzymes (e.g. iNOS, COX-2), (iii) increased synthesis and release of proteinases (e.g. MMP9), (iv) acidification of the environment, and (v) generation of free radicals. These responses are, in part, the reason why in an attempt to control inflammation after ICH, many studies focused on how to reduce microglia/macrophage activation and/or their phagocytosis function. However, as indicated above, phagocytosis is necessary for clearance of the hematoma[18,108]. Thus, it is necessary to find ways to tune-up the phagocytosis process, so that effective clearance can be generated under conditions that have minimal adverse effect to the surrounding brain tissue.
The anti-inflammatory role of PPARγ in ICH appears to be significant. Many studies using PPARγ activators showed a robust reduction in expression of pro-inflammatory mediators (TNF-α, IL-1β, iNOS, MMP9) in MMΦ with concurrently increased expression of anti-inflammatory cytokines (TGF-β and IL-10)[59,78,89,132,133]. In rat primary microglia in culture, PPARγ agonists not only increased microglia-mediated phagocytosis of RBC, but also reduced the production of H2O2 during the process of engulfment[78]. Treatment with PPARγ agonist is associated with increased production of anti-oxidative defense system enzymes such as catalase and superoxide dismutase that may explain reduced pro-oxidative responses in cells with activated PPARγ[72,74,77,78]. It appears that prevention of oxidative stress is obligatory in allowing microglia to show optimal cleanup capacity. We have demonstrated that exogenous application of catalase to primary microglia in culture can enhance internalization of RBC by these cells, suggesting that a self-protective mechanism (anti-oxidative defense) from the excessive oxidative stress is critical to ensure MMΦ survival and efficient clean-up function. Interestingly, one of key important gene targets of PPARγ is CD36. As mentioned above, PPARγ-induced CD36 expression may play an important role in stimulating phagocytotic efficacy of microglia[78]. While the process of phagocytosis is overall beneficial from the point of removal of toxic and pro-inflammatory cellular debris, it is well-recognized that microglia-mediated scavenging activities are associated with generation of massive amount of pro-oxidants[134] which could adversely affect surrounding brain cells. As such, it is intriguing to note the same transcription factor (PPARγ) not only up-regulates genes associated with enhanced phagocytosis (e.g. CD36), but also simultaneously up-regulates anti-oxidative genes (e.g. catalase) that permit more effective neutralization of oxidative stress associated with more robust scavenging activities. Interestingly, this cooperative generation of CD36 and anti-oxidative enzyme exists not only for PPARγ. In our ongoing research (unpublished results), we have determined that Nrf2, a transcription factor considered a master regulator of cellular anti-oxidative defense, is also capable of inducing CD36 expression in microglia. These findings strongly suggest that for optimal function of CD36 in hematoma resolution (and likely cleanup after ischemic stroke), the anti-oxidative defense system needs to be enhanced to eliminate the deleterious consequences (oxidative damage) associated with CD36-mediated phagocytosis/endocytosis.
Lastly, it should be mentioned that PPARγ is proposed to act as a signaling molecule downstream from the IL-4 receptor; a pathway that has a key role in an alternative activation of MMΦ [135,136,137], which results in formation of a “healing” trophic phenotype of MMΦ. In our ongoing research, we have established that IL-4 is generated locally in the brain and via IL-4 receptor activates STAT6 and PPARγ signaling leading to reduction of pro-inflammatory and induction of anti-inflammatory phenotype of microglia after stroke.
Taken together, PPARγ may benefit the inflammation in ICH by directly down-regulating the production of pro-inflammatory mediators and up-regulating anti-inflammatory mediators. This is in addition to its role in hematoma clearance, the process that leads to removal of the toxic and pro-inflammatory debris from the intraparenchymal tissue.
PPARγ activation and interaction of PPARγ and RXR
PPARγ and RXR, both are ligand-dependent pleiotropic transcription factors belonging to the nuclear hormone receptor family. Upon dimerization, PPARγ-RXR as “partners” regulate target gene expression by binding to conserved DNA sequences, PPRE[38]. There are three RXR isotypes, RXRα (NR2B1), RXRβ (NR2B2), and RXRγ (NR2B3), which have considerable tissue-specific differences in their expression[138] and are present in various cells of brain tissue[139]. The PPARγ-RXR heterodimer complex can be activated either by PPARγ ligands (e.g. TZD or 15d–PGJ2) and/or by RXR ligands (e.g. 9-cis retinoic acid)[140]; however, the occupancy of both (PPARγ plus RXR) ligand binding domains simultaneously could provide maximal receptor activity[32,141,142,143]. It is necessary to acknowledge that when comparing PPARγ activation in response to RXR- vs. PPARγ-activating ligand, RXR may dimerize with and activate other nuclear receptors (e.g. retinoic acid receptor, RAR; liver X receptor, LXR; pregnane X receptor, PXR; or farnesoid X receptor, FXR). As such, RXR agonists could have broader biological activity than PPARγ. However, it is often proposed that some key effects including anti-inflammatory effects of RXR agonists appear to be executed largely through a PPARγ pathway[144]. In our lab, we have demonstrated that the co-treatment of primary cortical cultured neurons with the combination of 15d–PGJ2 and 9-cis retinoic acid protected the cells from OGD-induced damage more robustly than each of the ligands alone. Also, primary rat microglia in response to combined PPARγ activator (e.g. pioglitazone) and RXR activator (e.g. Bexarotene), appear to demonstrate a significantly higher phagocytosis efficacy toward erythrocytes, as compared to each of the ligands alone (ongoing studies), further supporting the existence of synergy between PPARγ and RXR activators in various biological processes. These beneficial interactions of PPARγ and RXR ligands is consistent with an earlier report that 15d–PGJ2 plus 9-cis retinoic acid was superior in reducing behavioral dysfunction in a mouse model of experimental encephalomyelitis (EAE)[145]. Interestingly, it was recently demonstrated that retinoic acid at higher concentration can elicit different, even contrasting effect (to that seen with lower concentration) by activating PPARβδ/RXR heterodimers[146].
