

Abstract
Fosfazinomycin A is a phosphonate natural product in which the C-terminal carboxylate of a Val-Arg dipeptide is connected to methyl 2-hydroxy-2-phosphono-acetate (Me-HPnA) via a unique hydrazide linkage. We report here that Me-HPnA is generated from phosphonoacetaldehyde (PnAA) in three biosynthetic steps through the combined action of an O-methyltransferase (FzmB) and an α-ketoglutarate (α-KG) dependent non-heme iron dioxygenase (FzmG). Unexpectedly, the latter enzyme is involved in two different steps, oxidation of the PnAA to phosphonoacetic acid as well as hydroxylation of methyl 2-phosphonoacetate. The N-methyltransferase (FzmH) was able to methylate Arg-NHNH2 (3) to give Arg-NHNHMe (4), constituting the second segment of the fosfazinomycin molecule. Methylation of other putative intermediates such as desmethyl fosfazinomycin B was not observed. Collectively, our current data support a convergent biosynthetic pathway to fosfazinomycin.
Introduction
Phosphonate natural products are a class of compounds featuring a C-P bond.1 Many of these natural products show interesting and potent bioactivity. For instance, fosfomycin is an antibiotic in clinical use, while phosphonothricin tripeptide (PTT) is a widely used herbicide (Figure 1).1 The bioactivity of phosphonates is generally attributed to their C-P bond, which mimics, but is much more stable than, the corresponding O-P bond of phosphate esters.1 In recent years phosphonate natural products have drawn attention not only because of their bioactivity, but also because of the plethora of unusual and interesting chemistries associated with the biosynthesis of these molecules.2, 3
Figure 1.

Structures of select phosphonates.
Originally isolated from Streptomyces lavendofoliae 630, fosfazinomycins (Figure 1) are natural phosphonates with antifungal activity.4, 5 Compared to fosfazinomycin B (fosB), fosfazinomycin A (fosA) contains an extra Val residue at the N-terminus. The most interesting structural feature of fosfazinomycins is the unusual hydrazide linkage between the carboxylic acid of Arg and the phosphonic acid. Although the structures of these antibiotics were elucidated three decades ago, information on their biosynthesis was unavailable until our very recent description of the biosynthetic gene cluster.6
Very little is known about how nitrogen-nitrogen bonds are fashioned in Nature. In principle, three different pathways can be envisioned in the case of fosfazinomycin (Figure 2). The hydrazide moiety could be initially attached to the phosphonate and then coupled to the Arg or the Val-Arg dipeptide (retro-synthetic disconnection a, Figure 2). Alternatively, the hydrazide could be formed on the Arg or the Val-Arg dipeptide and then be condensed with the phosphonic acid (disconnection b), or the hydrazide could be formed from an amide and phosphonamide (disconnection c). Herein we report the first in vitro biochemical investigation of fosfazinomycin biosynthesis. Our studies assign the function of the enzymes involved in the formation of (S)-methyl 2-hydroxy-2-phosphono-acetate ((S)- Me-HPnA) from phosphonoacetaldehyde (PnAA, Scheme 1). FzmB is shown to be an O-methyltransferase that methylates phosphonoacetate, and the α-ketoglutarate (α-KG) dependent non-heme iron dioxygenase FzmG is demonstrated to catalyse the oxidation of two different substrates. Moreover, evaluation of various different potential substrates of the N-methyltransferase FzmH strongly suggests that fosfazinomycins are biosynthesized through a convergent pathway that is consistent with disconnection b (Figure 2).
Figure 2.
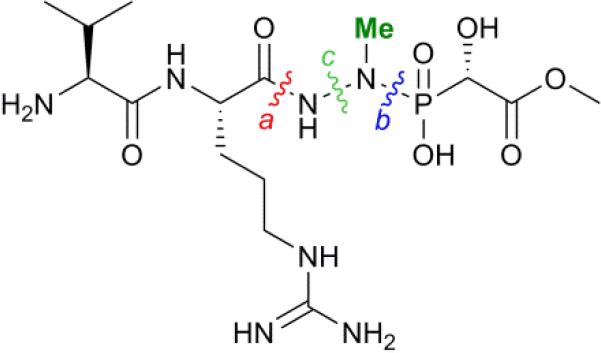
Three disconnections to generate the central hydrazide core of fosfazinomycins.
