

Abstract
In chronic serum sickness, glomerular immune complexes form, yet C57BL/6 mice do not develop glomerulonephritis unless plasma factor H (CfH) is absent, indicating the relevance of complement regulation. Complement receptor 3 (CD11b) and Fcγ receptors on leukocytes, and CfH on platelets, can bind immune complexes. Here we induced immune complex-mediated glomerulonephritis in CfH-/- mice chimeric for either wild type, CfH-/-, CD11b-/- and FcRγ-/- bone marrow stem cells. Glomerulonephritis was worse in CD11b-/- chimeras vs. all others, while disease in FcRγ-/- and wild type chimeras was comparable. Disease tracked strongly with humoral immune responses, but not glomerular immune complex deposits. Interstitial inflammation with M1 macrophages strongly correlated with glomerulonephritis scores. CD11b-/- chimeras had significantly more M1 macrophages and CD4+ T cells. The renal dendritic cell populations originating from bone marrow-derived CD11c+ cells were similar in all experimental groups. CD11b+ cells bearing colony stimulating factor 1 receptor were present in kidneys, including CD11b-/- chimeras; these cells correlated negatively with glomerulonephritis scores. Thus, experimental immune complex-mediated glomerulonephritis is associated with accumulation of M1 macrophages and CD4+ T cells in kidneys and functional renal insufficiency. Hence, CD11b on mononuclear cells is instrumental in generating an anti-inflammatory response in the inflamed kidney.
Keywords: glomerulonephritis, complement, inflammation, macrophages
Introduction
Complement activation through each of the three pathways leads to generation of C3 and C5 products. These mark infectious microorganisms as foreign and alert innate and adaptive immune cells to their presence. Spontaneous alternative pathway activation is regulated by complement factor H (CfH). When CfH is ineffective in the fluid phase, products of unrestricted C3 activation can deposit in the glomerular capillary wall, as in C3 glomerulopathies.1 Factor H also is retained by the glomerular and other capillary wall surfaces; when this is inefficient, endothelial cell injury and atypical hemolytic uremic syndrome can result.
Anaphylatoxin receptors C3aR and C5aR signal through a subset of pertussis toxin-sensitive GTP-binding protein α-subunits.2 The β2 integrin (CD18) heterodimers with αM (CD11b) and αX (CD11c) were first termed complement receptors (CR) 3 and 4 by virtue of their binding to inactivated (i) C3b.3 Although traditionally considered adherence receptors, CD11b/CD11c binding to ligand can lead to “outside-in” signals via immunoreceptor tyrosine-based activation motif (ITAM) proteins; these include the Fc receptor common γ-chain (FcRγ) and DNAX-activating protein of 12 kDa (DAP12) (encoded by Fcer1g and Tyrobp, respectively).4,5
Induction of chronic serum sickness (CSS) leads to deposition of immune complexes (ICs) in glomeruli.6 Yet, there is not uniform development of inflammation and glomerulonephritis (GN); for instance, C57BL/6 mice do not develop GN in CSS,7 while all CfH-deficient mice with CSS develop diffuse proliferative GN within 5 weeks.8 The GN in CSS is characterized by accumulation of IgG/C3-containing ICs together with F4/80+ macrophages.9,10 Disease requires signals through C5aR, as C5aR-/-CfH-/- mice with CSS do not develop GN.10
Humans utilize CR1 on erythrocytes for IC processing; in contrast, the rodent relies upon CfH on platelets.11,12 As such, CfH-/- mice lacking platelet CfH have markedly abnormal IC processing. Platelet CfH is restored in CfH-/- mice chimeric for wildtype bone marrow (BM).9 These animals still lack hepatic-produced plasma CfH. When CSS is induced in CfH-/- mice with wildtype BM, IC metabolism approaches that in wildtype mice, such that there are considerably fewer glomerular ICs. Yet, these animals still develop GN.9 In contrast, wildtype mice with CfH-/-BM failed to develop GN, despite the presence of large quantities of ICs (attributable to abnormal IC processing from CfH-deficient platelets).9 This illustrates the importance of complement regulation by plasma CfH in the setting of glomerular-bound ICs. The abundant ICs containing IgG and iC3b as ligands for inflammatory cell FcγRs and β2 integrins are inert. The presence of active C3b is required to generate C5a as the necessary signal that initiates inflammation in GN.9,10
Based on these data, we hypothesized that in CSS-induced GN: 1) leukocyte FcγRs and β2 integrins are activated by C5aR signaling; 2) all three are necessary, such that disease will not occur in the absence of any one; 3) while FcγRs and β2 integrins are relevant to systemic IC metabolism, this is of less consequence in this model; and, 4) FcγRIII and CD11b were the most likely relevant FcγRs and β2 integrins. To evaluate this, we studied CSS in CfH-/- mice chimeric for FcRγ-/- and CD11b-/- BM. Surprisingly, disease was considerably worse in the latter group.
Results
Increased anti-apoferritin IgG antibodies and circulating ICs in the absence of CfH or CD11b on BM-derived cells
We lethally irradiated CfH-/- mice and reconstituted their BM with CD117+ stem cells from wildtype (n = 8), CfH-/- (n = 12), FcRγ-/- (n = 7), and CD11b-/- (n = 7) mice. After stable hematopoeitic cell engraftment, CSS was induced by actively immunizing mice with a daily intraperitoneal dose of 4 mg horse spleen apoferritin. CfH-/- mice with CfH-/- BM receiving saline vehicle alone were used as control (n = 5). As expected, these control CfH-/- chimeras did not generate antibodies to apoferritin. All animals immunized with apoferritin generated an anti-apoferritin antibody response (Figure 1). As predicted from past data,8,9 CfH-/- chimeras had higher antibody levels than wildtype mice. Yet, the highest levels were observed in the CD11b-/- chimeras (Figure 1, P < 0.001).
Figure 1. Higher anti-apoferritin antibody levels in CSS induced in CfH-/- mice with CfH-/- and CD11b-/- bone marrow.