Interaction of PPARγ and Nrf2 and NF-κB
NF-κB is a transcription factor that regulates expression of many pro-inflammatory enzymes, cytokines, chemokines, proteases and adhesion molecules, contributing to amplification of the secondary inflammation response and neuronal damage after ICH[11,147,148,149,150]. The functional NF-κB exists as a dimer, which in neurons is composed primarily of the (NF-κB1) p50 and (RelA) p65 subunits. Other NF-κB members of the NF-κB/Rel family, include RelB, c-Rel, and p52 (NF-κB2)[151]. Numerous studies have confirmed that PPARγ may bind to the NF-κB subunits, p50 and p65, directly resulting in NF-κB inactivation[77,87,152]. PPARγ may also indirectly inhibit NF-κB by (1) competing for common transcription co-activators such as SRC-1[153] and p300/CBP (CREB-binding protein)[154,155]; (2) up-regulating the inhibitor kappa B (IκB), protein that prevents NF-κB nuclear translocation which is a prerequisite for NF-κB activation[88,156]; and (3) indirectly inhibiting NF-κB by activating transcription factor Nrf2, which reduces generation of pro-oxidative molecules which are required for NF-κB activation. Ultimately, inhibition of NF-κB by PPARγ agonists were reported to reduce generation of pro-inflammatory mediators/responses[56,57] and consequently the secondary brain damage associated with these pro-inflammatory responses[72,73,77,78].
Nrf2 is a ubiquitous pleiotropic transcription factor and a master genomic homeostatic regulator of intracellular stress[157,158,159]. Combining with Mif family proteins, Nrf2 forms heterodimeric complexes to transactivate the antioxidant response elements (ARE) within the regulatory region of many cytoprotective target genes [e.g. catalase, heme oxygenase-1 (HO-1), superoxide dismutase (SOD), glutathione-S-transferase (GST), thioredoxin and NAD(P)H dehydrogenase quinone 1 (NQO1)) [160] and also other proteins with important roles in neutralization of oxidative stress and detoxification of hemolytic products (e.g., haptoglobin, hemopexin, ferritin and hemooxygenase-1)[30,161]. In most cells, Nrf2 is present at low concentrations due to continuous Nrf2 degradation through the proteasome pathway. Nrf2 contributes to neuroprotection and amelioration of brain damage after cerebral ischemia[162,163], neurotrauma[164,165], neurodegenerative diseases[166,167,168] and ICH[30,161,169] primarily through reducing oxidative stress, inflammation, and generation of detoxifying molecules capable of neutralizing many noxious products generated in response to injury. The interaction between PPARγ and Nrf2 may involve multiple mechanisms. First, PPRE and ARE co-exist in the same genes, such as CD36[170] and catalase[171,172]; second, a reciprocal transcriptional regulation exists between Nrf2 and PPARγ genes, Nrf2 gene contains putative PPREs[173], and conversely, PPARγ gene appears to contain the ARE[174,175]; third, an interaction between PPARγ and Nrf2 may be through NF-κB inhibition. Since NF-κB activation highly depends upon the presence of oxidative stress, then the effect of Nrf2 in ameliorating oxidative stress was proposed to inhibit NF-κB[176]. As different mechanisms are used by Nrf2 and PPARγ in inhibiting NF-κB, it is likely that the simultaneous activation of both Nrf2 and PPARγ may have a synergistic effect. Due to the interactions among PPARγ, Nrf2 and NF-κB, it has been suggested that co-activation of PPARγ and Nrf2 may improve outcomes of several neurological disorders[177].
Neurotoxicity following PPARγ activation
Unlike synthetic thiazolidinediones (TZDs; e.g. pioglitazone and rosiglitazone) that have considerable levels of specificity toward PPARγ, prostaglandin D2 derivatives (primarily with cyclopentanone structure), including 15d–PGJ2, which is proposed to act as endogenous PPARγ ligands demonstrate rather limited selectivity toward PPARγ with some of its biological activities including activation of Nrf2[168] or inhibition of NF-κB[87]. There is existing evidence on the dose-dependent neurotoxicity of the 15d–PGJ2 in cerebellar granule cells, primary cortical neurons and spinal cord motor neurons[178,179] which originally were believed to be linked to PPARγ. The mechanism that underlies the neurotoxic effect of 15d–PGJ2 is chiefly linked to higher doses of the compound. In addition, it is primarily associated with induction of apoptosis and not likely associated with the activation of PPARγ[180,181]. On the other hand, the clinically relevant, more selective PPARγ agonist such as rosiglitazone, was linked to peripheral edema, increase in body weight, and cardiomyopathies and heart failure[182]. Again the relationship between these side effects and PPARγ is somewhat controversial, as another TZD PPARγ agonist, pioglitazone, may show beneficial effects. The PROACTIVE (PROspective pioglitAzone Clinical Trial In macroVascular Events; NCT00174993) randomized, double-blinded, placebo-controlled study looked at the impact of pioglitazone on total mortality and macrovascular morbidity in 5,238 patients with DM2 and macrovascular disease. This secondary prevention study showed safety and a macrovascular benefit with pioglitazone in terms of major adverse cardiovascular events including all-cause mortality, MI, and stroke[183,184]. Finally, it should be mentioned that the above side effects of rosiglitazone are described in patients taking TZDs long-term for DM2. It is likely that PPARγ agonist treatment for ICH will be short-term, potentially avoiding these side effects, although this needs careful testing.
Clinical trials of PPARγ agonists in ICH
Pre-clinical work has shown that PPARγ agonists are capable of promoting endogenous hematoma clearance, decreasing neuronal damage, and improving functional recovery in rodent model of ICH[77,78]. In addition, PPARγ agonists in vitro reduced the production of pro-inflammatory mediators and free radicals produced during phagocytosis[78]. Based on these studies, a clinical trial to evaluate the Safety of Pioglitazone for Hematoma Resolution in ICH has been launched (SHRINC)[108]. This is a prospective, randomized, blinded, placebo-controlled, dose-escalation safety trial in which patients with spontaneous intracerebral hemorrhage are randomly allocated to placebo or treatment. Pioglitazone, one of the PPARγ agonists that are approved by the Food and Drug Administration (FDA) for glycemic control in type II diabetes mellitus, was provided to the patients with escalating dosages. There was an evaluation period of 3∼6 months and the continual reassessment method for dose finding is used to determine the maximum tolerated dose of pioglitazone. Hematoma and edema resolution is evaluated with serial magnetic resonance imaging (MRI) at specified time points. The Phase 2 study with a planned sample size of 84 patients has preliminarily demonstrated safety regarding mortality[108], and is now in the next planning stages (ClinicalTrials.gov Identifier: NCT00827892).