Scheme 1.

Previously proposed initial steps of the biosynthetic pathway of fosfazinomycin A.
Results
The fzmB and fzmG genes were amplified by the polymerase chain reaction (PCR) from the fosmids MMG 358 and 360,7 respectively, that both harbour the fosfazinomycin gene cluster. The amplified genes were inserted into the expression vector pET-15b. Both proteins were expressed in Escherichia coli as N-terminal hexahistidine-tagged constructs and purified by immobilized metal affinity chromatography (IMAC) to near homogeneity (>90% purity) according to SDS/PAGE analysis (Figure S1 in the ESI†).
With the soluble proteins in hand, we first tested the activity of His6-FzmB with two putative substrates, phosphonoacetate (PnA) and rac-2-hydroxy-2-phosphono-acetate (rac-HPnA) (Figure 1). When PnA was used as substrate in the presence of S-adenosyl methionine (SAM), ca. 90% conversion to the methyl ester was achieved according to analysis by 31P NMR spectroscopy. The product was confirmed to be Me-PnA by spiking with an authentic synthetic standard (Figure S2). In the absence of either the enzyme or SAM, only the starting material was recovered. In addition, complete conversion of rac-HPnA to Me-HPnA was observed in the presence of His6-FzmB and SAM (Figure S3). Therefore His6-FzmB was able to methylate both enantiomers of HPnA. The high conversions in both reactions made it difficult to conclude whether PnA or HPnA would be the physiological substrate of FzmB. To further study its substrate tolerance, methylenebis(phosphonic acid) (Figure 1) and malonic acid were tested as close analogs of PnA, but product was not detected in either reaction.
Next we investigated the function of the putative α-KG dependent dioxygenase FzmG. Initially Me-PnA was tested, resulting in complete conversion of Me-PnA to Me-HPnA (Figure 3A,B). The reaction was enzyme-catalysed and α-KG was essential for catalysis (Figure 3C,E). In addition, the common reductant L-ascorbate accelerated the reaction (Figure 3A,D). Recently Hammerschmidt and co-workers have determined the absolute configuration of Me-HPnA in fosfazinomycins to be S by comparison of chemically synthesized standards to the fragments generated from acid-mediated hydrolysis of fosfazinomycins.8 In addition, they found that Me-HPnA was configurationally stable in water at slightly acidic pH, but completely racemized at pH 7-8. Given its ease to racemize at the pH we used for the enzymatic assays, we did not attempt to determine the stereochemistry of our enzymatic product. PnA was also incubated with His6-FzmG. Under the same conditions that resulted in complete consumption of Me-PnA, the conversion of PnA was only 30%; the product was confirmed to be HPnA by spiking with authentic synthetic standard (Figure S4).
Figure 3.

31P NMR spectra of the products obtained by incubation of His6-FzmG with Me-PnA. A. NMR spectrum of the reaction mixture containing Me-PnA, O2, α-KG, Fe(II), L-ascorbate, and His6-FzmG. B. NMR spectrum of the enzymatic reaction mixture spiked with authentic standard of Me-HPnA. C. NMR spectrum of the reaction mixture in the absence of α-KG. D. NMR spectrum of the reaction mixture in the absence of L-ascorbate. E. NMR spectrum of the reaction mixture in the absence of His6-FzmG. F. NMR spectrum of authentic Me-HPnA.
DhpA is another α-KG dependent non-heme iron dioxygenase that hydroxylates 2-hydroxyethylphosphonoacetate (2-HEP) to generate 1,2-dihydroxyethylphosphonoacetate (1,2-DHEP) (Figure 1) during the biosynthesis of dehydrophos.9, 10 FzmG and DhpA share 45% and 55% sequence identity and similarity, respectively.6 The high sequence identity prompted us to investigate whether these two enzymes could act on each other's substrates. His6-FzmG completely converted 2-HEP to 1,2-DHEP (Figure S5), whereas His6-DhpA did not accept Me-PnA as a substrate (Figure S6). 2-Aminoethylphosphonate (2-AEP) was also incubated with His6-FzmG, but no activity was observed.
