

Abstract
We previously identified quinoline-based oligoamide helical foldamers and a trimeric macrocycle as selective ligands of DNA quadruplexes. Their helical structure may permit the targeting of the backbone loops and grooves of G-quadruplexes instead of the G-tetrads. Given the vast array of morphologies G-quadruplex structures can adopt, this may be a way to elicit sequence selective binding. Herein, we describe the design and synthesis of molecules based on macrocyclic and helically folded oligoamides. We tested their ability to interact with the human telomeric G-quadruplex and an array of promoter G-quadruplexes using Förster Resonance Energy Transfer (FRET) melting assay and single molecule FRET. Our results show that they constitute very potent ligands, comparable to the best reported in the literature. Their mode of interaction differs from that of traditional tetrad binders, opening avenues for the development of molecules specific for certain G-quadruplex conformations.
Keywords: G-Quadruplex, Foldamers, DNA, FRET, single molecule fluorescence
Introduction
Chromatin is organized into distinct regions which are defined by their biochemical environment. Intricate protein networks are involved in maintaining chromatin structure and function and DNA can adapt distinct secondary structures besides the standard B-helix,[1] which adds to the structural repertoire controlling the functionality of chromatin. Certain G-rich sequences can adopt supramolecular structures called G-quadruplexes, which comprise tetrads of Hoogsten hydrogen bonded guanines that stack via π–π interactions. These structures are stabilized by monovalent cations such as Na+ and K+ in the central electron-rich channel. The conformation of the strands and loops connecting the tetrads vary, depending on the primary nucleic acid sequence and factors such as strand number, salt composition and concentration.[2] Genome-wide analyses revealed that putative G-quadruplex sequences are abundant in the human genome, with an enrichment in promoter regions.[3] They are also found at telomeres, which contain the repetitive G-rich sequence (TTAGGG)n that can fold into G-quadruplexes.[4] This suggests, that these elements may play an important role in controlling gene expression[5] and telomere maintenance. In a seminal paper, Zahler et al. demonstrated the formation of G-quadruplexes from telomeric sequences in vitro, rendering them resistant to extension by the reverse transcriptase telomerase.[6] In addition, certain G-rich RNA can also fold into G-quadruplexes and these may be important for the control of gene expression (e.g. translation, unsilencing imprinted genes) and telomere maintenance. G-quadruplex interacting molecules can stabilize RNA G-quadruplexes, and this can lead to a down-regulation of translational activity, which may implicate them as regulatory elements.[7] RNA G-quadruplexes thus certainly constitute interesting structures with possible biological consequences.[8] Recent evidence supports the formation of G-quadruplexes in vivo, including nucleic acid pull-down strategies,[9] in vivo labelling and genome-wide sequencing with small molecules,[10] and the use of specific antibodies. A recent report describing the visualization of RNA G-quadruplexes in the cytoplasm of human cells may provide new avenues to study RNA-G-quadruplexes and their effect on RNA secondary structure. This approach can be used to monitor the effects of small molecules on RNA G-quadruplexes in cells.[7c, 11] Indeed, small molecules have proven to be formidable tools to study G-quadruplexes, and they can exert various effects on cells. These include effects on gene expression patterns,[5b] induction of telomere shortening[12] and uncapping[13] and induction of DNA damage.[10, 13b] Whereas the DNA damage response can be activated through telomere uncapping,[14] whole-genome sequencing of γH2A.X upon treatment with the G-quadruplex interactor pyridostatin revealed hotspots dispersed over genomic DNA that contain clusters of putative G-quadruplex forming sequences.[10] Given the possible involvement of G-quadruplexes in a plethora of biological processes, there is scope for the development of therapeutic agents based on G-quadruplex ligands, in order to interfere with these processes in pathological situations.[15]
Most of the molecules designed to interact with G-quadruplexes comprise an electron poor flat aromatic surface surrounded by cationic charges,[16] in order to target the external tetrads of the nucleic acid structure,[13b, 17] putting forward the notion that such small molecules should ideally be flat and aromatic in nature. However, an increasing number of ligands has been reported with alternative modes of interactions, for instance a chiral cyclic helicene[18], distamycin A and some of its derivatives which were shown by NMR to be quadruplex groove binders,[19] the loop-binder DODC,[20] the groove binder Toxapy,[21] binol derivatives,[22] supramolecular complexes,[23] and synthetic G-quartets.[24] In order to exploit such interactions further, foldamers emerge as an interesting class of compounds as they are able to adopt well-defined structures stabilized by non-covalent interactions mimicking the structures of biopolymers.[25] Foldamers that have been developed for biological applications such as G-quadruplexes binding are, for example, peptidic nucleic acids (PNA), peptoids and β, γ, δ, and ε peptides.[26] These molecules are small to medium sized, often resistant to proteolytic cleavage[27] and show good cell permeability.[28]
Previously, we reported on quinoline-based macrocycle 1 and dimeric and tetrameric foldamers 2 and 3 (Scheme 1) as G-quadruplex interacting molecules.[29] Macrocycle 1 was reported as a very potent ligand and falls into the category of planar aromatic compounds with positive cationic side chains. In contrast, tetrameric foldamer 3 is helical, i.e. non planar, yet it also displayed remarkable stabilization of the human telomeric G-quadruplex and the c-kit promoter G-quadruplex. These interactions were shown to depend on helix handedness as CD experiments revealed that they resulted in a preferred helix sense of 3. Subsequently, we dissected the interactions of aromatic oligoamide foldamers with nucleic acids using directed-DNA evolution against a helical cationic foldamer. We confirmed that G-quadruplexes stand as preferred targets and found that foldamers specifically interact with the backbones of G-quadruplexes (loops or grooves) as opposed to the top or bottom tetrads.[30] We also observed preference for DNA-quadruplex binding over RNA-quadruplex binding, and foldamer helix handedness and sequence dependence of the foldamer-quadruplex interaction.[30] Thus, macrocyclic and folded helical oligoamides selectively interact with G-quadruplexes[17c,29,30]
Scheme 1.