Since hematoma absorption is an extremely important objective after ICH, the SHRINC study should provide important information regarding the safety and clinical outcome regarding PPARγ agonists in the endogenous hematoma absorption. Besides primary ICH, secondary brain hemorrhage following brain trauma and brain surgery, subarachnoid hemorrhage (SAH), and hemorrhagic transformation of the ischemic stroke treated with rtPA may also be managed through this endogenous blood reabsorption (clearance) mechanism. Therefore, we are expecting that PPARγ, as a promising therapeutic target, could open a new field for managing hematoma clearance through a non-surgical mechanism.
Figure 1.
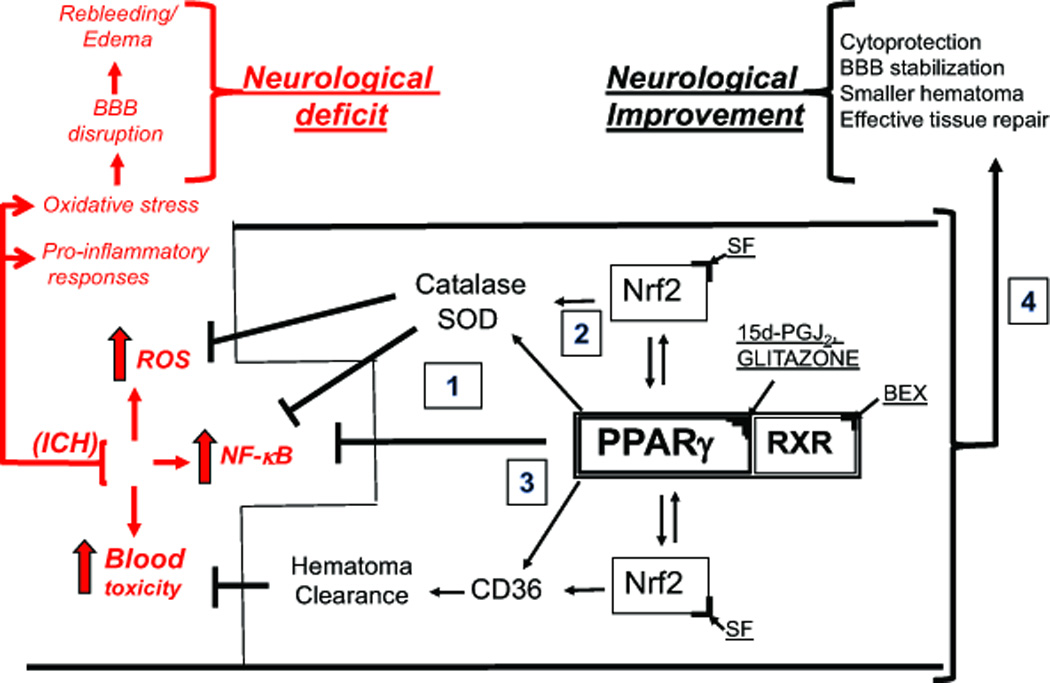
PPARγ as therapeutic targets for ICH. In response to ICH, the local generation of reactive oxygen species (ROS), accumulation of toxic blood components (e.g. hemolytic products) in brain parenchyma, and activation of pro-inflammatory transcription factor NF-κB (causing generation of pro-inflammatory cytokines and enzymes) leads to brain injury, often referred to as secondary brain damage, which manifest itself with blood brain barrier (BBB) disruption, rebleeding, edema and ultimately neurological deficit or death. Activation of PPARγ with, e.g., 15d–PGJ2 or thiazolidinediones (known as glitazones) leads to: upregulation of the anti-oxidative enzymes, catalase and superoxide dismutase (SOD); scavenger receptor (e.g. CD36 on macrophages/microglia MMΦ) for RBC and hematoma clearance. Both PPARγ and Nrf2 (which can be activated with sulforaphane, SF) regulate transcription of these genes. PPARγ suppresses NF-κB to limit the pro-inflammatory response. Also, activation of RXR, an obligatory heterodimeric partner for PPARγ activity (e.g. with 9-cis-retinoic acid or Bexarotene, BEX), could augment the effect of PPARγ ligand acting alone. Thus, PPARγ activation may benefit the acute ICH and post-ICH recovery by: (1) down-regulating the production of pro-inflammatory mediators, (2) up-regulating the anti-oxidative enzymes production, (3) promoting endogenous hematoma clearance thus eliminating the source of inflammation and allowing for more effective repair, and (4) direct and indirect cytoprotection.
Acknowledgments
Some of the studies indicated in this report were supported by NIH R01NS060768, 1R01NS064109 and R01NS084292 grants.