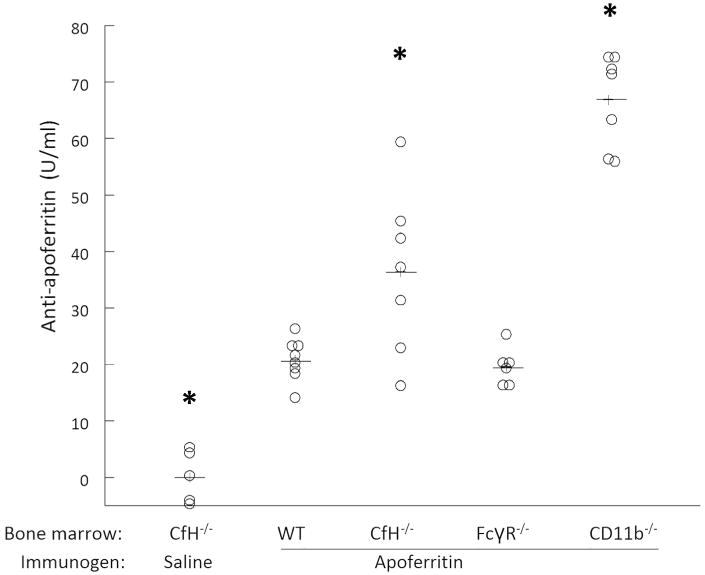
Animals were actively immunized with daily intraperitoneal doses of 4 mg horse spleen apoferritin in saline. Five weeks later, anti-apoferritin IgG levels were measured. Data from individual mice are shown. These data from each group were normally distributed (Anderson-Darling test P > 0.08) *P < 0.001 vs. all other groups, ANOVA followed by Tukey all-pairwise approach.
As expected, all animals with CSS had IgG-containing ICs in plasma. Anti-apoferritin IgG levels and ICs were positively correlated (Supplemental Figure S1a). As with the anti-apoferritin IgG, CfH-/- and CD11b-/- chimeric mice had the highest IC levels compared to wildtype and FcRγ-/- mice.
We were surprised that CD11b-/- chimeras had the highest anti-apoferritin IgG levels. To examine the role for absent plasma CfH in these findings, we induced CSS in wildtype, CfH-/-, and CD11b-/- mice (i.e., that were not BM chimeras; n = 6 each). Serum anti-apoferritin IgG levels were no different between wildtype and CD11b-/- mice (29.5 ± 1.4 and 31.2 ± 1.6 U/ml, respectively). As we have seen before,8 CfH-/- mice had higher anti-apoferritin IgG (52.4 ± 4.2, P < 0.001 by ANOVA). These anti-apoferritin IgG levels also strongly correlated with circulating C1q- and IgG-containing ICs (R = 0.73), with a slope comparable to the BM chimera studies (0.759 versus 0.743) (Supplemental Figure S1b). Thus, the absence of CD11b on BM-derived cells together with absent plasma CfH led to a heightened humoral immune response to the heterologous apoferritin.
Glomerular ICs in CSS
Because of the absence of plasma CfH in all BM chimeric animals, including controls receiving saline, all mouse glomeruli had strongly positive capillary wall staining for C3 (Figure 2a). The IgG deposits in wildtype chimeric mice were largely within the mesangium, while CfH-/- chimeras had strongly positive mesangial IgG (Figure 2a, asterisks) which extended to the peripheral capillary wall. This is attributable to the absence of CfH on platelets, resulting in impaired IC processing in these mice.9,11
Figure 2. Glomerular immune complex deposition in CSS.
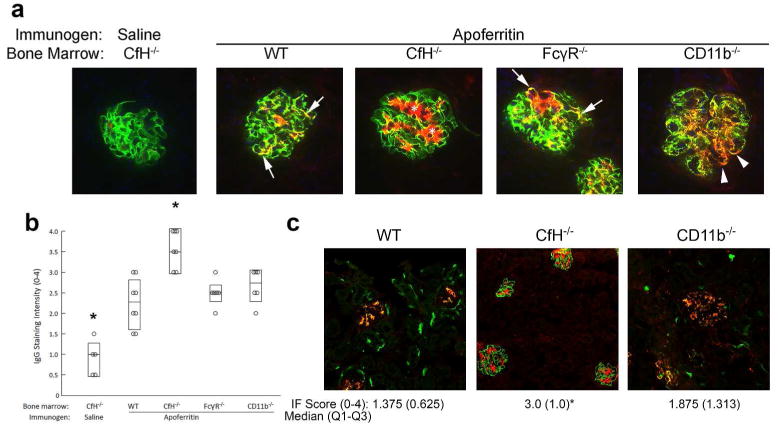
(a) Representative immunofluorescence photomicrographs from CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcγR-/-, and CD11b-/- bone marrow, or (c) native WT, CfH-/- and CD11b-/- mice (i.e., without bone marrow transfer) actively immunized with apoferritin to induce chronic serum sickness, or saline as control. Dual staining for C3 (green) and IgG (red) is shown; the yellow color indicates immune complexes containing IgG and C3. Because animals lacked plasma CfH, there was strongly positive glomerular C3 staining in all groups. The asterisks and arrows depict mesangial and peripheral capillary wall immune complex deposits, respectively. The arrowheads show the more granular peripheral capillary wall immune complex staining in CD11b-/- chimeras. (b, c) Semiquantitative scoring for IgG staining intensity from individual mice from all groups. In b, data are shown as median (horizontal line), and Q3-Q1 in the boxes, while in c these are presented below the figures. Group medians were significantly different (P = 0.02, Mood Median test). *P < 0.05 vs. all other groups.
The intensity of IgG staining was similar in the three groups with CSS but intact platelet CfH (i.e., wildtype, FcRγ-/- and CD11b-/-; quantified in Figure 2b), with extension of IgG from the mesangium to the peripheral capillary loops, where it appeared to be present in ICs, as evidenced by dual staining for IgG and C3 (Figure 2a, arrows). In the CD11b-/- chimeric mice, there was greater extension of granular IgG (and C3) deposits into the peripheral capillary wall (Figure 2a, arrowheads).
The intensity and localization of IgG within glomeruli in CSS was similar in wildtype, CfH-/-, and CD11b-/- mice relative to their respective BM chimeras (Figure 2c). As with CD11b-/-BM chimeras, CD11b-/- mice had greater peripheral glomerular capillary wall extension of IgG,13 though this was not statistically different from wildtype mice. Glomerular C3 staining in wildtype and CD11b-/- mice colocalized with IgG (yellow color in merged images in Figure 2c). In all experimental animals lacking plasma CfH, there was not C3 or IgG staining within the extraglomerular vessels or in the tubulointerstitium. The segmental linear staining for C3 seen in normal mouse peritubular and periglomerular regions was also absent, consistent with absence of plasma CfH.14,15
Worsened renal disease in the absence of plasma CfH and CD11b on BM-derived cells
Control mice did not develop renal disease, as evidenced by normal BUN values (26.1 ± 0.8 mg/dl, Figure 3a). All animals with CSS developed renal insufficiency, with increased BUN values (Figure 3a). Wildtype and FcRγ-/- chimeric mice had comparably increased BUN values (36.5 ± 1.2 and 37.3 ± 1.1 mg/dl, respectively). CfH-/- chimeras with CSS had BUN values (42.7 ± 1.2 mg/dl) that were significantly different than wildtype, indicating the relevance of CfH on BM-derived cells. Surprisingly, each of the seven CD11b-/- chimeric mice had higher BUN levels than any of the other 27 animals (Figure 3a) (mean ± SEM = 53.3 ± 1.5 mg/dl). As we have observed before, anti-apoferritin IgG levels strongly correlated with BUN values (Supplemental Figure S2).8
Figure 3. Worsened glomerulonephritis in CfH-/- mice with CfH-/- and CD11b-/- bone marrow.