Macrocyles and foldamers synthesized for this study, derived from 8-amino-quinolinecarboxylic acids and 5-aminomethyl-pyridinecarboxylic acid. Some secondary amide structures are shown as cis conformers for clarity but in fact exist as trans conformers in the folded helices.
Inspired by earlier studies showing that producing derivatives of G-quadruplex ligands can generate better ligands than the lead structures[12,31] and that controlled folding of a small molecule can enhance G-quadruplex selectivity,[32] we endeavored to synthesize a new series of aromatic amide ligands consisting of nine helical foldamers and a macrocycle. We evaluated their ability to stabilize the human telomeric G-quadruplex and a number of promoter G-quadruplexes using Förster Resonance Energy Transfer (FRET) and investigated the interaction of two of the best foldamers further by single molecule FRET. This method’s merit is to allow a comparison to other well established G-quadruplex ligands reported in the literature. We demonstrate that our new molecules interact specifically with G-quadruplex DNA over duplex DNA, and compare favorably with the best G-quadruplex stabilizing molecules reported in the literature so far.[13b, 29] Interestingly, we find that some of the helical foldamers we synthesized show a better stabilization potential than macrocycles in this family.
Results and Discussion
Synthesis and Design of Small Molecules
We previously reported the syntheses of macrocycle 1, of the dimeric and tetrameric foldamers 2 and 3[29] and of the foldamers 4[33] and 5.[34] All are based on quinolinecarboxamide sequences. Macrocycle 1 and dimer 2 adopt a flat conformation,[35] whereas tetramers 3 and 5 and octamer 4 form helices spanning 1.5 turn for the former two and over three turns for the latter. For the purpose of this study, we synthesized another macrocycle 6 to compare it to the lead macrocycle 1. Macrocycle 6 has the same ring atom number as 1, but one of the quinolines was replaced by an aminomethylpyridine derivative which was prepared using a previously described procedure.[34,36] The preparation of 1 involved the low yielding direct cyclotrimerization of a monomer precursor, a scheme that only allows to assemble three copies of the same monomer. In contrast, the preparation of macrocycle 6 involved the high yielding cyclization of an isolated trimeric non-cyclic precursor. This new scheme is compatible with the sequential incorporation of different monomers. The yield of cyclization is enhanced by the high nucleophilicity of the benzylic amine involved in this step (see Supporting Information). The exact conformation of the resulting macrocycle has not been investigated. It may adopt a planar conformation but a distortion at the additional sp3 centre may also favour non planer states, as observed in a related herringbone helical foldamer.[36]
Inspired by the previously reported promising ability of the tetrameric foldamer 3[29] to selectively stabilize G-quadruplex DNA, we varied its C-terminal group R2 and the nature of the side chains R1 to generate tetrameric foldamers 7–9 in order to assess their effects on G-quadruplex stabilization. The Boc-protected precursor of ester 7 was converted to the precursor of acid 3 by saponification, and also to the precursor of secondary amide 8 using excess methylamine (see Supporting Information). Side chain deprotection of these precursors using trifluoroacetic acid (TFA) yielded 3, 7 and 8. Compound 9 was synthesized by converting the propylammonium side chains of 7 to guanidinium using 1-H-pyrazol-1-carboxamide hydrochloride. The rationale behind this was to create a side chain with a delocalized cationic charge, which may allow the formation of an arginine fork motif with the phosphate groups of the nucleic acid backbones of the loops and grooves of G-quadruplexes.[37]
Next, we synthesized tetramer 10 with alternating quinoline and methylaminopyridine building blocks using simple coupling strategies (see Supporting Information). This type of backbone can fold in a similar manner as homo-quinoline multimers, but with decreased stability. We did not synthesize homo-methylaminopyridine multimers as these species do not fold in organic solvents[36] or water.[34] In order to assess the effect of foldamer length on G-quadruplex recognition, we also synthesized the octameric foldamers 11 and 12 (see Supporting Information), which carry cationic and anionic side chains to introduce a potential level of selectivity. These two molecules are made of quinoline building blocks but differ in the sequence of side chain substitution. These synthetic procedures highlight how complex foldamers can be synthesized in a few steps in good yield.
FRET-Melting Analyses
We assessed the interactions of the molecules with G-quadruplex DNA employing a Förster Resonance Energy Transfer (FRET) melting assay previously established by Mergny and Maurizot.[38] This assay is widely established in the field to assess the potency of G-quadruplex ligands and thus constitutes a good method to compare novel molecules with the plethora of ligands reported in the literature. It proved to be an efficient method in order to investigate our family of foldamers as G-quadruplex ligands in comparison with other studies. We focused on DNA quadruplex sequences and used the human telomeric G-quadruplex (H-telo)[6] and the sequences of the promoter quadruplexes of a selection of genes, namely c-kit (which contains the two G-quadruplex sequences c-kit1[39] and c-kit2[40]), c-myc,[5b, 41] bcl2[42] and k-ras.[43] These different sequences differ in composition, in the number of nucleotides involved in each loop, and adopt different quadruplex conformations, potentially allowing differential recognition by small molecules that do not target the tetrads per se. As a control we used a sequence that forms a duplex DNA in solution. We labeled each of these sequences with 6-carboxyfluorescein (FAM) and 6-carboxytetramethylrhodamine (TAMRA) at the 5′ and 3′ ends, respectively. Melting of the G-quadruplexes results in an increase of the distance between the fluorophores, which can be measured by changes in the FRET signal. We measured the changes in melting temperature (ΔTm) upon varying the concentration of added foldamer or macrocyle. The ΔTm values recorded in presence of 1 µM of ligand are summarized in Table 1. The concentrations of ligands required to produce the maximal stabilization of the DNA structures are summarized in Table 2 and the full melting profiles can be found in the Supporting Information.
