

Abstract
The rapid technological developments following the Human Genome Project have made possible the availability of personalized genomes. As the focus now shifts from characterizing genomes to making personalized disease associations, in combination with the availability of other omics technologies, the next big push will be not only to obtain a personalized genome, but to quantitatively follow other omics. This will include transcriptomes, proteomes, metabolomes, antibodyomes, and new emerging technologies, enabling the profiling of thousands of molecular components in individuals. Furthermore, omics profiling performed longitudinally can probe the temporal patterns associated with both molecular changes and associated physiological health and disease states. Such data necessitates the development of computational methodology to not only handle and descriptively assess such data, but also construct quantitative biological models. Here we describe the availability of personal genomes and developing omics technologies that can be brought together for personalized implementations and how these novel integrated approaches may effectively provide a precise personalized medicine that focuses on not only characterization and treatment but ultimately the prevention of disease.
Introduction
With the advent of high-throughput technologies genomic science has experienced great leaps, rapidly expanding its domain beyond the characterization of short genomic reads in the early days of sequencing to the possibility of obtaining personalized genomes, once considered the holy grail of genomic methodology and technology development. The value of personalized genomic analysis, and evaluation of variant associations to disease, is becoming more apparent, even spurring directly to consumer implementations. Further developments in the last few years now lead to a more ambitious goal: the longitudinal monitoring of multiple omics components in individuals and the characterization of the molecular changes associated with disease onset in individuals, at an unprecedented level. In this review we describe technological and methodological developments in personal genomics, and the new promise of multiple omics profiling, including transcriptomes, proteomes, metabolomes, autoantibodyomes and so forth, (sample omics analysis workflows shown in Figures 1–4). We then discuss a framework on how such data may be integrated with a view towards the application of a personalized precise and preventive medicine, and describe an implementation of this approach. The technological developments and methodology allow for inroads into the future of quantitative personal medicine, which we can now plan carefully by taking into account not only the scientific developments that need to be implemented, but also the social implications coupled to ethical and legal considerations.
Figure 1. Genomic variants.
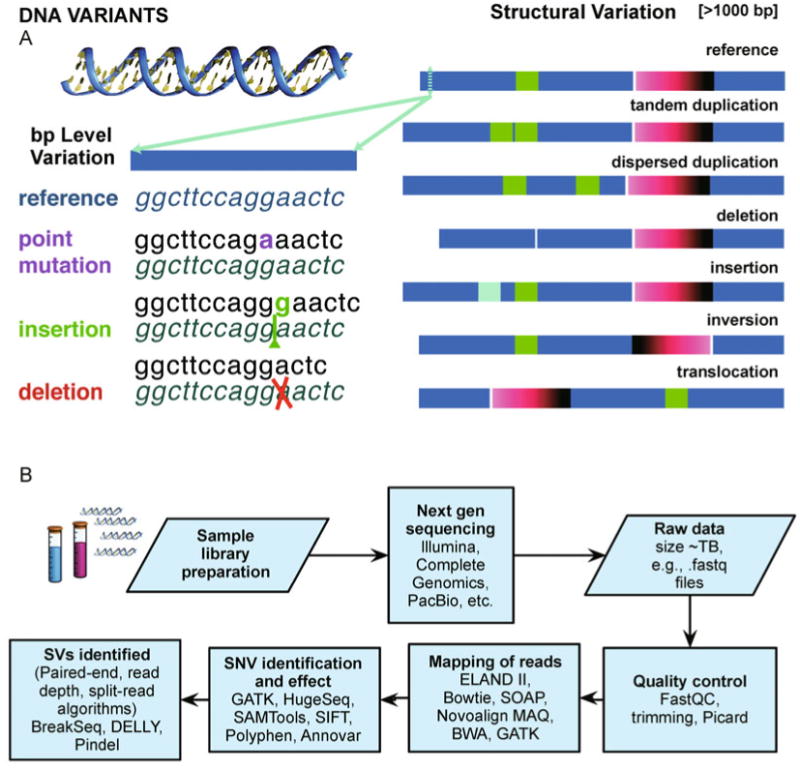
(A) Variation in the human genome. The personal genomic code can differ from the published reference genome. Basic examples of variation are shown on a single or few base variants (e.g., point mutations, insertions and deletions), or a larger scale for structural variants (>1000 bp, e.g., large insertions, deletions, inversions, tandem repeats, translocations). (B) Sample variant analysis workflow. In a genomic variant analysis, for example, after sample preparation and sequencing the raw files can be passed through quality control (e.g., using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) and removing PCR artifacts using tools as Picard (http://picard.sourceforge.net)). Reads are mapped to the genome and variants are assessed, e.g., mapping with several algorithms, including ELAND II (Illumina), SOAP [221], MAQ and Burrows-Wheeler