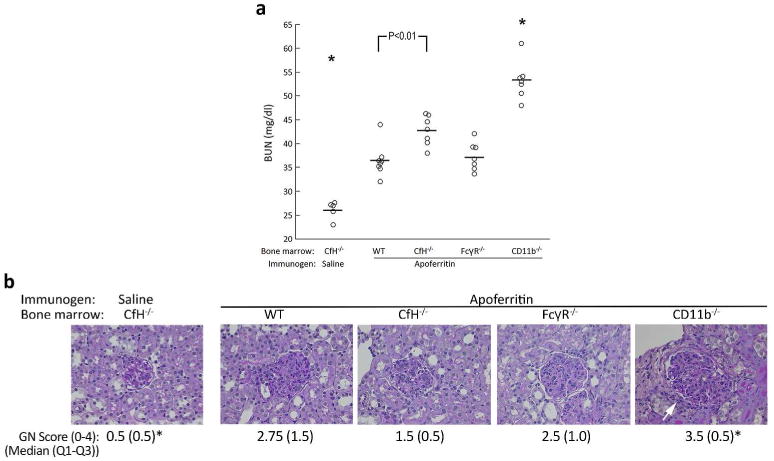
CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcγR-/-, and CD11b-/- bone marrow were actively immunized with apoferritin to induce chronic serum sickness, or saline as control. (a) After five weeks, BUN was measured. Data from individual mice are shown. These data from each group were normally distributed (Anderson-Darling test P > 0.04), while the population means were unequal (one-way ANOVA, P < 0.001). The Tukey all-pairwise approach was used to compare individual means. *P < 0.007 vs. all other groups. (b) Representative histopathology is shown. Diffuse endocapillary proliferative glomerulonephritis was evident in all groups receiving apoferritin. The white arrow shows periglomerular infiltrates in CD11b-/- chimeras. Semiquantitative scores for GN are provided as median, with Q3-Q1 interval in parentheses. Group medians were significantly different (P = 0.003, Mood Median test). *P < 0.05 vs. all other groups.
Control CfH-/- chimeras receiving saline instead of apoferritin had normal urinary albumin excretion (13.5 ± 1.5 μg/mg creatinine). Albuminuria was 30.5 ± 6.5, 44.3 ± 5.8, and 74.0 ± 4.7 μg/mg creatinine in wildtype, CfH-/- and CD11b-/- BM chimeras with CSS, respectively (P < 0.001, one-way ANOVA; P < 0.03, CD11b-/- vs. all other groups). In CSS induced in native wildtype, CfH-/- and CD11b-/- mice (i.e., that did not receive BM transfers), urinary albumin was 15.4 ± 1.7, 32.9 ± 2.5, and 26.6 ± 1.6 μg/mg creatinine, respectively (P < 0.001, one-way ANOVA; P < 0.005, wildtype vs. CfH-/- and CD11b-/-).
All animals lacking plasma CfH (i.e., all BM chimeras and CfH-/- mice without BM transfer) in which CSS was induced developed diffuse proliferative GN (Figure 3b). There was little to no interstitial inflammation in any of the groups, except for the CD11b-/- chimeras in which there were focal periglomerular mononuclear cell infiltrates (Figure 3b, arrow). There was no appreciable interstitial fibrosis, tubular atrophy, or arteritis in any kidney. Native wildtype and CD11b-/- mice did not develop GN in CSS (median GN scores 0 and 0.5, respectively; P = 0.002 vs. CfH-/- mice studied contemporaneously).
Notably, these three measures of renal disease - BUN concentrations, albuminuria and GN scores were highly related (R values ≥0.81, see legend to Supplemental Figure S2). BUN and urinary albumin were the most strongly correlated (BUN = 20.9 + (0.47 × UAlb), R = 0.97). It is notable that each measurement is derived from a distinct anatomic site (i.e., blood, urine and renal tissue). Thus, in this model, the extent of histopathological GN tracks with functional renal disease.
Increased interstitial mononuclear cell infiltrates in the absence of CD11b on BM-derived cells
In the CSS model, there is IC deposition, complement activation and inflammation in glomeruli. While there is not histopathological evidence of tubulointerstitial nephritis, the mononuclear cell compartment as measured by flow cytometry expands and is related to disease features in this model.10 Thus, we examined mononuclear cells in the renal interstitium 5 weeks after induction of CSS. There were F4/80+ and CD115+ cells, but not F4/80+CD115+ cells (Supplemental Figure S3). The F4/80+ cells were distinguished by CCR2 and Ly6C staining (Figure 4a). There were few Ly6ChiCCR2hi M1 (inflammatory) macrophages in the kidneys of CfH-/- mice with CfH-/- BM, with or without CSS (Figure 4a, red boxes). This is consistent with our past work with CfH-/- mice with CSS.9 Yet, the wildtype, FcRγ-/- and CD11b-/- chimeric mice had an increased number of Ly6ChiCCR2hi M1 macrophages (Figure 4a, red arrows), which was significantly greater in CD11b-/- chimeras (Figure 4b). As anticipated, these cells were CD11b-(Figure 4c, red arrowhead). The proportion of F4/80+ cells that were Ly6ChiCCR2hi M1 macrophages positively correlated with GN scores (Figure 5a), supporting glomerular and TI inflammation were linked.
Figure 4. Characterization of F4/80+ cells in the tubulointerstitium of kidneys from CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcγR-/-, and CD11b-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control.
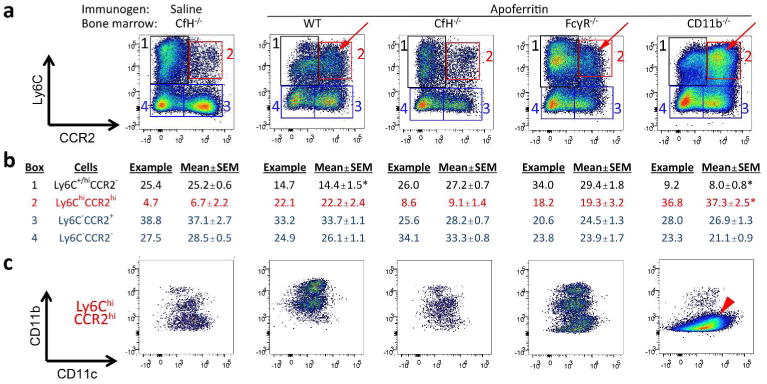
(a) Representative data for Ly6C (y-axis) and CCR2 (x-axis). The four quadrants are boxed and numbered; the percentage each group comprises of total F4/80+ cells from the representative graphs is provided in tabular form in b (in the columns “Example”). Data from all animal kidneys are also provided as “Mean ± SEM” in the adjacent columns. *P < 0.01 vs. all other groups (one-way ANOVA followed by Tukey all-pairwise testing). (c) Ly6ChiCCR2hi M1 macrophages (quadrant 2, red arrows) were analyzed for CD11c (y-axis) and CD11b (x-axis) staining. The red arrowhead depicts Ly6ChiCCR2hiCD11c+CD11b- cells in CD11b-/- chimeras.