Table 1.
ΔTm in 60 mM K+ at 1 µM compound in the FRET-melting assays.
| Ligand | duplex[a] | H-Telo | c-kit1 | c-kit2 | c-myc | bcl2 | k-ras |
|---|---|---|---|---|---|---|---|
| ΔTm max in °C[b] |
32.8 ±1.2 |
36.1 ±1.3 |
41.0 ±1.5 |
22.8 ±0.7 |
16.2 ±1.1 |
32.1 ±0.9 |
49.6 ±1.4 |
| 1 | 0.7 | 24.0 | 16.0 | 21.4 | 6.2 | 14.6 | 7.3 |
| 2 | 0.0 | 3.6 | 10.1 | 4.4 | 2.1 | 0.5 | 2.8 |
| 3 | 1.1 | 22.4 | 15.1 | 17.6 | 14.3 | 18.9 | 20.8 |
| 4 | 3.9 | 36.5 | 39.1 | 21.5 | 17.0 | 30.7 | 48.4 |
| 5 | 0.0 | 0.5 | 2.7 | 0.0 | 0.2 | 0.0 | 0.0 |
| 6 | 1.6 | 29.0 | 13.4 | 4.8 | 16.4 | 13.4 | 10.5 |
| 7 | 1.3 | 36.4 | 37.9 | 16.8 | 16.4 | 30.8 | 24.1 |
| 8 | 0.0 | 35.1 | 24.0 | 15.9 | 17.0 | 19.9 | 14.2 |
| 9 | 0.6 | 22.1 | 20.2 | 9.9 | 8.5 | 12.3 | 18.4 |
| 10 | 3.1 | 34.8 | 25.8 | 21.0 | 16.5 | 27.7 | 26.8 |
| 11 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.2 |
| 12 | 0.0 | 2.8 | 6.1 | 2.8 | 1.4 | 0.2 | 1.8 |
Duplex formed by two complementary strands linked via hexaethyleneglycol (HEG) loop.
This value corresponds to 95.5°C - Tm (in °) for quadruplex alone as 95.5°C is the maximal temperature measurable in this assay.
Table 2.
Concentration (in µM) required for maximal stabilization in the FRET- melting assays.
| Ligand | duplex[a] | H-Telo | c-kit1 | c-kit2 | c-myc | bcl2 | k-ras |
|---|---|---|---|---|---|---|---|
| 1 | 6.3 | 4.1 | 2.9 | 1.6 | 8.1 | 4.6 | 6.5 |
| 2 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| 3 | 9.7 | 4.4 | 4.3 | 2.9 | 2.1 | 4.7 | 9.0 |
| 4 | 6.10 | 0.36 | 0.34 | 0.8 | 0.34 | 0.56 | 0.69 |
| 5 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| 6 | > 10 | 1.41 | 6.01 | 5.3 | 1.32 | 2.29 | > 10 |
| 7 | 4.70 | 0.92 | 1.94 | 2.0 | 1.07 | 1.23 | 6.83 |
| 8 | 5.23 | 1.25 | 3.69 | 3.1 | 0.85 | 3.09 | 6.42 |
| 9 | > 10 | 2.4 | 3.8 | 5.4 | 2.2 | 2.1 | 4.2 |
| 10 | 6.77 | 1.18 | 4.39 | 1.3 | 0.77 | 1.58 | 5.57 |
| 11 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| 12 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
Duplex formed by two complementary strands linked via hexaethyleneglycol (HEG) loop.
Because of their differences in sequence and structure, the various G-quadruplexes possess different Tm values. The maximal ΔTm values that can be envisaged thus also differ substantially, from 16.2°C for c-myc to 49.6 for k-ras (Table 1). Changes in melting temperature as presented below provide some information about the potential of the ligands to interact with G-quadruplexes. Nevertheless, the differences mentioned above along with the fact that G-quadruplexes may change conformation upon binding a ligand and not another ligand call for great caution when drawing comparisons. In addition, interactions between ligands and quadruplexes that do not lead to quadruplex stabilization are overlooked in these assays. As a complement to ΔTm values, we attempted to measure dissociation constants (Kd) using surface plasmon resonance (SPR). However, SPR measurements with DNA sequences attached to the chip failed because of non-specific interactions between the foldamers and the substrate of the chip. Better results may be obtained by attaching the foldamers to the chip.30 But slow dissociation kinetics, possible quadruplex aggregation and conformational changes complicate the measurements which cannot be fitted to a simple 1:1 binding model. Nevertheless, it can reasonably be inferred from the ΔTm values presented below that the lowest dissociation constants are in the low or sub-micromolar range. Accurate determination of binding constants is possible using specific techniques that are beyond the scope of the present study.44 It should also be kept in mind that neither Kd nor ΔTm values provide sufficient information to ascertain-quadruplex binding in cells and the triggering of a biological response.