REFERENCES
- 1.Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 2009;8:355–369. doi: 10.1016/S1474-4422(09)70025-0. [DOI] [PubMed] [Google Scholar]
- 2.van Asch CJ, Luitse MJ, Rinkel GJ, et al. Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol. 2010;9:167–176. doi: 10.1016/S1474-4422(09)70340-0. [DOI] [PubMed] [Google Scholar]
- 3.Sangha N, Gonzales NR. Treatment targets in intracerebral hemorrhage. Neurotherapeutics. 2011;8:374–387. doi: 10.1007/s13311-011-0055-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qureshi AI, Tuhrim S, Broderick JP, et al. Spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344:1450–1460. doi: 10.1056/NEJM200105103441907. [DOI] [PubMed] [Google Scholar]
- 6.Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373:1632–1644. doi: 10.1016/S0140-6736(09)60371-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brouwers HB, Goldstein JN. Therapeutic strategies in acute intracerebral hemorrhage. Neurotherapeutics. 2012;9:87–98. doi: 10.1007/s13311-011-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keep RF, Xi G, Hua Y, Hoff JT. The deleterious or beneficial effects of different agents in intracerebral hemorrhage: think big, think small, or is hematoma size important? Stroke. 2005;36:1594–1596. doi: 10.1161/01.STR.0000170701.41507.e1. [DOI] [PubMed] [Google Scholar]
- 9.Keep RF, Xi G, Hua Y, Xiang J. Clot formation, vascular repair and hematoma resolution after ICH, a coordinating role for thrombin? Acta Neurochir Suppl. 2011;111:71–75. doi: 10.1007/978-3-7091-0693-8_12. [DOI] [PubMed] [Google Scholar]
- 10.Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- 11.Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27:268–279. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- 12.Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwang BY, Appelboom G, Ayer A, et al. Advances in neuroprotective strategies: potential therapies for intracerebral hemorrhage. Cerebrovasc Dis. 2011;31:211–222. doi: 10.1159/000321870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belur PK, Chang JJ, He S, Emanuel BA, Mack WJ. Emerging experimental therapies for intracerebral hemorrhage: targeting mechanisms of secondary brain injury. Neurosurg Focus. 2013;34:E9. doi: 10.3171/2013.2.FOCUS1317. [DOI] [PubMed] [Google Scholar]
- 15.Broderick J, Connolly S, Feldmann E, et al. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke. 2007;38:2001–2023. doi: 10.1161/STROKEAHA.107.183689. [DOI] [PubMed] [Google Scholar]
- 16.Koeppen AH. The history of iron in the brain. 1995;134(Suppl):1–9. doi: 10.1016/0022-510x(95)00202-d. [DOI] [PubMed] [Google Scholar]
- 17.Xi G, Fewel ME, Hua Y, et al. Intracerebral hemorrhage: pathophysiology and therapy. Neurocrit Care. 2004;1:5–18. doi: 10.1385/ncc:1:1:5. [DOI] [PubMed] [Google Scholar]
- 18.Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke. 2009;40:S92–S94. doi: 10.1161/STROKEAHA.108.533158. [DOI] [PubMed] [Google Scholar]
- 19.Lok J, Leung W, Murphy S, et al. Intracranial hemorrhage: mechanisms of secondary brain injury. Acta Neurochir Suppl. 2011;111:63–69. doi: 10.1007/978-3-7091-0693-8_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen-Roetling J, Sinanan J, Regan RF. Effect of Iron Chelators on Methemoglobin and Thrombin Preconditioning. Transl Stroke Res. 2012;3:452–459. doi: 10.1007/s12975-012-0195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Dore S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007;27:894–908. doi: 10.1038/sj.jcbfm.9600403. [DOI] [PubMed] [Google Scholar]
- 22.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Regan RF, Panter SS. Hemoglobin potentiates excitotoxic injury in cortical cell culture. J Neurotrauma. 1996;13:223–231. doi: 10.1089/neu.1996.13.223. [DOI] [PubMed] [Google Scholar]
- 24.Xi G, Keep RF, Hoff JT. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg. 1998;89:991–996. doi: 10.3171/jns.1998.89.6.0991. [DOI] [PubMed] [Google Scholar]
- 25.Huang FP, Xi G, Keep RF, et al. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Mori T, Sumii T, Lo EH. Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke. 2002;33:1882–1888. doi: 10.1161/01.str.0000020121.41527.5d. [DOI] [PubMed] [Google Scholar]
- 27.Kuramatsu JB, Huttner HB, Schwab S. Advances in the management of intracerebral hemorrhage. J Neural Transm. 2013 doi: 10.1007/s00702-013-1040-y. [DOI] [PubMed] [Google Scholar]
- 28.Babu R, Bagley JH, Di C, Friedman AH, Adamson C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg Focus. 2012;32:E8. doi: 10.3171/2012.1.FOCUS11366. [DOI] [PubMed] [Google Scholar]
- 29.Brouwers HB, Greenberg SM. Hematoma expansion following acute intracerebral hemorrhage. Cerebrovasc Dis. 2013;35:195–201. doi: 10.1159/000346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao X, Aronowski J. Nrf2 to pre-condition the brain against injury caused by products of hemolysis after ICH. Transl Stroke Res. 2013;4:71–75. doi: 10.1007/s12975-012-0245-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 1994;91:7355–7359. doi: 10.1073/pnas.91.15.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 33.Fajas L, Auboeuf D, Raspe E, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- 34.Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu Rev Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- 35.Cristiano L, Bernardo A, Ceru MP. Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. J Neurocytol. 2001;30:671–683. doi: 10.1023/a:1016525716209. [DOI] [PubMed] [Google Scholar]
- 36.Greene ME, Blumberg B, McBride OW, et al. Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expr. 1995;4:281–299. [PMC free article] [PubMed] [Google Scholar]
- 37.Elbrecht A, Chen Y, Cullinan CA, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun. 1996;224:431–437. doi: 10.1006/bbrc.1996.1044. [DOI] [PubMed] [Google Scholar]
- 38.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lemberger T, Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol. 1996;12:335–363. doi: 10.1146/annurev.cellbio.12.1.335. [DOI] [PubMed] [Google Scholar]
- 40.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272:5128–5132. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 41.Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaud SE, Renier G. Direct regulatory effect of fatty acids on macrophage lipoprotein lipase: potential role of PPARs. Diabetes. 2001;50:660–666. doi: 10.2337/diabetes.50.3.660. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 44.Forman BM, Tontonoz P, Chen J, et al. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 45.Lehmann JM, Moore LB, Smith-Oliver TA, et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 46.Stumvoll M, Haring HU. Glitazones: clinical effects and molecular mechanisms. Ann Med. 2002;34:217–224. [PubMed] [Google Scholar]