To examine the possibility that hydroxylation at the alpha carbon of the phosphonate moiety of fosfazinomycins occurs before the oxidation at the beta carbon, phosphonoacetaldehyde (PnAA) was incubated with His6-FzmG. In this model, 2-hydroxy-2-phosphono-acetaldehyde (HPnAA) was expected to be the product. Upon incubation, the PnAA peak (9 ppm) in the 31P NMR spectrum indeed disappeared and a new peak appeared at 13.7 ppm (Figure 4A,B). A 1H-31P HMBC NMR spectrum suggested the newly formed product might be PnA rather than the anticipated HPnAA. This assignment was confirmed by spiking with authentic PnA (Figure 4C). This unexpected result was quite interesting, because in our proposed biosynthetic pathway (Scheme 1) a canonical aldehyde dehydrogenase was invoked to convert PnAA to PnA, but such a gene is absent in the gene cluster. The current data suggest that FzmG could catalyse two distinct steps in fosfazinomycin biosynthesis, oxidation of PnAA to PnA and hydroxylation of Me-PnA.
Figure 4.

31P NMR spectra analysis of the incubation of His6-FzmG with PnAA. A. NMR spectrum of the reaction mixture containing PnAA, O2, α-KG, Fe(II), L-ascorbate, and His6-FzmG. B. NMR spectrum of the enzymatic reaction mixture spiked with authentic standard of PnAA. C. NMR spectrum of the reaction mixture spiked with authentic standards of PnAA and PnA. D. NMR spectrum of the reaction mixture in the absence of His6-FzmG. The chemical shifts of phosphonates in 31P NMR are very sensitive to pH near their pKa values, accounting for the changes in chemical shifts between experiments and requiring spiking with standards for assignments.
To further test this hypothesis, we determined the kinetic parameters of His6-FzmG towards the substrates. By monitoring the O2 consumption rate using a Hansatech O2 electrode, the apparent steady-state kinetic parameters for several substrates were determined (Table 1). Clearly, MePnA is a much better substrate than PnA, suggesting that methylation occurs before hydroxylation. Furthermore, PnAA is also a reasonably good substrate (Figure S7), consistent with FzmG catalysing two steps in the biosynthesis of fosfazinomycin.
Table 1.
Apparent steady-state kinetic parameters of His6-FzmG towards various of substrates.
| kcat (s−1) | Km (mM) | kcat/Km (M−1 s−1) | |
|---|---|---|---|
| Me-PnA | 0.27 ± 0.01 | 0.08 ± 0.02 | 3.3 × 103 |
| PnA | 0.17 ± 0.01 | 7.5 ± 1.3 | 23 |
| 2-HEP | 0.35 ± 0.02 | 0.21 ± 0.04 | 1.7 × 103 |
| PnAA | 0.46 ± 0.08 | 0.84 ± 0.29 | 5.5 × 102 |
After the successful in vitro reconstitution of the biosynthesis of Me-HPnA, we next sought to decipher how this moiety might be incorporated into fosfazinomycins. At least two possible routes can be envisioned from (S)-Me-HPnA to fosfazinomycin B (Scheme 2). In route a, (S)-Me-HPnA is first transformed into the phosphonamide 1, then converted to the phosphonylhydrazide 2, which would condense with arginine to afford desmethyl fosfazinomycin B (desmethyl-fosB) in a linear pathway. Compound 2 could also be formed by condensation of MeHPnA with a hydrazine derivative. Alternatively, arginine could be converted to the arginine hydrazide 3 (Arg-NHNH2) and joined with (S)-Me-HPnA to generate desmethyl-fosB in a convergent pathway (route b). Finally, compound 1 could be linked to arginine amide by unprecedented chemistry (not shown). To date, we have not been successful in reconstituting the activity of the enzymes that have been proposed to be involved in N-N bond formation and the condensation reactions that install the linkages to the Arg and Me-HPnA.6 However, we were able to obtain important information on the timing of these processes in an indirect way. The fosfazinomycins contain a methyl group on the nitrogen directly attached to the phosphonate, and in principle, the methyl group could be introduced on one of four putative intermediates, compounds 1-3, and desmethyl-fosB (Scheme 2). Determination of the substrate of the putative methyltransferase FzmH could therefore provide key insights on the manner in which the central core of fosfazinomycin is assembled.