Aligner (BWA) [222] and Novoalign by © Novocraft Technologies (http://www.novocraft.com). Read re-alignment can be performed, e.g., using Genome Analysis Toolkit (GATK) [223], or HugeSeq [211], to call variants, including implementations with Sequence Alignment Map format Tools (SAMtools) [224], annotation using Annovar [225], SIFT [226] and Polyphen [227] for determining variant effects on proteomic translation [228]. Furthermore, using a variety of methods the structural variants can be determined. For example the paired-end mapping method considers how paired-end reads mapped to the reference to assign deletions and insertions, from reads whose mapped span is longer or shorter than the average span; inversions, from position and relative orientations of the ends of reads [39,40]. The read depth method allows the possibility to identify the proportional genomic copy number variation. In the approach of Abyzov et al. [229] the read depth considered as an image is analyzed using image processing techniques, viz. mean-shift-theory [230]. Programs such as Pindel [231] and BreakSeq [232] consider split-read analysis to determine breakpoints of insertions and deletions. DELLY [233] by Rausch et al. takes into account paired-end and split-read methods for determining structural variants. Many packages for analysis are available through the Bioconductor [234] project as implemented in the freely available R statistical analysis platform (http://www.R-project.org).
Figure 4. Metabolome analysis.
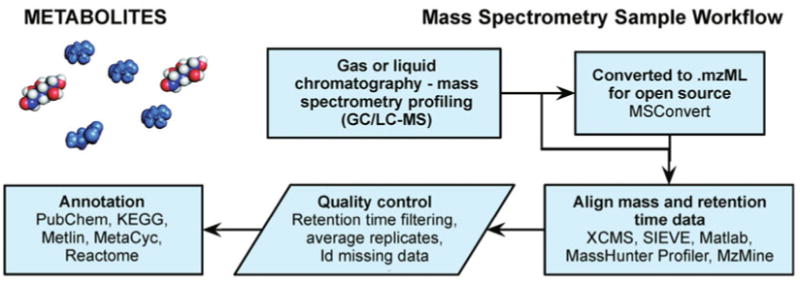
In metabolomics analysis chromatography columns are used for purification and preparation of samples coupled to mass spectrometry (gas chromatography (GC) or liquid chromatography (LC)-MS); standards for specific compounds may also be used in parallel for positive identification. Raw files may be analyzed using vendor software or converted to open formats (such as .mzXML, .mzData or the current standard .mzML [247–249], e.g., using MSConvert). The spectral data may be aligned for retention time and mass intensity calibration, e.g., using XCMS [265–267], SIEVE by Thermo Scientific, Matlab toolboxes by MathWorks, MassHunterProfiler by Agilent, MzMine [268,269]. After quality control and statistical analysis, masses of interest can be annotated using databases, e.g., Metlin [155,156], KEGG [151], MetaCyc [153,270,271], Reactome [157–161].
Genomic Sequencing
In 2001 the completion of the Human Genome Project (HGP) was announced effectively with the publication of the first complete human genome sequence. The HGP came at a hefty $2.7 billion cost using the best technology of the time, making it seemingly prohibitive to expect personal genome sequences to be achieved shortly thereafter. Yet the immense technological advancement, spurred by motivation by the National Institute of Health (NIH) and the National Human Genome Research Institute (NHGRI) to bring down genomic costs, led to an unprecedented growth in technology and methodology, enabling the drop in sequencing costs (http://www.genome.gov/sequencingcosts) to continue at a rate beyond the most optimistic projections of 2001 (< $4000 currently). While initially the human genome was a combination of multiple individual genomic data [1–3], the developments by 2008 had allowed the determination of genomic individual makeup [4–7]. It is now possible to personalize Whole Genome Sequencing (WGS), and the dwindling sequencing costs promise the possibility of affordability for all in the near future [8]. These developments encouraged efforts to characterize disease on a genomic level, towards the application of an all-encompassing genomic medicine, at the molecular level. The initial goals were the characterization of populations for large studies, now shifting to the individual.