Figure 5. Correlation between TI mononuclear cells and glomerulonephritis scores.
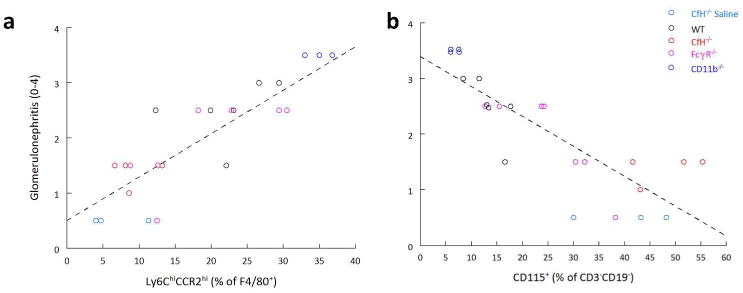
(a) The number of Ly6ChiCCR2hi M1 macrophages as a proportion of all F4/80+ cells positively correlates with GN. The line was fit by least squares analysis, with the derived equation, GN = 0.51 + (0.078 × Ly6ChiCCR2hi), R = 0.88, P < 0.001. (b) The number of CD115+F4/80-CD11b+ cells as a percentage of all CD3-CD19- mononuclear cells negatively correlated with GN. The line was fit by least squares analysis, with the derived equation, GN = 3.4 - (0.054 × CD115+), R = -0.84, P < 0.001.
There were also Ly6C+CCR2- cells (Figure 4a, black boxes) comprising two distinct populations; namely, Ly6C+CD11b+CD11c- (except in CD11b-/- chimeras) and Ly6ChiCD11b-CD11clo (Supplemental Figure S4). The former is consistent with M2 macrophages, while the identity of the latter is unclear.
The Ly6C- cellular population contained both CCR2- and CCR2+ cells (Figure 4a, blue boxes). Overall, these cells are typical for native kidney dendritic cells.16,17 The majority were CD11b+CD11c+, though there were different patterns in the different groups (Supplemental Figure S5a). In the CD11b-/- chimeras, these cells were CD11b- and had greater surface CD11c staining than wildtype (Supplemental Figure S5b).
In all groups, the CD115+F4/80- cells were CD11b+CD11c- (Supplemental Figure S3b), Ly6C- and CCR2- (data not shown). Interestingly, even CD11b-/- chimeras had these CD115+F4/80-CD11b+ cells, albeit in lesser quantities (Supplemental Figure S3b). Thus, this particular cellular population is radioresistant. The number of CD115+F4/80-CD11b+ cells negatively correlated with GN scores (Figure 5b), and M1 macrophages (R = -0.86) suggesting this is an anti-inflammatory population. Morever, GN scores in a given animal could be accurately predicted by the two-component model, GN = 1.37 - (0.044 × CD115+) + (0.047 × BUN) (R = 0.92, P < 0.001).
There were CD3+ T lymphocytes in all kidneys, which were either CD4+ or CD8+ (Figure 6a). Relative to all other groups, the kidneys of CD11b-/- chimeras had an expanded population of CD3+CD4+ cells (Figure 6a, red arrow). This was unique to the kidney in these animals, as the spleens had comparable numbers of lymphocytes (Figure 6b). Thus, the absence of CD11b in either macrophages and/or dendritic cells in mice with experimental GN was associated with increased numbers of CD4+ T lymphocytes within the kidney.
Figure 6. Characterization of CD3+ cells in CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcγR-/-, and CD11b-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control.
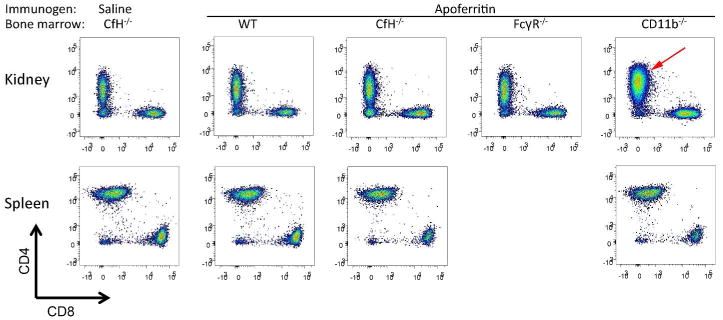
Representative data for CD4 (y-axis) and CD8 (x-axis) are shown from kidneys (a) and spleens (b). The red arrow depicts the expanded CD3+CD4+ population in CD11b-/- chimeras.
Discussion
In these studies, we induced CSS in CfH-/- BM chimeras. Immune complexes deposited in glomeruli, around which C3 was activated. In the absence of plasma CfH, active C3b remained, capable of contributing to C5 cleavage.10 In this setting, all animals developed renal functional impairment and histopathological features of GN. The presence of iC3b and F4/80+ macrophages in inflamed glomeruli9,10 supported our hypothesis CD11b would be pathologic in this model. Unexpectedly, CD11b-/- BM chimeric mice had significantly worse renal disease than any other group. This required the concomitant absence of plasma CfH, as native CD11b-/- mice did not develop GN in CSS.
The CSS model used here relies upon repetitive administration with horse spleen apoferritin over 5 weeks, without use of adjuvant (hence, a non-accelerated chronic active autologous model). Actively immunized animals generated anti-apoferritin IgG antibodies, which formed ICs with the ongoing source of apoferritin antigen. Thus, it is not surprising the quantities of circulating ICs were related to anti-apoferritin IgG. The measurement of the former relied upon C1q binding, and was independent from effects of CfH or CD11b deficiencies on C3.
Complement activation in ICs leads to their incorporating C3b which bind to cells with specific membrane receptors, a phenomenon termed immune adherence by Nelson.18 Rodents, including mice, rely upon CfH on platelets, while primates diverged to utilize CR1 on erythrocytes for this particular immune function. Immune complexes bound to platelet CfH or erythrocyte CR1 are transported to hepatic macrophages for elimination.19 This transfer is believed to occur because these cells bear CR3 and FcγRs. CR3 has greater binding affinity for ICs bearing iC3b than does CfH (or CR1). In IC processing, it is postulated that FcγRII is the most relevant, as it is inhibitory and does not associate with FcRγ.20 Consistent with this premise, here we showed FcRγ-/- BM chimeras had antibody and IC responses comparable to wildtype chimeras.