The results show that all fully cationic oligomers, be they cyclic (1, 6), flat and non cyclic (2), or helical (3, 4, 7–10), be they short or long (4), bearing ammonium or guanidinium (9) side chains, show from good to high stabilization potential of DNA-quadruplexes and selectivity against duplex DNA. In contrast, the presence of anionic residues alternating with cationic residues (5, 11) or clustered at one end of a sequence (12) is strongly detrimental to any kind of DNA binding. Earlier studies have shown that the high conformational stability of the helices do not depend on the sequence of monomers.[34] Thus different behaviors cannot be assigned to different foldamer conformations that would result from a change of sequence. Instead, electrostatic repulsions between foldamer and DNA negative charges are probably responsible for this dramatic effect. It is worth to note that octamer 12 contains a tetrameric cationic N-terminal segment which is a good ligand when taken independently. Nevertheless, this compound shows very small G-quadruplex stabilization. The neighboring negative charges do not allow strong binding of the cationic segment.
The two macrocycles 1 and 6 both strongly stabilize G-quadruplexes and have minimal effects on duplex DNA, confirming earlier results on the stabilization of h-telo by 1.[29] The sp3 center of 6 appears not to be an impediment. Data for the two compounds are overall comparable but some notable differences are worth pointing out. Macrocycle 1 displayed a remarkable stabilization potential for c-kit2 whereas 6 is limited in this respect. Conversely, 6 fares much better than 1 and seems to behave like helical oligomers concerning the stabilization of c-myc. This might be related to the fact that c-myc adopts an unusual propeller-type parallel-stranded conformation.[45]
Upon comparing flat dimer 2, helical tetramer 3 and helical octamer 4, a major effect of length is evidenced. Compound 2 exhibited weak stabilization potentials for all G-quadruplex targets investigated. Tetramer 3 displayed very good stabilization properties towards the same target nucleic acids and matches or surpasses macrocycle 1 in most cases. Strikingly, octamer 4 stabilizes all the G-quadruplex targets to the maximal possible ΔTm at 1 µM compound (Table 1) and the concentration required to stabilize these targets is between 0.36 and 0.80 µM (Table 2), whereas the duplex stabilization is still negligible. This makes this octameric foldamer by far the most potent G-quadruplex stabilizing ligand in the series, comparable to the most potent small molecules reported in the literature.[13b]
Changing the C-terminal negatively charged carboxylate function of 3 to a neutral ester (7) or methyl-amide (8) significantly enhances G-quadruplex stabilization potential in almost all cases, confirming the detrimental effect of negative charges observed in the side chains. Interestingly, the minor structural difference between 7 and 8 nevertheless results in some substantial variations of ΔTm (see c-kit1, Bcl2 and k-ras entries in Table 1). The ester appears to be more efficient than the methyl-amide.
The introduction of guanidinium side chains on foldamer 9 reduced G-quadruplex targeting efficiency despite their ability to form salt bridges with phosphate ions enhanced by bidentate hydrogen bonding. However, the DNA-melting profiles in presence of 9 (see Supporting Information) show that this foldamer exhibits a formidable selectivity for G-quadruplexes over duplex DNA, even when maximal stabilization occurs above 1 µM. In this respect, 9 is more selective than all other foldamers and macrocycles of this family.
To finish, tetramer 10 has G-quadruplex stabilization properties similar to that of tetramer 7. Both are methyl esters and bear four ammonium side chains. But 10 is much more flexible and not expected to fold well due to its aminomethyl-pyridine units.[34] Compared to quinoline rings, these units possess a reduced surface for aromatic stacking. Compound 10 also has an N-terminal ammonium function, but these features have weak or compensating effects.
Altogether, these FRET-melting results demonstrate that we improved on the G-quadruplex stabilization potential of the lead compounds 1 and 3. We also showed the potential of some selectivity of helical oligoamides for various G-quadruplex targets. This shows that these foldamers constitute a very potent family of G-quadruplex ligands.
Single Molecule FRET Analyses
To further dissect the conformations and dynamics of G-quadruplex DNA with foldamers and to get an insight into the binding mode, we investigated the interactions of foldamers 3 and 4 with the human telomeric G-quadruplex by single molecule FRET. We tethered biotinylated DNA onto a PEG-passivated quartz surface by utilizing the specific biotin- neutravidin interaction, as depicted in Figure 1.
Figure 1.

Experimental scheme for single molecule FRET.
This technique allows the monitoring three different conformational states of the H-telo sequence: unfolded U, and resolvable folded conformations F1 and F2 which show FRET efficiencies of E=0.43, 0.63 and 0.80, respectively. We reported previously that macrocycle 1 binds tightly to an unfolded telomeric strand even in the absence of K+ and selectively stabilizes conformation F1 over the naturally favored conformation F2 of the human telomeric G-quadruplex.[17c] Unlike macrocycle 1, the tetrameric foldamer 3 is unable to fold the human telomeric sequence into a G-quadruplex in the absence of K+ (Figure 2). The octameric foldamer 4 equally does not induce a folding of the sequence into a G-quadruplex (data not shown).
Figure 2.