Scheme 2.

Two proposed routes of converting (S)-Me-HPnA to fosB.
The gene encoding the putative N-methyltransferase FzmH was amplified by PCR from the genomic DNA of another fosfazinomycin producing strain, Streptomyces sp. WM6372, and ligated into the expression vector pET-15b. The corresponding protein was expressed in E. coli as an N-terminal hexahistidine-tagged construct and purified by IMAC (Figure S8). All four putative substrates for FzmH were chemically synthesized (see ESI for details). Compounds 1, 2 or desmethyl-fosB were incubated with His6-FzmH in the presence of SAM and S-adenosyl homocysteine (AdoHcy) nucleosidase to prevent product inhibition by AdoHcy. With all three compounds, methylated products were not formed (Figures S9-S13). In contrast, a clear new methyl peak was observed in the 1H NMR spectrum when Arg-NHNH2 (3) was incubated with His6-FzmH in the presence of SAM. The product was confirmed to be Arg-NHNHMe (4) by spiking with a synthetic authentic standard (Figure 5B,C) and this assignment was also supported by high resolution Fourier transform MS (Figure S14). In the absence of His6-FzmH, no methylation occurred (Figure 5A). These results are consistent with a convergent pathway (route b) and not with a linear pathway and provide important insights into the potential identity of substrates for N-N bond formation (e.g. this process does not occur on phosphonate substrates). To further study the substrate specificity of FzmH, Ac-Arg-NHNH2 (5) and Val-Arg-NHNH2 (6) (Scheme 2) were synthesized. The enzyme methylated both of these compounds on the terminal nitrogen and the product identity was again confirmed by spiking with synthetic authentic standards (Figures S15 and S16) and FT-MS (Figures S17 and S18).
Figure 5.

1H NMR spectra of products generated upon incubation of His6-FzmH with Arg-NHNH2 (3). A. NMR spectrum of the reaction mixture in the absence of His6-FzmH. B. NMR spectrum of the enzymatic reaction mixture containing Arg-NHNH2, SAM, AdoHcy nucleosidase, and His6-FzmH. C. NMR spectrum of the reaction mixture spiked with authentic standard of Arg-NHNHMe. The peaks marked with asterisks originate from SAM and SAM-related products.
Discussion
The O-methyltransferase His6-FzmB methylated both PnA and HPnA with high efficiency in vitro, preventing assignment of the physiological substrate. On the other hand, His6-FzmG hydroxylated Me-PnA and PnA with very different catalytic efficiencies. The Km of Me-PnA was 0.08 ± 0.02 mM, almost 100-fold lower than that of PnA (7.5 ± 1.3 mM). Combined with the similar kcat values (0.27 ± 0.01 s-1 versus 0.17 ± 0.01 s−1), these results strongly suggest Me-PnA is the physiological substrate of FzmG. Therefore, methylation of PnA by FzmB likely occurs before hydroxylation. This order of methylation and hydroxylation also matches well with the fact that Me-PnA is one of the observed metabolites produced by fosfazinomycin producing strains.6
PnAA is a common intermediate in phosphonate biosynthesis and it is processed in three different ways in various characterized pathways.11 PnAA can be converted to 2-aminoethylphosphonate (2-AEP) by a transaminase,12 or reduced to 2-HEP by an alcohol dehydrogenase.10, 13-15 Finally it can also condense with oxaloacetate to give 2-keto-4-hydroxy-5-phosphonopentanoic acid in rhizocticin biosynthesis.11, 16 To our knowledge, the fosfazinomycin pathway is the first example in which PnAA is transformed to PnA in a phosphonate biosynthetic pathway. We note that the same reaction catalysed by an aldehyde dehydrogenase PhnY was reported in phosphonate catabolic pathways.11 Our current findings that FzmG was able to generate PnA from PnAA with only 6-fold lower catalytic efficiency compared to its oxidation of Me-PnA (kcat/Km = 5.5 × 102 M−1 s−1 versus kcat/Km = 3.3 × 103 M−1 s−1) suggests that FzmG possesses a relaxed substrate specificity. Hence, it is possibly the missing enzyme in our previously proposed fosfazinomycin biosynthetic pathway (Scheme 1). However, we cannot rule out the possibility that a housekeeping aldehyde dehydrogenase outside the gene cluster might oxidize PnAA to PnA. There are other examples of α-KG dependent non-heme iron dioxygenases catalysing multiple reactions in a single biosynthetic pathway.17 One such example is the clavaminate synthase, which catalyses three separate oxidative steps during clavulanic acid biosynthesis, involving hydroxylation, cyclization and desaturation reactions.17, 18