In this disease model, glomerular ICs contain apoferritin, IgG and C3 components.21 As in our past studies,9 excessive glomerular IC deposition occurred when mice lacked platelet CfH, supporting these formed from deposited ICs. Anti-apoferritin IgG and IgG-containing ICs were highest in CD11b-/- BM chimeric mice; while this did not translate into a higher quantity of total glomerular ICs, qualitatively there was greater extension into the peripheral capillary wall. Yet, CD11b-/- mice with intact CfH were largely indistinguishable from wildtype mice. Similar CD11b-independent IC handling and exaggerated GN was observed by the Mayadas group in an accelerated CSS model.13 In their studies, neutrophils preceded macrophages in glomeruli in a CD11b-dependent fashion,13 a phenomenon we have not examined. Nonetheless, taking these data together, glomerular IC deposition appears to be largely independent from macrophage IC handling. CfH-/- mice have an abnormally active humoral immune response, which is exaggerated in the absence of CD11b on BM-derived cells.
Diverse glomerular diseases are associated with substantial accumulations of mononuclear cells in the tubulointerstitium (TI)I. In fact, often the strongest predictive variable of glomerular disease outcome is the state of the TI.22-25 A limitation to assessing the relative extent of GN in both animal models and in human disease is the inherent subjectivity that goes into estimating disease activity. In proliferative GN, such as our model used here, higher scores largely reflect increased glomerular cellularity. In contrast, the ability to accurately quantify cells within the TI by flow cytometry has provided considerable insight into the makeup of these cells in various disease processes. Ly6ChiCCR2hi cells present in diseased tissue are considered M1 (classically activated) macrophages.26,27 In this study, there were increased numbers of M1 macrophages in the TI of animals with CSS. The relationship between glomerular and TI inflammation is supported by the strong positive relationship between TI Ly6ChiCCR2hi cell numbers and GN scores.
The surface marker staining of M1 macrophages as Ly6ChiCCR2hiF4/80+CD11c+ was consistent across the groups. In contrast, there were wide variations in CD11b staining, with cells being CD11b-, CD11b+ or CD11bhi. The clearest example of all three occurred in FcRγ-/- BM chimeras, in which approximately equal numbers of each was present in the TI (c.f. Figure 4c). The CD11b-/- BM chimeric mice had solely CD11b- M1 macrophages. From our data presented here, we are unable to explain these differences. Overall, we do feel this illustrates the importance of CD11b in macrophages to shape inflammatory disease, for which we have evidence from other models.28
In our studies and those of several other groups, the resident renal dendritic cell population is F4/80hiLy6C-CD11b+CD11c+;16,28,29 and, as true here, they can be separated into two groups with and without CCR2.28 The F4/80hiLy6C- population in CfH-/- mice chimeric for wildtype BM were CD11b+, while those with CD11b-/- BM were CD11b-, consistent with these dendritic cells being radiosensitive and terminally differentiated. In these cells lacking CD11b, there was greater surface expression of CD11c. While this may reflect that CD11c is the sole binding partner for CD18in these cells, it also may represent a more activated phenotype.
The F4/80+Ly6C+CCR2- cells were composed of two distinct cellular populations, namely Ly6C+CD11b+CD11c- and Ly6ChiCD11b-CD11c-. The former has features consistent with alternatively activated macrophages.26,27,30 The latter cell type was prominent in CfH-/- and FcRγ-/- BM chimeras, including in the CfH-/- BM controls. Thus, the physiological relevance of these F4/80+Ly6Chi cells is unclear. Yet, it does provide another example of the interesting diversity of CD11b expression among mononuclear cells.
The CD115+ cellular population was Ly6C-F4/80-CD11c-, but CD11b+ in all BM chimeras. This extended to the CD11b-/- BM chimeras. While there were lesser quantities in the latter animal kidneys, their phenotypic characteristics were otherwise comparable to the other groups. Thus, this appears to be a radioresistant population newly recruited from BM. Potentially this represents a population within the myeloid-derived suppressor cell family.31 As noted, these negatively predict disease. Here, GN can be accurately predicted by the number of TI CD115+ cells (in an inverse fashion) and measured BUN values; of note is that each of these three variables was arrived at by entirely separate means. Overall, this does support the anti-inflammatory nature of these cells.
There was not a remarkable expansion of CD3+ or CD19+ lymphocytes in the kidneys or spleens of experimental or control animals. The exception occurred with CD3+CD4+ T cells in CD11b-/- BM chimeras. This did not occur in the spleens of these animals, indicating the specificity of this expansion to the kidney. In general, even though CD4+ T cells can accumulate in the diseased sterile kidney, whether this is antigen-dependent or -independent is unclear.32 In this model, potential antigen-specific responses could occur to apoferritin (accumulated in glomerular ICs) and/or to native renal antigens from a break in tolerance. Alternatively, T cell accumulation could occur independent of antigen and T cell receptor.
There is growing appreciation that β2 integrins have roles beyond cellular adhesion. The three β2 integrin (CD18) partners are αL (CD11a), αM (CD11b), and αX (CD11c). CD11b has the broadest set of potential ligands, including iC3b and intercellular adhesion molecules (ICAM)-1 and 2, which encompass those of CD11a and CD11c.33 Anaphylatoxin and chemokine receptor activation, including C5aR, recruit Giα leading to “inside-out” activation of CD11b and increased avidity for ligand34 such as iC3b.35 When CD11b binds its ligand it can generate “outside-in” tyrosine kinase signals mediated by ITAM proteins, DAP12 and FcγR. From these two, mononuclear phagocytic cells primarily rely on DAP12.36 Of considerable interest is the anti-inflammatory nature of these signals through CD11b. For example, CD11b can limit pro-inflammatory signals through Toll-like receptor 4 activation by E3 ubiquitin-protein ligase CBL-B-mediated removal of activated proteins.37,38 Taken together, it seems likely CD11b binding to ligand is necessary for any anti-inflammatory role. Plausible explanations include CD11b-ICAM1 binding to divert CD11c-iC3b binding; CD11b occupying ICAM1 or ICAM2 sites on dendritic cells which block T cell CD11a binding; and, or active signaling through macrophage CD11b. The relevance of anti-inflammatory pathways brought about by CD11b signaling is the topic of ongoing investigations by us and others.
In summary, here we examined IC-mediated GN in CfH-/- BM chimeric mice. The absence of plasma CfH in all animals conferred susceptibility to GN. Compared to wildtype, CfH-/- chimeric mice had increased glomerular ICs and higher BUN values, consistent with the role for platelet CfH to process ICs. Disease in FcRγ-/- chimeras was no different than wildtype, implying the ITAM-bearing FcγRs (FcγRI and FcγRIII) are dispensible in this model. In contrast, CD11b-/- chimeric mice had worsened histopathological and functional disease compared to the other groups; this required plasma CfH be absent, in which setting greater quantities of iC3b ligand were generated. Disease in CfH-/- mice chimeric for CD11b-/- BM was associated with increased numbers of M1 macrophages and CD4+ T lymphocytes in the renal interstitium. This occurred together with an enhanced humoral immune response to the heterologous antigen, suggesting local immune responses. The population of interstitial CD115+CD11b+ cells was associated with protection from GN. Thus, CD11b on mononuclear cells is instrumental in generating an anti-inflammatory response in the inflamed kidney.