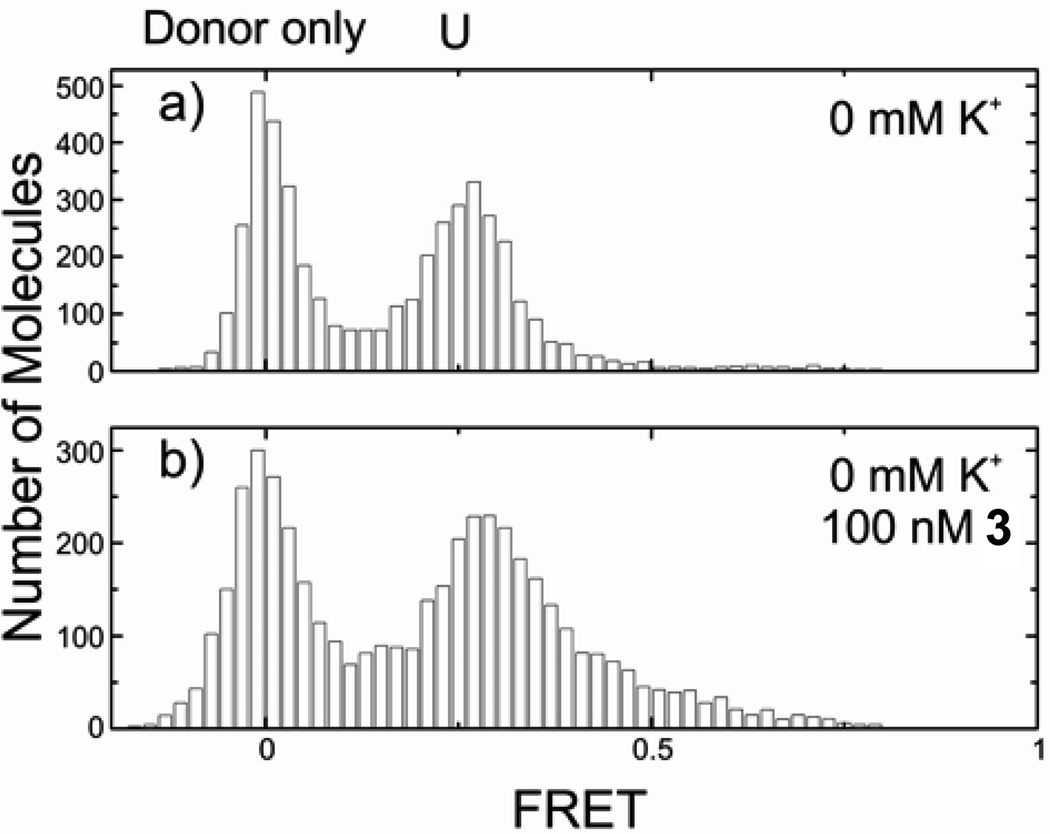
FRET histogram of human telomeric quadruplex (a) unfolded in absence of K+ and (b) upon addition of 100 nM of tetramer 6.
Addition of 10 mM K+ to the single stranded human telomeric sequence leads to the coexistence of two folded conformations F1 and predominantly F2 (Figure 3a–b). Further addition of 100 nM 3 induced a rapid increase in the proportion of F1 (Figure 3c). To a lesser extent, 4 behaved in the same way (data not shown). After 20 minutes an equilibrium was reached with both foldamers (Figure 3d–e). Strikingly, unlike previously observed for macrocycle 1, the folded state F2 is not completely shifted to F1 with the tetrameric foldamer 3 (Figure 3d). This effect is even more pronounced for the octameric foldamer 4 (Figure 3e). When free foldamer 4 and K+ are removed (Figure 3f), a mixed population of F1 and F2 remains present, suggesting a strong stabilizing effect of still associated foldamer 4 with the H-Telo G-quadruplex. Altogether, these data suggest a remarkably different mode of interaction between the macrocycle and the helical foldamers tested, also highlighting a change in dynamics of interaction between the tetrameric and octameric foldamer with the human telomeric G-quadruplex. This is in agreement with the very shape of the molecules which allow (for 1) or not (for 4) direct stacking on top of the G-tetrads. The different behavior of the helices and of the macrocycle is also in agreement with earlier circular dichroism data which suggested that 1 stabilizes the anti-parallel conformation of H-Telo,[29] and that 4 stabilizes the parallel conformation of all G-quadruplex aptamer sequences to which it was exposed.[30]
Figure 3.

FRET histogram of H-Telo (a) in absence of K+ in the unfolded state, (b) folded in F1 and F2 states upon addition of 10 mM K+, (c) addition of tetramer 3 after 2 min, (d) 20 min, (e) addition of octamer 4 after 20 min, (f) 30 min. after removing free K+ and 4.
Conclusions
We introduced a facile synthesis of a variety of foldamers based on helical oligoamides, which enabled us to generate a library of small molecules in a few simple synthetic steps. Most of the molecules we generated showed very good stabilization potentials for a variety of G-quadruplex tested by FRET-melting experiments. We established helical oligoamides as a potent class of G-quadruplex interacting molecules and showed that their mode of interaction is different from our previously reported macrocycle, which presumably interacts with the tetrads of the target nucleic acid structure. The helical foldamers thus possibly interact with the backbones loops and grooves G-quadruplexes.[17c, 30] The nature of the side chains as well as foldamer length play an important role in the stabilization potential of these foldamers with G-quadruplex nucleic acids. We found that the octameric foldamer 4 was even more potent as a G-quadruplex stabilizing compound than macrocycle 1, placing it in the range of the best molecules reported in the literature so far. Given the relatively easy synthetic procedure and its modular nature, this family of ligands can easily be expanded further in order to generate even more potent molecules. Since their mode of interaction with G-quadruplex DNA differs to that of traditional tetrad binders, this opens new avenues for the further development of molecules specific for certain G-quadruplex conformations. For this purpose, a detailed NMR and crystallographic structural investigation of a foldamer-quadruplex complex would be most helpful and is currently in progress. Preliminary results came from a co-crystal between tetrameric foldamer 3 and the model DNA sequence G4T4G4 the structure of which showed no contact between the foldamer and G-tetrads.