Although most secondary metabolites are biosynthesized in a linear fashion, some small molecules, mostly polyketides and non-ribosomal peptides, are generated via a convergent manner.19, 20 For example, the chromophore of maduropeptin, an enediyne antitumor compound, was installed from four components using a convergent strategy.19 In contrast, until this study, all known phosphonates with characterized biosynthetic pathways were shown to be generated in a linear pathway. Our observation that FzmH can convert Arg-NHNH2 (3) and related compounds 5 and 6 to their N-methylated products, whereas other potential substrates were not accepted, strongly supports methylation of an acylhydrazine during the biosynthesis of fosfazinomycins. In turn, these results indicate that fosfazinomycin B is biosynthesized from two advanced intermediates, Arg-NHNHMe (4) and (S)-Me-HPnA following a convergent manner. The enzymes involved in the formation of 3 and in the condensation reaction remain to be identified among the proteins encoded in the cluster whose functions have not yet been assigned.6 Such studies can now commence with the knowledge gained about potential substrates in this study.
Methods
General Methods
For reagents, molecular biology procedures, enzyme purifications, synthetic procedures for the preparation of substrates and standards, and their spectroscopic characterization, see the ESI.
FzmB activity assays
The reaction mixture (500 μL) contained 5 mM of the appropriate phosphonic acid, 6 mM S-adenosyl methionine (SAM), 0.9 mg/mL AdoHcy nucleosidase, and 74 μM FzmB in 50 mM sodium phosphate (NaPi) buffer, pH 7.7. After 5.5 h incubation at ambient temperature, the proteins were removed by passing the reaction mixture through an Amicon spin column of 10 kDa molecular weight cut-off (MWCO) and 150 μL D2O was added into the sample before NMR analysis.
FzmG activity assays
The reaction mixture (500 μL) contained 5 mM of the appropriate phosphonic acid, 10 mM α-ketoglutarate, 1 mM L-ascorbic acid, 0.2 mM (NH4)2Fe(SO4)2 and 37 μM FzmG in 50 mM NaPi, pH 7.7. Typically the enzyme had been reconstituted in an anaerobic glove box (Coy, Grass Lake, MI) with 1.2 equivalent of Fe(II) for ca. 10 min on ice prior to its use. After 5.5 h incubation at ambient temperature ca. 100 μL of Chelex resin was added to the reaction tube. The resin was left to chelate metal ions at room temperature for ca. 15 min to reduce line broadening in the NMR spectra. The protein was removed by passing the reaction mixture through an Amicon spin column (10 kDa MWCO) and 150 μL D2O was added into the sample before NMR analysis. The assay with PnAA contained 1 mM PnAA, 2 mM α-ketoglutarate, 0.2 mM L-ascorbic acid, 0.03 mM (NH4)2Fe(SO4)2, and 7.4 μM FzmG in 50 mM NaPi, pH 7.7 and was incubated for 7.5 h.
Kinetic assays with FzmG
A Hansatech O2 electrode was used to generate the Michaelis-Menten curves by varying the concentration of the appropriate phosphonic acid while keeping α-KG at a saturating concentration (1 mM) and at a constant FzmG concentration (3.7 μM for Me-PnA, PnAA and 2-HEP assays, and 9.25 μM for PnA assay). All reactions were initiated by the addition of FzmG and the initial rate of O2 consumption was measured in triplicate. Nonlinear regressions were calculated using Igor Pro version 6.1.
FzmH activity assays
The reaction mixture (500 μL) contained 2.5 mM of the appropriate substrate, 3.5 mM S-adenosyl methionine (SAM), 0.9 mg/mL AdoHcy nucleosidase, and 26 μM FzmH in 50 mM NaPi, pH 7.7. After 6.5 h incubation at ambient temperature, the proteins were removed by passing the reaction mixture through an Amicon spin column (10 kDa MWCO) and 150 μL D2O was added into the sample before NMR analysis.