Materials and Methods
Animals
CD11b-/- mice generated by Mayadas et al39 were obtained from Jackson Laboratories. FcRγ-/- mice generated by Ravetch et al40 were obtained from Taconics Laboratories. CfH-/- mice were provided by Pickering, Botto, et al.14 All animals were backcrossed > 10 generations onto normal wild-type C57BL/6 mice (Jackson). The use of animals in these studies was approved by the University of Chicago and University at Buffalo Institutional Animal Care and Use Committees.
Bone marrow chimeric mice
CfH-/- mice between 4 and 6 weeks of age received 1050 cGy (irradiation).9 Bone marrow cells were isolated by standard techniques from femurs of wild-type, CfH-/-, FcRγ-/-, or CD11b-/- mice, and CD117+ (c-kit+) progenitor cells were isolated from BM cells using a monoclonal antibody magnetic positive selection technique (Miltenyi CD117 MicroBeads, Auburn, CA). One day after irradiation, mice received 1 × 106 CD117+ progenitor cells intravenously. In our prior studies, this protocol rescued animals from lethality and led to full hematopoietic reconstitution within 3 weeks.9
Chronic serum sickness model of immune complex-mediated glomerulonephritis
Immune complex disease was induced by actively immunizing chimeric mice 4 weeks after BM transfer with a daily intraperitoneal dose of 4 mg of horse spleen apoferritin (Calzyme Laboratories, San Luis Obispo, CA). Controls were CfH-/- mice into which CfH-/- mouse BM was transferred immunized with the same schedule of saline vehicle alone. After 5 weeks, mice were killed and renal disease was characterized.
Measurements from sera
Blood urea nitrogen concentrations were measured with a Beckman Autoanalyzer (Beckman Coulter, Fullerton, CA). Serum anti-apoferritin IgG antibodies were measured by ELISA.9 Briefly, 96-well plates were coated with apoferritin. Serial dilutions of sera were plated and incubated at room temperature for 2 hours, followed by HRP-conjugated goat anti-mouse IgG (Kierkegard & Perry Laboratories, Gaithersburg, MD) and OPD peroxidase substrate (Sigma-Aldrich, St. Louis, MO). The OD450 was then measured.
Circulating IC levels were measured by C1q binding ELISA as described previously.41 Briefly, 96-well plates were coated with 10 μg/ml human C1q (Sigma) in carbonate buffer. After blocking with 1% BSA, sera samples were loaded in serial dilutions starting at 1/1000, followed by HRP-goat anti-mouse IgG (Sigma, 1:2000) and then TMB (Jackson ImmunoResearch Laboratories, West Grove, PA). The OD450 was then measured. Sera samples from unmanipulated wildtype mice were used as negative control.
Immunofluorescence microscopy
Four-micrometer sections from frozen mouse kidneys were fixed in ethanol:ether (1:1) for 10 min followed by 95% ethanol for 20 min and washed with PBS. They were then stained with fluorescein isothiocyanate-conjugated anti-mouse C3 (Cappel, MP Biomedicals, Solon, OH) and Alexa 594-conjugated goat anti-mouse IgG (Invitrogen, Life Technologies, Carlsbad, CA). Slides were viewed with an Olympus BX-60 microscope (Melville, NY) and scored in a masked manner from 0 to 4. Photomicrographs of images representative of the group score were taken with a Zeiss LSM 510 microscope (Carl Zeiss Microscopy, Jena, Germany) and merged using ZEN imaging software at identical settings. Images were imported as TIFF files into Photoshop CC (Adobe, San Jose, CA). The “transform scale” function was performed on all layers together, with width-to-height proportions constrained.
Renal pathology
Tissues were fixed in 10% buffered formalin and embedded in paraffin, from which 4 μm-thick sections were cut and stained with periodic acid-Schiff. Each slide was scored in a blinded manner by a renal pathologist (T.C.) for the extent of GN on a scale of 0 to 4 (in increments of 0.5) according to the schema of Passwell et al42 as described previously.10 In addition, the extent of interstitial inflammation, interstitial fibrosis/tubular atrophy, and arteritis was determined. Images representative of the group score were taken similarly to those for immunofluorescence microscopy.
Flow cytometry
Single-cell suspensions were prepared from kidneys and spleens. This approach is directed towards isolating cells within the kidney wide interstitium; this anatomic space within the TI is between tubules and vessels. Tissue was minced into small pieces (<1 mm) in ice-cold 1× HBSS media and incubated with 1 mg/ml collagenase (type IV, Sigma) and 0.1 mg/ml deoxyribonuclease (type I, Sigma) at 37°C for 25 min with gentle shaking. The suspension was centrifuged at 250 g × 5 minutes and the pellet was resuspended in 2 ml of 150 mM NH4Cl, 10 mM KHCO3, 0.5 M EDTA, pH 8 (erythrocyte lysing reagent) and incubated for 5 min at room temperature. The suspension was again centrifuged at 250 g × 5 minutes, and the supernatant was discarded. The cells were resuspended in 1–2 ml of ice-cold PBS containing 2% calf serum, 5 mM EDTA and 0.1% sodium azide (FACS buffer) and passed through a 40 μm cell-strainer. Cells were resuspended at approximately 107/mL FACS buffer and incubated with anti-CD16/32 (2.4G2) for 5 minutes on ice prior to staining with specific antibodies. Cells were then washed with 1 mL FACS buffer, centrifuged, resuspended in 200 μl FACS buffer and analyzed on a flow cytometer (LSRII, BD, Franklin Lakes, NJ). Data were analyzed using FlowJo software (v. 10).
Antibodies used for flow cytometry were APC-Cy7-conjugated anti-mouse CD19, Brilliant Violet 421-conjugated anti-mouse CD3, Brilliant Violet 605-conjugated anti-mouse CD4, Alexa fluor 488-conjugated anti-mouse CD115, PerCP5.5-conjugated anti-mouse Ly6C, Alexa fluor 700-conjugated anti-CD11c (Biolegend, San Diego, CA), V500-conjugated anti-mouse CD11b (BD), PE-Texas Red conjugated anti-mouse CD8 (Life Technologies, Carlsbad, CA), phycoerythrin-conjugated anti-mouse CCR2 (R&D Systems Minneapolis, MN), and Alexa fluor 647-conjugated anti-F4/80 (AbD Serotec, Kidlington, England, UK).