- 47.Ou Z, Zhao X, Labiche LA, et al. Neuronal expression of peroxisome proliferator-activated receptor-gamma (PPARgamma) and 15d–prostaglandin J2--mediated protection of brain after experimental cerebral ischemia in rat. Brain Res. 2006;1096:196–203. doi: 10.1016/j.brainres.2006.04.062. [DOI] [PubMed] [Google Scholar]
- 48.Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARgamma expression and activation after transient focal ischemia in rats. Eur J Neurosci. 2006;24:1653–1663. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- 49.Etminan N, Beseoglu K, Turowski B, Steiger HJ, Hanggi D. Perfusion CT in Patients With Spontaneous Lobar Intracerebral Hemorrhage: Effect of Surgery on Perihemorrhagic Perfusion. Stroke. 2011 doi: 10.1161/STROKEAHA.111.616730. [DOI] [PubMed] [Google Scholar]
- 50.Zazulia AR, Diringer MN, Videen TO, et al. Hypoperfusion without ischemia surrounding acute intracerebral hemorrhage. J Cereb Blood Flow Metab. 2001;21:804–810. doi: 10.1097/00004647-200107000-00005. [DOI] [PubMed] [Google Scholar]
- 51.Qureshi AI, Wilson DA, Hanley DF, Traystman RJ. No evidence for an ischemic penumbra in massive experimental intracerebral hemorrhage. Neurology. 1999;52:266–272. doi: 10.1212/wnl.52.2.266. [DOI] [PubMed] [Google Scholar]
- 52.Ardizzone TD, Lu A, Wagner KR, et al. Glutamate receptor blockade attenuates glucose hypermetabolism in perihematomal brain after experimental intracerebral hemorrhage in rat. Stroke. 2004;35:2587–2591. doi: 10.1161/01.STR.0000143451.14228.ff. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Bueno B, Caso JR, Perez-Nievas BG, Lorenzo P, Leza JC. Effects of peroxisome proliferator-activated receptor gamma agonists on brain glucose and glutamate transporters after stress in rats. Neuropsychopharmacology. 2007;32:1251–1260. doi: 10.1038/sj.npp.1301252. [DOI] [PubMed] [Google Scholar]
- 54.Sarruf DA, Yu F, Nguyen HT, et al. Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology. 2009;150:707–712. doi: 10.1210/en.2008-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neve BP, Fruchart JC, Staels B. Role of the peroxisome proliferator-activated receptors (PPAR) in atherosclerosis. Biochem Pharmacol. 2000;60:1245–1250. doi: 10.1016/s0006-2952(00)00430-5. [DOI] [PubMed] [Google Scholar]
- 56.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 57.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 58.Heneka MT, Landreth GE. PPARs in the brain. Biochim Biophys Acta. 2007;1771:1031–1045. doi: 10.1016/j.bbalip.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 59.Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;13:1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ballesteros I, Cuartero MI, Pradillo JM, et al. Rosiglitazone-induced CD36 up-regulation resolves inflammation by PPARgamma and 5-LO-dependent pathways. J Leukoc Biol. 2013 doi: 10.1189/jlb.0613326. [DOI] [PubMed] [Google Scholar]
- 61.Cuartero MI, Ballesteros I, Moraga A, et al. N2 Neutrophils, Novel Players in Brain Inflammation After Stroke: Modulation by the PPARgamma Agonist Rosiglitazone. Stroke. 2013;44:3498–3508. doi: 10.1161/STROKEAHA.113.002470. [DOI] [PubMed] [Google Scholar]
- 62.Zhao X, Aronowski J. The role of PPARγ in stroke. Immunological Mechanisms and Therapies in Brain Injuries and Stroke. Springer Series in Translational Stroke Research. 2013 [Google Scholar]
- 63.Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer's disease. Neurobiol Aging. 2001;22:937–944. doi: 10.1016/s0197-4580(01)00296-2. [DOI] [PubMed] [Google Scholar]
- 64.Feinstein DL. Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabetes Technol Ther. 2003;5:67–73. doi: 10.1089/152091503763816481. [DOI] [PubMed] [Google Scholar]
- 65.Randy LH, Guoying B. Agonism of Peroxisome Proliferator Receptor-Gamma may have Therapeutic Potential for Neuroinflammation and Parkinson's Disease. Curr Neuropharmacol. 2007;5:35–46. doi: 10.2174/157015907780077123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clark J, Simon DK. Transcribe to survive: transcriptional control of antioxidant defense programs for neuroprotection in Parkinson's disease. Antioxid Redox Signal. 2009;11:509–528. doi: 10.1089/ars.2008.2241. [DOI] [PubMed] [Google Scholar]
- 67.Feinstein DL, Galea E, Gavrilyuk V, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 68.Racke MK, Gocke AR, Muir M, et al. Nuclear receptors and autoimmune disease: the potential of PPAR agonists to treat multiple sclerosis. J Nutr. 2006;136:700–703. doi: 10.1093/jn/136.3.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sundararajan S, Gamboa JL, Victor NA, et al. Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130:685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 70.Zhao Y, Patzer A, Gohlke P, Herdegen T, Culman J. The intracerebral application of the PPARgamma-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur J Neurosci. 2005;22:278–282. doi: 10.1111/j.1460-9568.2005.04200.x. [DOI] [PubMed] [Google Scholar]
- 71.Vemuganti R. Therapeutic Potential of PPARgamma Activation in Stroke. PPAR Res. 2008;2008:461981. doi: 10.1155/2008/461981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao X, Strong R, Zhang J, et al. Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci. 2009;29:6186–6195. doi: 10.1523/JNEUROSCI.5857-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park SW, Yi JH, Miranpuri G, et al. Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther. 2007;320:1002–1012. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- 74.Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 2008;1244:164–172. doi: 10.1016/j.brainres.2008.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sauerbeck A, Gao J, Readnower R, et al. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol. 2011;227:128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thal SC, Heinemann M, Luh C, et al. Pioglitazone reduces secondary brain damage after experimental brain trauma by PPAR-gamma-independent mechanisms. J Neurotrauma. 2011;28:983–993. doi: 10.1089/neu.2010.1685. [DOI] [PubMed] [Google Scholar]
- 77.Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2006;26:811–820. doi: 10.1038/sj.jcbfm.9600233. [DOI] [PubMed] [Google Scholar]
- 78.Zhao X, Sun G, Zhang J, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61:352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- 79.Tureyen K, Kapadia R, Bowen KK, et al. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem. 2007;101:41–56. doi: 10.1111/j.1471-4159.2006.04376.x. [DOI] [PubMed] [Google Scholar]