Multiple technologies development/dropping costs
The HGP relied on technology using Sanger-based capillary sequencing [1] with an estimated production of 115k base pairs per day (kbp/day) [9]. The NHGRI spurred progress by encouragement through the $1000 genome program (http://www.genome.gov/11008124-al-4), leading to the industry development of multiple massively parallel [10] sequencing platforms (e.g., Roche/454, based on pyrosequencing [11–13]; Life Technologies SOLiD [14–16]; Illumina [5,6]; Complete Genomics based on DNA nanoball sequencing [17]; Helicos Biosciences [18]; and recently single molecule real-time technology [19,20] by Pacific Biosciences). These next generation sequencing platforms are now being supplemented but what has been termed as third-generation sequencing, [21], including such nanopore technologies as announced early in 2012 by Oxford Nanopore Technologies [22]. The technological developments and competition resulted in a drastic and continuing drop in sequencing cost, processing times and exponential increases in number of reads produced.
An alternative to sequencing the whole genome has been whole exome sequencing (WES) [23]. This technology aims to study the exonic regions of the genome (∼2%–3%), which are associated to several Mendelian disorders. It offers a lower cost option (e.g., Illumina, Agilent, and Niblegen platforms, see Clark et al. for a comparison of the latter two [24]) and has received immense attention, including the Exome Sequencing Project (ESP) (see the Exome Variant Server at http://evs.gs.washington.edu/EVS/), supported by the National Heart, Lung and Blood Institute (NHLBI).
Quantitating genomic variation
Concurrently with the technological developments, our understanding of the human genome has grown immensely since the publication of the reference genome in 2003. The aim was to determine the precise role of each base in the genome and identify genomic variants (Figure 1). Several collaborative large-scale efforts pursued such investigations. The International HapMap Consortium [25,26] tried to identify common population variants and led to the development of public databases, such as dbSNP [27] (http://www.ncbi.nlm.nih.gov/SNP/), which catalogues Single Nucleotide Polymorphisms (SNPs) (defined as occurring in >1% of the population to differentiate from Single Nucleotide Variants (SNVs)). This has revealed great genomic variation both in global populations [28,29] and populations of admixed ancestry [30–33].
Typically the technologies involve the assignment of reads to the reference genome to determine the structure of the underlying sequence, including variation (Figure 1). Beyond nucleotide variation, other genomic differences have been investigated, including small insertions and deletions (indels), copy number variations (CNVs) indicating varying numbers of segments and longer chromosomal segments that contribute to Structural Variation (SVs) — SVs are defined for segments of chromosomes larger than 1000 bp (Figure 1A). Such efforts have been based on microarray methodology [34–37] and even higher-resolution in structural variants may be achieved with other methods [38–41]. Structural variants have been publically made available in the database of Genomic Structural Variation (dbVAR; http://www.ncbi.nlm.nih.gov/dbvar/).
Furthermore, functional elements have been extensively catalogued by the Encyclopedia of DNA Elements consortium (ENCODE; http://genome.gov/encode ∼10 production projects), with funding from the NHGRI. ENCODE data, including regulatory elements and RNA and protein level elements, have now been released and the project has received widespread attention [42–45]. The ENCODE project aims at a biochemical genomic characterization, with a thorough mapping of transcribed regions, transcription factor binding sites, open chromatin signatures, chromatin modification and DNA methylation. Such extensive data still needs to be annotated [46] interpreted in terms of biological significance, mechanisms and connections to phenotype and will likely prove invaluable in our interpretation of personalized genomic differences.