Supplementary Material
Acknowledgements
We thank Spencer Peck (UIUC) for helpful discussion and Dr. Despina Bougioukou (UIUC) for critical reading of this manuscript. This work was supported by the National Institute of Health (GM PO1 GM077596 to W.A.V.). NMR spectra were recorded on a 600 MHz instrument purchased with support from NIH Grant S10 RR028833.
Footnotes
Electronic Supplementary Information (ESI) available: Supplementary figures, molecular biology procedures, enzyme purifications, and synthetic procedures for the preparation of substrates and standards and their spectroscopic characterization.
References
- 1.Metcalf WW, van der Donk WA. Annu. Rev. Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peck SC, van der Donk WA. Curr. Opin. Chem. Biol. 2013;17:580–588. doi: 10.1016/j.cbpa.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cicchillo RM, Zhang H, Blodgett JAV, Whitteck JT, Li G, Nair SK, van der Donk WA, Metcalf WW. Nature. 2009;459:871–874. doi: 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuroda Y, Okuhara M, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. J. Antibiot. (Tokyo) 1980;33:29–35. doi: 10.7164/antibiotics.33.29. [DOI] [PubMed] [Google Scholar]
- 5.Ogita T, Gunji S, Fukuzawa Y, Terahara A, Kinoshita T, Nagaki H, Beppu T. Tetrahedron Lett. 1983;24:2283–2286. [Google Scholar]
- 6.Gao J, Ju KS, Yu X, Velasquez JE, Mukherjee S, Lee J, Zhao C, Evans BS, Doroghazi JR, Metcalf WW, van der Donk WA. Angew. Chem. Int. Ed. 2014;53:1334–1337. doi: 10.1002/anie.201308363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu XM, Doroghazi JR, Janga SC, Zhang JK, Circello B, Griffin BM, Labeda DP, Metcalf WW. Proc. Natl. Acad. Sci. USA. 2013;110:20759–20764. doi: 10.1073/pnas.1315107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiessl K, Roller A, Hammerschmidt F. Org. Biomol. Chem. 2013;11:7420–7426. doi: 10.1039/c3ob41574k. [DOI] [PubMed] [Google Scholar]
- 9.Circello BT, Eliot AC, Lee JH, van der Donk WA, Metcalf WW. Chem. Biol. 2010;17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bougioukou DJ, Mukherjee S, van der Donk WA. Proc. Natl. Acad. Sci. U. S. A. 2013;110:10952–10957. doi: 10.1073/pnas.1303568110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agarwal V, Peck SC, Chen JH, Borisova SA, Chekan JR, van der Donk WA, Nair SK. Chem. Biol. 2014;21:125–135. doi: 10.1016/j.chembiol.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim AD, Baker AS, Dunaway-Mariano D, Metcalf WW, Wanner BL, Martin BM. J. Bacteriol. 2002;184:4134–4140. doi: 10.1128/JB.184.15.4134-4140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peck SC, Kim SY, Evans BS, van der Donk WA. Medchemcomm. 2012;3:967–970. doi: 10.1039/C2MD20009K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao Z, Blodgett JA, Circello BT, Eliot AC, Woodyer R, Li G, van der Donk WA, Metcalf WW, Zhao H. J. Biol. Chem. 2008;283:23161–23168. doi: 10.1074/jbc.M801788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodyer RD, Li G, Zhao H, van der Donk WA. Chem. Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 16.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Chem. Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hausinger RP. Crit. Rev. Biochem. Mol. Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 18.Lloyd MD, Merritt KD, Lee V, Sewell TJ, Wha-Son B, Baldwin JE, Schofield CJ, Elson SW, Baggaley KH, Nicholson NH. Tetrahedron. 1999;55:10201–10220. [Google Scholar]
- 19.Van Lanen SG, Oh TJ, Liu W, Wendt-Pienkowski E, Shen B. J. Am. Chem. Soc. 2007;129:13082–13094. doi: 10.1021/ja073275o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooke HA, Guenther EL, Luo YG, Shen B, Bruner SD. Biochemistry. 2009;48:9590–9598. doi: 10.1021/bi901257q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.