- 80.Aleshin S, Grabeklis S, Hanck T, Sergeeva M, Reiser G. Peroxisome proliferator-activated receptor (PPAR)-gamma positively controls and PPARalpha negatively controls cyclooxygenase-2 expression in rat brain astrocytes through a convergence on PPARbeta/delta via mutual control of PPAR expression levels. Mol Pharmacol. 2009;76:414–424. doi: 10.1124/mol.109.056010. [DOI] [PubMed] [Google Scholar]
- 81.Wang HM, Zhao YX, Zhang S, et al. PPARgamma agonist curcumin reduces the amyloid-beta-stimulated inflammatory responses in primary astrocytes. J Alzheimers Dis. 2010;20:1189–1199. doi: 10.3233/JAD-2010-091336. [DOI] [PubMed] [Google Scholar]
- 82.Hucke S, Flossdorf J, Grutzke B, et al. Licensing of myeloid cells promotes central nervous system autoimmunity and is controlled by peroxisome proliferator-activated receptor gamma. Brain. 2012;135:1586–1605. doi: 10.1093/brain/aws058. [DOI] [PubMed] [Google Scholar]
- 83.Ramirez SH, Heilman D, Morsey B, et al. Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. J Immunol. 2008;180:1854–1865. doi: 10.4049/jimmunol.180.3.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Petrova TV, Akama KT, Van Eldik LJ. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci U S A. 1999;96:4668–4673. doi: 10.1073/pnas.96.8.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- 86.Mandrekar-Colucci S, Karlo JC, Landreth GE. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer's disease. J Neurosci. 2012;32:10117–10128. doi: 10.1523/JNEUROSCI.5268-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rossi A, Kapahi P, Natoli G, et al. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 88.Cernuda-Morollon E, Rodriguez-Pascual F, Klatt P, Lamas S, Perez-Sala D. PPAR agonists amplify iNOS expression while inhibiting NF-kappaB: implications for mesangial cell activation by cytokines. J Am Soc Nephrol. 2002;13:2223–2231. doi: 10.1097/01.asn.0000025786.87646.b1. [DOI] [PubMed] [Google Scholar]
- 89.Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 90.Shimazu T, Inoue I, Araki N, et al. A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke. 2005;36:353–359. doi: 10.1161/01.STR.0000152271.21943.a2. [DOI] [PubMed] [Google Scholar]
- 91.Zhao X, Ou Z, Grotta JC, Waxham N, Aronowski J. Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res. 2006;1073–1074:460–469. doi: 10.1016/j.brainres.2005.12.061. [DOI] [PubMed] [Google Scholar]
- 92.Romera C, Hurtado O, Mallolas J, et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. Journal of Cerebral Blood Flow & Metabolism. 2007;27:1327–1338. doi: 10.1038/sj.jcbfm.9600438. [DOI] [PubMed] [Google Scholar]
- 93.Moore KJ, Rosen ED, Fitzgerald ML, et al. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 94.Patel SN, Serghides L, Smith TG, et al. CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis. 2004;189:204–213. doi: 10.1086/380764. [DOI] [PubMed] [Google Scholar]
- 95.Majai G, Sarang Z, Csomos K, Zahuczky G, Fesus L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37:1343–1354. doi: 10.1002/eji.200636398. [DOI] [PubMed] [Google Scholar]
- 96.Shimazu T, Greenberg JH. A PPAR gamma agonist reduces infarct size in transient but not in permanent ischemia. Society for Neuroscience Abstracts. 2003;883:4. doi: 10.1161/01.STR.0000152271.21943.a2. [DOI] [PubMed] [Google Scholar]
- 97.Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev. 2001;21:185–210. doi: 10.1002/med.1006. [DOI] [PubMed] [Google Scholar]
- 98.Moreno S, Mugnaini E, Ceru MP. Immunocytochemical localization of catalase in the central nervous system of the rat. J Histochem Cytochem. 1995;43:1253–1267. doi: 10.1177/43.12.8537642. [DOI] [PubMed] [Google Scholar]
- 99.Koeppen AH, Dickson AC, McEvoy JA. The cellular reactions to experimental intracerebral hemorrhage. J Neurol Sci. 1995;134(Suppl):102–112. doi: 10.1016/0022-510x(95)00215-n. [DOI] [PubMed] [Google Scholar]
- 100.Hall NC, Packard BA, Hall CL, de Courten-Myers G, Wagner KR. Protein oxidation and enzyme susceptibility in white and gray matter with in vitro oxidative stress: relevance to brain injury from intracerebral hemorrhage. Cell Mol Biol (Noisy-le-grand) 2000;46:673–683. [PubMed] [Google Scholar]
- 101.Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005;1039:30–36. doi: 10.1016/j.brainres.2005.01.036. [DOI] [PubMed] [Google Scholar]
- 102.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 103.Broderick JP, Brott TG, Duldner JE, Tomsick T, Huster G. Volume of intracerebral hemorrhage A powerful and easy-to-use predictor of 30-day mortality. Stroke. 1993;24:987–993. doi: 10.1161/01.str.24.7.987. [DOI] [PubMed] [Google Scholar]
- 104.Jordan LC, Kleinman JT, Hillis AE. Intracerebral hemorrhage volume predicts poor neurologic outcome in children. Stroke. 2009;40:1666–1671. doi: 10.1161/STROKEAHA.108.541383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rincon F, Mayer SA. Novel therapies for intracerebral hemorrhage. Curr Opin Crit Care. 2004;10:94–100. doi: 10.1097/00075198-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 106.Mendelow AD, Gregson BA, Rowan EN, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet. 2013;382:397–408. doi: 10.1016/S0140-6736(13)60986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mould WA, Carhuapoma JR, Muschelli J, et al. Minimally invasive surgery plus recombinant tissue-type plasminogen activator for intracerebral hemorrhage evacuation decreases perihematomal edema. Stroke. 2013;44:627–634. doi: 10.1161/STROKEAHA.111.000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gonzales NR, Shah J, Sangha N, et al. Design of a prospective, dose-escalation study evaluating the Safety of Pioglitazone for Hematoma Resolution in Intracerebral Hemorrhage (SHRINC) Int J Stroke. 2012 doi: 10.1111/j.1747-4949.2011.00761.x. [DOI] [PubMed] [Google Scholar]
- 109.Bhattathiri PS, Gregson B, Prasad KS, Mendelow AD, Investigators S. Intraventricular hemorrhage and hydrocephalus after spontaneous intracerebral hemorrhage: results from the STICH trial. Acta Neurochir Suppl. 2006;96:65–68. doi: 10.1007/3-211-30714-1_16. [DOI] [PubMed] [Google Scholar]
- 110.Abdu E, Hanley DF, Newell DW. Minimally invasive treatment for intracerebral hemorrhage. Neurosurg Focus. 2012;32:E3. doi: 10.3171/2012.1.FOCUS11362. [DOI] [PubMed] [Google Scholar]
- 111.Hua Y, Schallert T, Keep RF, et al. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- 112.Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3) J Immunol. 1998;161:6250–6257. [PubMed] [Google Scholar]