Statistical analyses
Numeric data were analyzed with Minitab software (v. 16.2.4). Normality of data was evaluated by Anderson-Darling testing. Data are given as means ± SEM for parametric data and medians with Q1-Q3 intervals for non-parametric data. In the figures, data from individual mice are presented.
Univariate ANOVA was used to compare experimental parametric data. When the variances were significantly different (P < 0.05), follow-up comparisons were made with the Tukey all-pairwise approach. To compare multiple groups of non-parametric data, the Mood median test was used. Potential correlations among data were determined by calculating Pearson product moment correlation coefficients. Fitted line plots were generated using the least squares method. Subset models that provided highest R2 values with least predictors were determined by best subsets regression.
Supplementary Material
Figure S1. Positive correlation between plasma free immune complexes and anti-apoferritin IgG levels in CSS induced in CfH-/- mice chimeric for the indicated bone marrow (a) and in those without BM transfers (b).
The lines fit by least squares analysis were:
(a) ICs = 17.3 + 0.743 × anti-apoferritin, R = 0.71, P = 0.002;
(b) ICs = 4.8 + 0.759 × anti-apoferritin, R = 0.73, P = 0.001.
Figure S2. Positive correlation between measured BUN values and anti-apoferritin IgG levels. The line fit by least squares analysis was BUN = 29.3 + (35.4 × anti-apoferritin). R = 0.91, P < 0.001.
The other significant correlations among disease measures were:
BUN = 20.9 + (0.47 × urinary albumin). R = 0.97, P < 0.001;
BUN = 26.8 + (8.16 × GN score). R = 0.86, P = 0.002; and,
Urinary albumin = 15.3 + (16.1 × GN score). R = 0.81, P = 0.004.
Figure S3. Analysis of CD3-CD19- cells by flow cytometry. Cells were from kidneys of CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcyR-/-, and CDllb-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control. (a) Data for CD115 (y-axis) and F4/80 (x-axis). (b) CD115+ cells were analyzed for CDllb (y-axis) and CDllc (x-axis).
Figure S4. Analysis of F4/80+Ly6C+CCR2- cells by flow cytometry. Cells were from kidneys of CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcyR-/-, and CDllb-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control. Representative data for CDllb (y-axis) and CDllc (x-axis) are shown. Ly6Chi cells were CDllb-, while Ly6C+ cells were CDllb+, except in the CDllb-/- chimeras.
Figure S5. Analysis of F4/80+Ly6C cells for CDllb and CDllc by flow cytometry. Cells were from kidneys of CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcyR-/-, and CDllb-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control. CCR2+ and CCR2- cells were analyzed separately; these are from quadrants 3 and 4, respectively, from Figure 4. (a) Representative data for CDllb (y-axis) and CDllc (x-axis) are shown, (b) CDllc expression is shown in histograms for all wildtype and CDllb-/- chimeras.
Acknowledgments
This work was supported by US National Institutes of Health grants DK007510 (L.D.C.) and DK041873 (R.J.Q.), and a grant from Kidneeds to J.J.A.
Footnotes
Disclosure: All the authors declared no conflict of interests.
References
- 1.Fakhouri F, Fremeaux-Bacchi V, Noel LH, et al. C3 glomerulopathy: a new classification. Nat Rev Nephrol. 2010;6:494–499. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 2.Amatruda TT, III, Gerard NP, Gerard C, Simon MI. Specific interactions of chemoattractant factor receptors with G-proteins. J Biol Chem. 1993;268:10139–10144. [PubMed] [Google Scholar]
- 3.Ross GD, Vetvicka V. CR3 (CD11b, CD18): a phagocyte and NK cell membrane receptor with multiple ligand specificities and functions. Clin Exp Immunol. 1993;92:181–184. doi: 10.1111/j.1365-2249.1993.tb03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han C, Jin J, Xu S, et al. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11:734–742. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 5.Mocsai A, Abram CL, Jakus Z, et al. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stilmant MM, Couser WG, Cotran RS. Experimental glomerulonephritis in the mouse associated with mesangial deposition of autologous ferritin immune complexes. Lab Invest. 1975;32:746–756. [PubMed] [Google Scholar]
- 7.Quigg RJ, Lim A, Haas M, et al. Immune complex glomerulonephritis in C4- and C3-deficient mice. Kidney Int. 1998;53:320–330. doi: 10.1046/j.1523-1755.1998.00723.x. [DOI] [PubMed] [Google Scholar]
- 8.Alexander JJ, Pickering MC, Haas M, et al. Complement factor H limits immune complex deposition and prevents inflammation and scarring in glomeruli of mice with chronic serum sickness. J Am Soc Nephrol. 2005;16:52–57. doi: 10.1681/ASN.2004090778. [DOI] [PubMed] [Google Scholar]
- 9.Alexander JJ, Aneziokoro OGB, Chang A, et al. Distinct and separable roles of the complement system in factor H-deficient bone marrow chimeric mice with immune complex disease. J Am Soc Nephrol. 2006;17:1354–1361. doi: 10.1681/ASN.2006020138. [DOI] [PubMed] [Google Scholar]
- 10.Alexander JJ, Chaves L, Chang A, Quigg RJ. The C5a receptor has a key role in immune complex glomerulonephritis in complement factor H-deficient mice. Kidney Int. 2012;82:961–968. doi: 10.1038/ki.2012.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander JJ, Hack BK, Cunningham PN, Quigg RJ. A protein with characteristics of factor H is present on rodent platelets and functions as the immune adherence receptor. J Biol Chem. 2001;276:32129–32135. doi: 10.1074/jbc.M101299200. [DOI] [PubMed] [Google Scholar]
- 12.Ren G, Doshi M, Hack BK, et al. Rat glomerular epithelial cells produce and bear factor H on their surface which is upregulated under complement attack. Kidney Int. 2003;64:914–922. doi: 10.1046/j.1523-1755.2003.00188.x. [DOI] [PubMed] [Google Scholar]
- 13.Rosetti F, Tsuboi N, Chen K, et al. Human lupus serum induces neutrophil-mediated organ damage in mice that is enabled by Mac-1 deficiency. J Immunol. 2012;189:3714–3723. doi: 10.4049/jimmunol.1201594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickering MC, Cook HT, Warren J, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424–428. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 15.Alexander JJ, Wang Y, Chang A, et al. Mouse podocyte complement factor H - The functional analogue to human complement receptor 1. J Am Soc Nephrol. 2007;18:1157–1166. doi: 10.1681/ASN.2006101125. [DOI] [PubMed] [Google Scholar]