It is noteworthy that foldamer/G-quadruplex adducts may trigger a different signaling response in a cellular environment compared to traditional tetrad binders providing a tool to shed light on G-quadruplex location and function in the genome. In addition, with the advent of the development of G-quadruplex interacting small molecules as therapeutic agents,[15] and their good cell penetration ability,[28] these small molecules can potentially be used in biological assays and exploited for therapeutic development.
Experimental Section
FRET melting experiments
Oligonucleotides were initially dissolved as a 100 µM stock solution in MilliQ water; further dilutions were carried out in 60 mM potassium cacodylate buffer, pH 7.4 and FRET experiments were carried out with a 200 nM oligonucleotide solution. Seven DNA oligonucleotides were used in these experiments which were dual fluorescently labelled. K-ras is a dual-labelled 32-mer oligonucleotide comprising a quadruplex forming region in the promoter region of the human K-ras gene, 5′-FAM-AGG GCG GTG TGG GAA GAG GGA AGA GGG GGA GG-TAMRA-3′. c-kit1 was a dual-labelled 21-mer oligonucleotide comprising one of the quadruplex forming regions in the promoter region of the human c-kit oncogene, 5′-FAM-GGG AGG GCG CTG GGA GGA GGG-TAMRA-3′. H-Telo was a dual-labelled 21-mer oligonucleotide comprising the minimum human telomeric G-overhang sequence required to fold into an intramolecular quadruplex, 5′-FAM-GGG TTA GGG TTA GGG TTA GGG-TAMRA-3′. C-kit2 is a dual-labelled 20-mer oligonucleotide comprising one of the quadruplex forming regions in the promoter region of the human c-kit oncogene, 5′-FAM-GGG CGG GCG CGA GGG AGG GG-TAMRA-3′. C-myc is a dual-labelled 22-mer oligonucleotide comprising one of the quadruplex forming regions in the promoter region of the human c-myc oncogene, 5′-FAM-TGA GGG TGG GTA GGG TGG GTA A-TAMRA-3′. Bcl2 is a dual-labelled 27-mer oligonucleotide comprising the quadruplex forming region in the promoter of the human bcl2 gene. 5′ CGG-GCG-CGG-GAG-GAA-GGG-GGC-GGG-AGC 3′ ds-DNA was a dual-labelled 20-mer oligonucleotide comprising a self-complementary sequence with a central polyethylene glycol linker able to fold into a hairpin, 5′-FAM-TAT AGC TAT A HEG TAT AGC TAT A-TAMRA-3′. The donor fluorophore was 6-carboxyfluorescein, FAM, and the acceptor fluorophore was 6-carboxytetramethylrhodamine, TAMRA. Dual-labelled DNA was annealed at a concentration of 400 nM by heating at 94 °C for 10 min followed by cooling to room temperature at a rate of 0.1 K/min. 96-well plates were prepared by addition of 50 µl of the annealed DNA solution to each well, followed by 50 µl of a solution of the respective molecule at an appropriate concentration. Measurements were made in triplicate with an excitation wavelength of 483 nm and a detection wavelength of 533 nm using a LightCycler® 480 System RT-PCR machine (Roche). Final analysis of the data was carried out using OriginPro 7.5 data analysis and graphing software (OriginLab®).
Single Molecule FRET
Single Molecule FRET studies were carried out as described previously.[17c] In brief, prism-type total internal reflection fluorescence (TIRF) microscopy was performed with 532 nm laser excitation to detect the conformation changes of surface immobilized human telomeric DNA molecules, labeled with a Cy5-TMR FRET pair. The detected intensity of TMR and Cy5 from each individual h-telo molecule was used to derive the FRET efficiency, and combined to obtain the FRET histograms shown. Other experimental parameters remained identical to our previous single molecule FRET study on H-telo.
Supplementary Material
Acknowledgements
We thank the “Cancer Research UK” for doctoral funding (SM) and “Association pour la recherche sur le cancer” for a postdoctoral fellowship (KLR). The Balasubramanian laboratory is core-funded by a programme grant from Cancer Research UK. TH acknowledges the NIH grant GM065367.
Footnotes
Supporting information for this article is available.
References
- 1.a) Ghosh A, Bansal M. Acta Crystallogr.D. 2003;59:620–626. doi: 10.1107/s0907444903003251. [DOI] [PubMed] [Google Scholar]; b) Zhao J, Bacolla A, Wang G, Vasquez KM. Cell Mol. Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis JT. Angew. Chem. Int. Ed. 2004;43:668–698. doi: 10.1002/anie.200300589. Angew. Chem. Int. Ed.2004,116, 684–716. [DOI] [PubMed] [Google Scholar]
- 3.a) Huppert JL, Balasubramanian S. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Todd AK, Johnston M, Neidle S. Nucleic Acids Res. 2005;33:2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Parkinson GN, Lee MP, Neidle S. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]; b) Wang Y, Patel DJ. J. Mol. Biol. 1993;234:1171–1183. doi: 10.1006/jmbi.1993.1668. [DOI] [PubMed] [Google Scholar]
- 5.a) Cogoi S, Xodo LE. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Siddiqui–Jain A, Grand CL, Bearss DJ, Hurley LH. Proc. Natl. Acad. Sci.(USA) 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zahler AM, Williamson JR, Cech TR, Prescott DM. Nature. 1991;350:718–720. doi: 10.1038/350718a0. [DOI] [PubMed] [Google Scholar]
- 7.a) Gomez D, Guedin A, Mergny JL, Salles B, Riou JF, Teulade–Fichou MP, Calsou P. Nucleic Acids Res. 2010;38:7187–7198. doi: 10.1093/nar/gkq563. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kumari S, Bugaut A, Huppert JL, Balasubramanian S. Nat.Chem. Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Di Antonio M, Biffi G, Mariani A, Raiber E-A, Rodriguez R, Balasubramanian S. Angew. Chem. Int. Ed. 2012;51:11073–11078. doi: 10.1002/anie.201206281. Angew. Chem.2012, 124, 11235–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For a review, see: Millevoi S, Moine H, Vagner S. Wiley Interdiscip Rev RNA. 2012;3:495–507. doi: 10.1002/wrna.1113.