Though initially limited by the number of complete genomic sequences, such data are now continuously updated and expanded by information from other projects such as the 1000 Genomes Project [47] as discussed below, which has allowed us to have a better view of the great variability in each individual genome (∼3–4 × 106 SNPs, > 200000 SVs of varying sizes, ∼1500 SVs>2 kbp), with much of the variation considered rare (1%–5%). Genome-Wide Association Studies (GWAS) try to associate the common variants to disease, by combining the now readily available extensive variant information and allelic variability, with linkage disequilibrium (a description of the correlation patterns between proximal variants). The NHGRI provides a publically available catalogue of published GWAS (http://www.genome.gov/gwastudies) [48]. The early expectations of finding common traits and genomic features unique to diseases have proven more complicated, as the genomic variability turns out to be higher than expected and additionally the genetic variants need further validation.
Use of WGS and WES has been successful in the identification of somatic mutations. Mendelian disorders including neurological disorders, and cancer have been characterized using WES [49–58], including some recent single-cell studies [59,60]. Genomics may help classifying cancer subtypes, and possible treatment, and such research is at the center of WGS, with projects such as the Cancer Genome Atlas [61] (http://cancer-genome.nih.gov/), and the International Cancer Genome Consortium (http://www.icgc.org). Additionally, cancer specific public databases already are available [62], including a cancer cell line encyclopedia [63], and genome characterization has been carried out, for example in ovarian cancer [61], melanoma [64], lymphocytic leukemia [65], breast cancer [66-69] and acute myeloid leukemia (AML) [70,71].
Personalized risk evaluation
One of the goals of personalized genome interpretation is the evaluation of disease risk factors based on an individual's variant and allelic distribution composition. Such information may be compared to similar individuals with known disease associations to assess whether an individual shows increased or decreased risk compared to the control group. A combination of know SNPs and personalized variants has been found to be effective [72–75] and has been used in clinical studies; more recently, a seminal study by Ashley et al. [76] evaluated disease risk for a patient with family history of vascular disease.
Personalized evaluation of potential drug responses can be based on the effects of variants [77,78], including drug selection, sensitivity and dosage estimation, e.g., cardiovascular drugs [79], schizophrenia related medications [80]. For example, PharmGKB (http://www.pharmgkb.org) provides a curated database of possible genomics information [81,82], exploring the impact of genomic variation on drug responses as these relate to expressed genes and associated pathways and disorders. The future applications are to include a precise drug dosage for an individual, avoiding trial and error methods and providing more effective treatment.
The evaluation of personalized risk based on genomes is now appearing in direct-to-consumer services. Companies like 23andMe, deCODEme, (and previously Navigenics), offer to assess individual genotypes and offer disease based interpretation services based on Mendelian disorder evaluation and including pharmacogenomics responses. These are mostly based on SNPs evaluation and the tests though limited in scope do offer interpretation attractive to multiple consumers.
Personal Genomes Project
Presently thousands of genomes have been completely sequenced. One of the first large scale projects has been the 1000 Genomes Project [47], that has made its data publically available, and has encouraged the development of streamlined bioinformatics tools to analyze the variation in the individual genomes (Figure 1). This project aims to combine data from 2500 individuals from multiple populations, at a 4 × coverage.
Another grand scale effort driven by George Church's group at Harvard University is the Personal Genome Project (PGP) [83–85]. The project has been recruiting individuals who can share their medical and other information together with genomic information online (http://www.personalgenomes.org). The volunteers share full DNA sequences, RNA and protein profile information in addition to extensive phenotype information including medical records and environmental considerations, with all the data made publically available, and plans to expand to 100000 individuals [86]. One of the rather unique features of the PGP project is that it differs in consent of participants as compared to traditional studies. The ownership of the data is to be open and publically available without restrictions, not only for the initial perspective of the study, but open to follow-up or additional investigations. The scope is participatory, with the volunteers for the project interacting directly with the researchers. To address informed consent, participants pass a basic genetic literacy exam and must understand the project's scope. Additionally, they provide complete medical history, immunization and medications history, which becomes part of the publically available subject information. The access to the individual's data in the project can be either private to the participant and researchers only or completely public, depending on the participant's choice. The availability of extensive patient and omic information will be invaluable to researchers in developing robust analysis models for characterizing genomes and disease and the PGP project, and its publically open structure model, will be at the forefront of such efforts.
Beyond the Genome: Other Omics
Transcriptomics
Though the genetic code in DNA is the almost identical (besides cellular variation), different cells have different gene expressions, corresponding to the kind of cell, developmental stage and physiological state. The collection of the transcripts in a cell (e.g., mRNA, non-coding RNA and small RNAs), the transcriptome, is essential in our understanding of cell function, and response to disease. Considerations must include start and end sites of genes, and coding, alternative splicing and post-transcriptional modifications.