- 113.Stolzing A, Grune T. Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. FASEB J. 2004;18:743–745. doi: 10.1096/fj.03-0374fje. [DOI] [PubMed] [Google Scholar]
- 114.Ren Y, Silverstein RL, Allen J, Savill J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med. 1995;181:1857–1862. doi: 10.1084/jem.181.5.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Smith TG, Serghides L, Patel SN, et al. CD36-mediated nonopsonic phagocytosis of erythrocytes infected with stage I and IIA gametocytes of Plasmodium falciparum. Infect Immun. 2003;71:393–400. doi: 10.1128/IAI.71.1.393-400.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ren Y, Savill J. Proinflammatory cytokines potentiate thrombospondin-mediated phagocytosis of neutrophils undergoing apoptosis. J Immunol. 1995;154e:2366–2374. [PubMed] [Google Scholar]
- 117.Navazo MD, Daviet L, Savill J, et al. Identification of a domain (155–183) on CD36 implicated in the phagocytosis of apoptotic neutrophils. J Biol Chem. 1996;271:15381–15385. doi: 10.1074/jbc.271.26.15381. [DOI] [PubMed] [Google Scholar]
- 118.Erwig LP, Gordon S, Walsh GM, Rees AJ. Previous uptake of apoptotic neutrophils or ligation of integrin receptors downmodulates the ability of macrophages to ingest apoptotic neutrophils. Blood. 1999;93:1406–1412. [PubMed] [Google Scholar]
- 119.Nicholson AC. Expression of CD36 in macrophages and atherosclerosis: the role of lipid regulation of PPARgamma signaling. Trends Cardiovasc Med. 2004;14:8–12. doi: 10.1016/j.tcm.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 120.Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–35722. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 121.Han S, Sidell N. Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid. Immunology. 2002;106:53–59. doi: 10.1046/j.1365-2567.2002.01404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Babaev V, Yancey P, Ryzhov S, et al. Conditional knockout of macrophage PPARg increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;8:1648–1653. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- 123.Ottnad E, Parthasarathy S, Sambrano GR, et al. A macrophage receptor for oxidized low density lipoprotein distinct from the receptor for acetyl low density lipoprotein: partial purification and role in recognition of oxidatively damaged cells. Proc Natl Acad Sci U S A. 1995;92:1391–1395. doi: 10.1073/pnas.92.5.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 125.Shehab AM, MacFadyen RJ, McLaren M, et al. Sudden unexpected death in heart failure may be preceded by short term, intraindividual increases in inflammation and in autonomic dysfunction: a pilot study. Heart. 2004;90:1263–1268. doi: 10.1136/hrt.2003.028399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Aibiki M, Ohtsubo S, Nishiyama T, et al. Elevated serum beta-D-glucan level and depressed neutrophil phagocytosis in a heatstroke patient. Resuscitation. 2005;65:115–117. doi: 10.1016/j.resuscitation.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 127.Fadok VA. Clearance: the last and often forgotten stage of apoptosis. J Mammary Gland Biol Neoplasia. 1999;4:203–211. doi: 10.1023/a:1011384009787. [DOI] [PubMed] [Google Scholar]
- 128.Ekdahl CT, Kokaia Z, Lindvall O. Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience. 2009;158:1021–1029. doi: 10.1016/j.neuroscience.2008.06.052. [DOI] [PubMed] [Google Scholar]
- 129.MacLellan CL, Colbourne F. Mild to moderate hyperthermia does not worsen outcome after severe intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. 2005;25:1020–1029. doi: 10.1038/sj.jcbfm.9600099. [DOI] [PubMed] [Google Scholar]
- 130.Gong C, Hoff JT, Keep RF. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res. 2000;871:57–65. doi: 10.1016/s0006-8993(00)02427-6. [DOI] [PubMed] [Google Scholar]
- 131.Cheret C, Gervais A, Lelli A, et al. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J Neurosci. 2008;28:12039–12051. doi: 10.1523/JNEUROSCI.3568-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sundararajan S, Jiang Q, Heneka M, Landreth G. PPARgamma as a therapeutic target in central nervous system diseases. Neurochem Int. 2006;49:136–144. doi: 10.1016/j.neuint.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 133.Pisanu A, Lecca D, Mulas G, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson's disease. Neurobiol Dis. 2014 doi: 10.1016/j.nbd.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 134.Splettstoesser WD, Schuff-Werner P. Oxidative stress in phagocytes--”the enemy within”. Microsc Res Tech. 2002;57:441–455. doi: 10.1002/jemt.10098. [DOI] [PubMed] [Google Scholar]
- 135.Szanto A, Balint BL, Nagy ZS, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARgamma-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712. doi: 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Derecki NC, Quinnies KM, Kipnis J. Alternatively activated myeloid (M2) cells enhance cognitive function in immune compromised mice. Brain Behav Immun. 2011;25:379–385. doi: 10.1016/j.bbi.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pelidou SH, Kostulas N, Matusevicius D, et al. High levels of IL-10 secreting cells are present in blood in cerebrovascular diseases. Eur J Neurol. 1999;6:437–442. doi: 10.1046/j.1468-1331.1999.640437.x. [DOI] [PubMed] [Google Scholar]
- 139.Cullingford TE, Bhakoo K, Peuchen S, et al. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. J Neurochem. 1998;70:1366–1375. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- 140.Leblanc BP, Stunnenberg HG. 9-cis retinoic acid signaling: changing partners causes some excitement. Genes Dev. 1995;9:1811–1816. doi: 10.1101/gad.9.15.1811. [DOI] [PubMed] [Google Scholar]
- 141.Mukherjee R, Davies PJ, Crombie DL, et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 142.Mukherjee R, Jow L, Croston GE, Paterniti JR., Jr Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J Biol Chem. 1997;272:8071–8076. doi: 10.1074/jbc.272.12.8071. [DOI] [PubMed] [Google Scholar]
- 143.Tontonoz P, Singer S, Forman BM, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci U S A. 1997;94:237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sanz MJ, Albertos F, Otero E, et al. Retinoid X receptor agonists impair arterial mononuclear cell recruitment through peroxisome proliferator-activated receptor-gamma activation. J Immunol. 2012;189:411–424. doi: 10.4049/jimmunol.1102942. [DOI] [PubMed] [Google Scholar]
- 145.Diab A, Hussain RZ, Lovett-Racke AE, et al. Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148:116–126. doi: 10.1016/j.jneuroim.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 146.Liao SL, Chen WY, Raung SL, Chen CJ. Ethanol attenuates ischemic and hypoxic injury in rat brain and cultured neurons. Neuroreport. 2003;14:2089–2094. doi: 10.1097/00001756-200311140-00016. [DOI] [PubMed] [Google Scholar]