- 9.Müller S, Kumari S, Rodriguez R, Balasubramanian S. Nat. Chem. 2010;2:1095–1098. doi: 10.1038/nchem.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP. Nat. Chem. Biol. 2012;8:301–310. doi: 10.1038/nchembio.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Biffi G, Di Antonio M, Tannahill D, Balasubramanian S. Nat. Chem. 2014;6:75–80. doi: 10.1038/nchem.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Henderson A, Wu Y, Huang YC, Chavez EA, Platt J, Johnson FB, Brosh RM, Jr, Sen D, Lansdorp PM. Nucleic Acids Res. 2014;42:860–869. doi: 10.1093/nar/gkt957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müller S, Sanders DA, Di Antonio M, Matsis S, Riou JF, Rodriguez R, Balasubramanian S. Org. Biomol. Chem. 2012;10:6537–6546. doi: 10.1039/c2ob25830g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Gomez D, Wenner T, Brassart B, Douarre C, O'Donohue MF, El Khoury V, Shin-Ya K, Morjani H, Trentesaux C, Riou JF. J. Biol. Chem. 2006;281:38721–38729. doi: 10.1074/jbc.M605828200. [DOI] [PubMed] [Google Scholar]; b) Rodriguez R, Müller S, Yeoman JA, Trentesaux C, Riou JF, Balasubramanian S. J Am Chem Soc. 2008;130:15758–15759. doi: 10.1021/ja805615w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. J. Med. Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 15.a) Balasubramanian S, Hurley LH, Neidle S. Nat. Rev. Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller KM, Rodriguez R. Expert Rev. Clin. Pharmacol. 2011;4:139–142. doi: 10.1586/ecp.11.4. [DOI] [PubMed] [Google Scholar]
- 16.Monchaud D, Teulade-Fichou MP. Org. Biomol. Chem. 2008;6:627–636. doi: 10.1039/b714772b. [DOI] [PubMed] [Google Scholar]
- 17.a) De Cian A, Delemos E, Mergny JL, Teulade-Fichou MP, Monchaud D. J. Am. Chem. Soc. 2007;129:1856–1857. doi: 10.1021/ja067352b. [DOI] [PubMed] [Google Scholar]; b) Sparapani S, Haider SM, Doria F, Gunaratnam M, Neidle S. J. Am. Chem. Soc. 2010;132:12263–12272. doi: 10.1021/ja1003944. [DOI] [PubMed] [Google Scholar]; c) Jena PV, Shirude PS, Okumus B, Laxmi-Reddy K, Godde F, Huc I, Balasubramanian S, Ha T. J. Am. Chem. Soc. 2009;131:12522–12523. doi: 10.1021/ja903408r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Laguerre A, Desbois N, Stefan L, Richard P, Gros CP, Monchaud D. ChemMedChem. 2014 doi: 10.1002/cmdc.201300526. [DOI] [PubMed] [Google Scholar]; e) Bejugam M, Gunaratnam M, Müller S, Sanders DA, Sewitz S, Fletcher JA, Neidle S, Balasubramanian S. ACS Med. Chem. Lett. 2010;1:306–310. doi: 10.1021/ml100062z. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chung WJ, Heddi B, Hamon F, Teulade-Fichou MP, Phan AT. Angew. Chem. Int. Ed. 2014;53:999–1002. doi: 10.1002/anie.201308063. Angew. Chem.2014, 126, 1017–1020. [DOI] [PubMed] [Google Scholar]
- 18.Shinohara K, Sannohe Y, Kaieda S, Tanaka K, Osuga H, Tahara H, Xu Y, Kawase T, Bando T, Sugiyama H. J. Am. Chem. Soc. 2010;132:3778–3782. doi: 10.1021/ja908897j. [DOI] [PubMed] [Google Scholar]
- 19.a) Martino L, Virno A, Pagano B, Virgilio A, Di Micco S, Galeone A, Giancola C, Bifulco G, Mayol L, Randazzo A. J. Am. Chem. Soc. 2007;129:16048–16056. doi: 10.1021/ja075710k. [DOI] [PubMed] [Google Scholar]; b) Cosconati S, Marinelli L, Trotta R, Virno A, De Tito S, Romagnoli R, Pagano B, Limongelli V, Giancola C, Baraldi PG, Mayol L, Novellino E, Randazzo A. J. Am. Chem. Soc. 2010;132:6425–6433. doi: 10.1021/ja1003872. [DOI] [PubMed] [Google Scholar]
- 20.Chen Q, Kuntz ID, Shafer RH. Proc. Natl. Acad. Sci.(USA) 1996;93:2635–2639. doi: 10.1073/pnas.93.7.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamon F, Largy E, Guedin-Beaurepaire A, Rouchon-Dagois M, Sidibe A, Monchaud D, Mergny JL, Riou JF, Nguyen CH, Teulade-Fichou MP. Angew. Chem. Int. Ed. 2011;50:8745–8749. doi: 10.1002/anie.201103422. Angew. Chem.2011, 123, 8904–8908. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H, Xiang JF, Hu HY, Li L, Jin X, Liu Y, Li PF, Tang Y, Chen CF. Biochemistry. 2010;49:10351–10353. doi: 10.1021/bi1001773. [DOI] [PubMed] [Google Scholar]
- 23.Zhao C, Geng J, Feng L, Ren J, Qu X. Chemistry Eur. J. 2011;17:8209–8215. doi: 10.1002/chem.201100272. [DOI] [PubMed] [Google Scholar]
- 24.Haudecoeur R, Stefan L, Denat F, Monchaud D. J. Am. Chem. Soc. 2013;135:550–553. doi: 10.1021/ja310056y. [DOI] [PubMed] [Google Scholar]
- 25.Guichard G, Huc I. Chem. Commun. 2011;47:5933–5941. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]
- 26.For reviews, see: Horne WS. Expert Opin. Drug. Discov. 2011;6:1247–1262. doi: 10.1517/17460441.2011.632002. Panyutin IG, Onyshchenko MI, Englund EA, Appella DH, Neumann RD. Curr. Pharm. Des. 2012;18:1984–1991. doi: 10.2174/138161212799958440.