- 16.Soos TJ, Sims TN, Barisoni L, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70:591–596. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 17.Hochheiser K, Tittel A, Kurts C. Kidney dendritic cells in acute and chronic renal disease. Int J Exp Pathol. 2011;92:193–201. doi: 10.1111/j.1365-2613.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson DS. Immune adherence. Adv Immunol. 1963;3:131–180. [Google Scholar]
- 19.Hebert LA. The clearance of immune complexes from the circulation of man and other primates. Am J Kidney Dis. 1991;27:352–361. doi: 10.1016/s0272-6386(12)80488-4. [DOI] [PubMed] [Google Scholar]
- 20.Dijstelbloem HM, van de Winkel JG, Kallenberg CG. Inflammation in autoimmunity: receptors for IgG revisited. Trends Immunol. 2001;22:510–516. doi: 10.1016/s1471-4906(01)02014-2. [DOI] [PubMed] [Google Scholar]
- 21.Alexander JJ, Hack BK, Jacob A, et al. Abnormal immune complex processing and spontaneous glomerulonephritis in complement factor H-deficient mice with human complement receptor 1 on erythrocytes. J Immunol. 2010;185:3759–3767. doi: 10.4049/jimmunol.1000683. [DOI] [PubMed] [Google Scholar]
- 22.Cameron JS. Tubular and interstitial factors in the progression of glomerulonephritis. Pediatr Nephrol. 1992;6:292–303. doi: 10.1007/BF00878382. [DOI] [PubMed] [Google Scholar]
- 23.Nangaku M. Mechanisms of tubulointerstitial injury in the kidney: final common pathways to end-stage renal failure. Intern Med. 2004;43:9–17. doi: 10.2169/internalmedicine.43.9. [DOI] [PubMed] [Google Scholar]
- 24.Roberts IS, Cook HT, Troyanov S, et al. The Oxford classification of IgA nephropathy: pathology definitions, correlations, and reproducibility. Kidney Int. 2009;76:546–556. doi: 10.1038/ki.2009.168. [DOI] [PubMed] [Google Scholar]
- 25.Hsieh C, Chang A, Brandt D, et al. Predicting outcomes of lupus nephritis with tubulointerstitial inflammation and scarring. Arthritis Care Res (Hoboken) 2011;63:865–874. doi: 10.1002/acr.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson PJ, Rees AJ, Griffin MD, et al. The renal mononuclear phagocytic system. J Am Soc Nephrol. 2012;23:194–203. doi: 10.1681/ASN.2011070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaves LD, Bao L, Wang Y, et al. Loss of CD11b exacerbates murine complement-mediated tubulointerstitial nephritis. PLoS One. 2014;9:e92051. doi: 10.1371/journal.pone.0092051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kruger T, Benke D, Eitner F, et al. Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J Am Soc Nephrol. 2004;15:613–621. doi: 10.1097/01.asn.0000114553.36258.91. [DOI] [PubMed] [Google Scholar]
- 30.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kelly CJ. Development and expression of nephritogenic T cells, chap. 12. In: Neilson EG, Couser WG, editors. Immunologic Renal Diseases. 2. Philadelphia: Lippincott-Raven; 2001. pp. 241–256. [Google Scholar]
- 33.Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol. 2012;24:107–115. doi: 10.1016/j.ceb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones SL, Knaus UG, Bokoch GM, Brown EJ. Two signaling mechanisms for activation of alphaM beta2 avidity in polymorphonuclear neutrophils. J Biol Chem. 1998;273:10556–10566. doi: 10.1074/jbc.273.17.10556. [DOI] [PubMed] [Google Scholar]
- 35.Chen X, Yu Y, Mi LZ, et al. Molecular basis for complement recognition by integrin alphaXbeta2. Proc Natl Acad Sci U S A. 2012;109:4586–4591. doi: 10.1073/pnas.1202051109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227:150–160. doi: 10.1111/j.1600-065X.2008.00720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Gordon RA, Huynh L, et al. Indirect inhibition of Toll-like receptor and type I interferon responses by ITAM-coupled receptors and integrins. Immunity. 2010;32:518–530. doi: 10.1016/j.immuni.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Means TK, Luster AD. Integrins limit the Toll. Nat Immunol. 2010;11:691–693. doi: 10.1038/ni0810-691. [DOI] [PubMed] [Google Scholar]
- 39.Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the β2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 40.Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- 41.Bao L, Haas M, Kraus DM, et al. Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr mice. J Am Soc Nephrol. 2003;14:670–679. doi: 10.1097/01.asn.0000051597.27127.a1. [DOI] [PubMed] [Google Scholar]
- 42.Passwell J, Schreiner GF, Nonaka M, et al. Local extrahepatic expression of complement genes C3, factor B, C2 and C4 is increased in murine lupus nephritis. J Clin Invest. 1988;82:1676–1684. doi: 10.1172/JCI113780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Positive correlation between plasma free immune complexes and anti-apoferritin IgG levels in CSS induced in CfH-/- mice chimeric for the indicated bone marrow (a) and in those without BM transfers (b).
The lines fit by least squares analysis were:
(a) ICs = 17.3 + 0.743 × anti-apoferritin, R = 0.71, P = 0.002;
(b) ICs = 4.8 + 0.759 × anti-apoferritin, R = 0.73, P = 0.001.
Figure S2. Positive correlation between measured BUN values and anti-apoferritin IgG levels. The line fit by least squares analysis was BUN = 29.3 + (35.4 × anti-apoferritin). R = 0.91, P < 0.001.
The other significant correlations among disease measures were:
BUN = 20.9 + (0.47 × urinary albumin). R = 0.97, P < 0.001;
BUN = 26.8 + (8.16 × GN score). R = 0.86, P = 0.002; and,
Urinary albumin = 15.3 + (16.1 × GN score). R = 0.81, P = 0.004.
Figure S3. Analysis of CD3-CD19- cells by flow cytometry. Cells were from kidneys of CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcyR-/-, and CDllb-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control. (a) Data for CD115 (y-axis) and F4/80 (x-axis). (b) CD115+ cells were analyzed for CDllb (y-axis) and CDllc (x-axis).
Figure S4. Analysis of F4/80+Ly6C+CCR2- cells by flow cytometry. Cells were from kidneys of CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcyR-/-, and CDllb-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control. Representative data for CDllb (y-axis) and CDllc (x-axis) are shown. Ly6Chi cells were CDllb-, while Ly6C+ cells were CDllb+, except in the CDllb-/- chimeras.
Figure S5. Analysis of F4/80+Ly6C cells for CDllb and CDllc by flow cytometry. Cells were from kidneys of CfH-/- mice chimeric for wildtype (WT), CfH-/-, FcyR-/-, and CDllb-/- bone marrow immunized for 5 weeks with apoferritin, or saline as control. CCR2+ and CCR2- cells were analyzed separately; these are from quadrants 3 and 4, respectively, from Figure 4. (a) Representative data for CDllb (y-axis) and CDllc (x-axis) are shown, (b) CDllc expression is shown in histograms for all wildtype and CDllb-/- chimeras.