- 147.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 148.Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J. Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke. 1999;30:2472–2477. doi: 10.1161/01.str.30.11.2472. discussion 77–8. [DOI] [PubMed] [Google Scholar]
- 149.Zhao X, Zhang Y, Strong R, et al. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. J Neurochem. 2007;101:652–663. doi: 10.1111/j.1471-4159.2006.04414.x. [DOI] [PubMed] [Google Scholar]
- 150.Wagner KR, Beiler S, Dean C, et al. NF-kappaB activation and pro-inflammatory cytokine gene upregulation in white matter following porcine intracerebral hemorrhage. In: Kriglstein, Klumpp, editors. Pharmacology of cerebral ischemia 2004. Stutgart: Wissenschafttliche Varlsgsesellschaft; 2004. pp. 185–194. [Google Scholar]
- 151.Verma IM. Nuclear factor (NF)-kappaB proteins: therapeutic targets. Ann Rheum Dis. 2004;63(Suppl 2):i57–ii61. doi: 10.1136/ard.2004.028266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Straus DS, Pascual G, Li M, et al. 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF- kappa B signaling pathway. Proc Natl Acad Sci U S A. 2000;97:4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chung SW, Kang BY, Kim SH, et al. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- 154.Dowell P, Ishmael JE, Avram D, et al. p300 functions as a coactivator for the peroxisome proliferator-activated receptor alpha. J Biol Chem. 1997;272:33435–33443. doi: 10.1074/jbc.272.52.33435. [DOI] [PubMed] [Google Scholar]
- 155.Nolte RT, Wisely GB, Westin S, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 156.Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem. 2000;275:36703–36707. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 157.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–286. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 159.van Muiswinkel FL, Kuiperij HB. The Nrf2-ARE Signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord. 2005;4:267–281. doi: 10.2174/1568007054038238. [DOI] [PubMed] [Google Scholar]
- 160.Ishii T, Itoh K, Yamamoto M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002;348:182–190. doi: 10.1016/s0076-6879(02)48637-5. [DOI] [PubMed] [Google Scholar]
- 161.Zhao X, Sun G, Zhang J, et al. Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke. 2007;38:3280–3286. doi: 10.1161/STROKEAHA.107.486506. [DOI] [PubMed] [Google Scholar]
- 162.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- 163.Giudice A, Montella M. Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays. 2006;28:169–181. doi: 10.1002/bies.20359. [DOI] [PubMed] [Google Scholar]
- 164.Dash PK, Zhao J, Orsi SA, Zhang M, Moore AN. Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci Lett. 2009;460:103–107. doi: 10.1016/j.neulet.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu HL, Feng YP. Inhibitory effects of chiral 3-n-butylphthalide on inflammation following focal ischemic brain injury in rats. Acta Pharmacol Sin. 2000;21:433–438. [PubMed] [Google Scholar]
- 166.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10235. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Leonard MO, Kieran NE, Howell K, et al. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. Faseb J. 2006;20:2624–2626. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- 168.Satoh T, Okamoto SI, Cui J, et al. Activation of the Keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc Natl Acad Sci U S A. 2006;103:768–773. doi: 10.1073/pnas.0505723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang J, Fields J, Zhao C, et al. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ishii T, Itoh K, Ruiz E, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 171.Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 2001;7:135–145. [PMC free article] [PubMed] [Google Scholar]
- 172.Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. 2002;16:2793–2801. doi: 10.1210/me.2002-0020. [DOI] [PubMed] [Google Scholar]
- 173.Shih AY, Imbeault S, Barakauskas V, et al. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005;280:22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 174.Cho HY, Gladwell W, Wang X, et al. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. 2010;182:170–182. doi: 10.1164/rccm.200907-1047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Park EY, Cho IJ, Kim SG. Transactivation of the PPAR-responsive enhancer module in chemopreventive glutathione S-transferase gene by the peroxisome proliferator-activated receptor-gamma and retinoid X receptor heterodimer. Cancer Res. 2004;64:3701–3713. doi: 10.1158/0008-5472.CAN-03-3924. [DOI] [PubMed] [Google Scholar]
- 176.Bowie A, O'Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 177.Whalen MJ, Carlos TM, Dixon CE, et al. Effect of traumatic brain injury in mice deficient in intercellular adhesion molecule-1: assessment of histopathologic and functional outcome. J Neurotrauma. 1999;16:299–309. doi: 10.1089/neu.1999.16.299. [DOI] [PubMed] [Google Scholar]
- 178.Smith SA, Monteith GR, Holman NA, et al. Effects of peroxisome proliferator-activated receptor gamma ligands ciglitazone and 15-deoxy-delta 12,14-prostaglandin J2 on rat cultured cerebellar granule neuronal viability. J Neurosci Res. 2003;72:747–755. doi: 10.1002/jnr.10613. [DOI] [PubMed] [Google Scholar]
- 179.Rohn TT, Wong SM, Cotman CW, Cribbs DH. 15-deoxy-delta12,14-prostaglandin J2, a specific ligand for peroxisome proliferator-activated receptor-gamma, induces neuronal apoptosis. Neuroreport. 2001;12:839–843. doi: 10.1097/00001756-200103260-00043. [DOI] [PubMed] [Google Scholar]
- 180.Kondo M, Shibata T, Kumagai T, et al. 15-Deoxy-Delta(12,14)-prostaglandin J(2): the endogenous electrophile that induces neuronal apoptosis. Proc Natl Acad Sci U S A. 2002;99:7367–7372. doi: 10.1073/pnas.112212599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yagami T, Ueda K, Asakura K, et al. Novel binding sites of 15-deoxy-Delta(12,14)-prostaglandin J(2) in plasma membranes from primary rat cortical neurons. Exp Cell Res. 2003;291:212–227. doi: 10.1016/s0014-4827(03)00369-0. [DOI] [PubMed] [Google Scholar]
- 182.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 183.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 184.Betteridge DJ, DeFronzo RA, Chilton RJ. PROactive: time for a critical appraisal. Eur Heart J. 2008;29:969–983. doi: 10.1093/eurheartj/ehn114. [DOI] [PubMed] [Google Scholar]