- 27.Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, Nielsen PE. Biochem. Pharmacol. 1994;48:1310–1313. doi: 10.1016/0006-2952(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 28.Okuyama M, Laman H, Kingsbury SR, Visintin C, Leo E, Eward KL, Stoeber K, Boshoff C, Williams GH, Selwood DL. Nat. Methods. 2007;4:153–159. doi: 10.1038/nmeth997. [DOI] [PubMed] [Google Scholar]
- 29.Shirude PS, Gillies ER, Ladame S, Godde F, Shin-Ya K, Huc I, Balasubramanian S. J. Am. Chem. Soc. 2007;129:11890–11891. doi: 10.1021/ja073775h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delaurière L, Dong Z, Laxmi-Reddy K, Godde F, Toulmé JJ, Huc I. Angew. Chem. Int. Ed. 2012;51:473–477. doi: 10.1002/anie.201106208. 2012, 124, 488–492. [DOI] [PubMed] [Google Scholar]
- 31.Lombardo CM, Welsh SJ, Strauss SJ, Dale AG, Todd AK, Nanjunda R, Wilson WD, Neidle S. Bioorg. Med. Chem. Lett. 2012;22:5984–5988. doi: 10.1016/j.bmcl.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 32.Müller S, Pantos GD, Rodriguez R, Balasubramanian S. Chem. Commun. 2009:80–82. doi: 10.1039/b816861j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillies ER, Deiss F, Staedel C, Schmitter JM, Huc I. Angew. Chem. Int. Ed. 2007;46:4081–4084. doi: 10.1002/anie.200700301. Angew. Chem.2007, 119, 4159–4162. [DOI] [PubMed] [Google Scholar]
- 34.Baptiste B, Douat-Casassus C, Laxmi-Reddy K, Godde F, Huc I. J. Org. Chem. 2010;75:7175–7185. doi: 10.1021/jo101360h. [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, Léger JM, Guionneau P, Huc I. Org. Lett. 2004;6:2985–2988. doi: 10.1021/ol0488643. [DOI] [PubMed] [Google Scholar]
- 36.Delsuc N, Godde F, Kauffmann B, Léger JM, Huc I. J. Am. Chem. Soc. 2007;129:11348–11349. doi: 10.1021/ja074285s. [DOI] [PubMed] [Google Scholar]
- 37.Calnan BJ, Tidor B, Biancalana S, Hudson D, Frankel AD. Science. 1991;252:1167–1171. doi: 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 38.Mergny JL, Maurizot JC. ChemBioChem. 2001;2:124–132. doi: 10.1002/1439-7633(20010202)2:2<124::AID-CBIC124>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 39.Phan AT, Kuryavyi V, Burge S, Neidle S, Patel DJ. J. Am. Chem. Soc. 2007;129:4386–4392. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.a) Rangan A, Fedoroff OY, Hurley LH. J. Biol. Chem. 2001;276:4640–4646. doi: 10.1074/jbc.M005962200. [DOI] [PubMed] [Google Scholar]; b) Seenisamy J, Rezler EM, Powell TJ, Tye D, Gokhale V, Joshi CS, Siddiqui- A, Hurley LH. J. Am. Chem. Soc. 2004;126:8702–8709. doi: 10.1021/ja040022b. [DOI] [PubMed] [Google Scholar]
- 42.a) Dai J, Dexheimer TS, Chen D, Carver M, Ambrus A, Jones RA, Yang D. J. Am. Chem. Soc. 2006;128:1096–1098. doi: 10.1021/ja055636a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Agrawal P, Lin C, Mathad RI, Carver M, Yang D. J. Am. Chem. Soc. 2014;136:1750–1753. doi: 10.1021/ja4118945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cogoi S, Paramasivam M, Spolaore B, Xodo LE. Nucleic Acids Res. 2008;36:3765–3780. doi: 10.1093/nar/gkn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koirala D, Dhakal S, Ashbridge B, Sannohe Y, Rodriguez R, Sugiyama H, Balasubramanian S, Mao H. Nat. Chem. 2001;3:782–787. doi: 10.1038/nchem.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phan AT, Modi YS, Patel DJ. J. Am. Chem. Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.