Initially inroads were made using high-density oligo microarrays, and in-house custom made microarrays [87], with high-density arrays having resolutions up to 100 bp [88–91]. While relatively inexpensive, these methods suffered from relying on prior knowledge of the genome, and faced technical issues such as background and saturation effects [92]. Hybridization interactions between probe sets in short oligo microarrays lead to spurious correlations [92,93].
The development of RNA sequencing (RNA-Seq) brought higher coverage, better precision and quantitation, and higher resolution and sensitivity, bringing RNA-Seq technology and transcriptomics on par with genomic sequencing [94–98]. RNA-Seq considers reads that correspond to millions of transcriptomic fragments that are mapped to the reference genome, to provide information on transcripts that may not be in the existing genomic annotation, allowing the search for novel transcripts, and even identification of SNPs and other variants, while showing remarkable reproducibility (Figure 2). Transcriptome profiling has included looking at cancers [99–101], including breast cancer [102], gastrointestinal tumors [103] and prostate cancer [104].
Figure 2. RNA-Seq analysis.
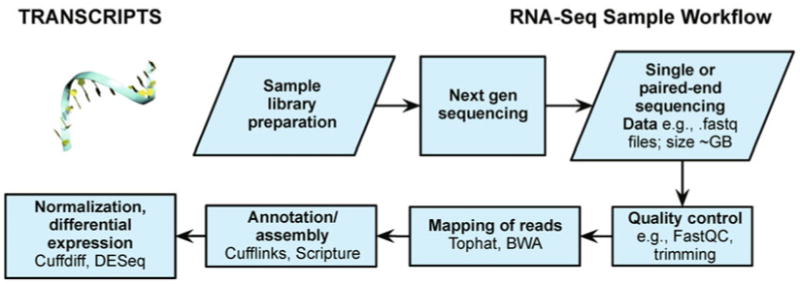
In RNA-Seq analysis, short reads can be assembled and then mapped to the reference genome (with tools such as Illumina's ELAND, MAQ and BWA [222], Bowtie [235–237], SOAP [221], and others). A recent protocol by Trapnell et al. [238] describes in detail the use of dedicated RNA-Seq programs from the Tuxedo suite, such as TopHat [239], Cufflinks [240,241] and an R implementation called CummeRBund as a Bioconductor package (an alternative is to run these directly or using GenePattern [242,243], which also includes possible reconstruction by Scripture [244]). Other programs such as DESeq, another package in Bioconductor, can also help test for differential expression [245]. The numerous analyses availabilities are now publically discussed online, in a forum (http://SEQanswers.com/) that discusses many other examples and all aspects of the mapping process [246].
Mass spectrometry, proteomics and metabolomics
Gene expression was expected to correlate with protein levels in a cell and it was thought that methods such as RNA-Seq would be enough to ascertain the proteomic expression corresponding to gene expression. Proteins are expected to be closer to phenotype, as they participate in every aspect of cellular biology, but their expression levels are difficult to quantitate, partly because of translational control in cells, possible degradation and sampling issues [105–107]. The development of electrospray ionization brought mass spectrometry (MS) to the field of proteomics and the possible identification of thousands of molecules based on mass [108–112]. This has enabled not only the cataloguing of proteins, but also querying post-translational modifications [113,114]. As the techniques matured, liquid chromatography tandem mass spectrometry (LC-MS/MS) has become standard, and novel instruments (e.g., Velos family [115] by Thermo Scientific; quadrupole time-of-flight mass spectrometers (QTOFs) by Agilent) allow unprecedented precision to enable the development of methods to identify thousands of proteins (∼4000–6000 over 2 days), and quantitate protein levels [73,116] (Figure 3). One set of methods uses stable isotopic labeling by amino acids in cell culture (SILAC) to label cell in light and heavy isotopes of amino acids providing double spectral peaks in MS for identification and quantitation [117–120] — this method is now supplemented by ‘spike-in’/‘super’ SILAC which has been used to measure biopsy tumor proteomes [121]. Another possibility is to use isobaric tags for relative and absolute quantitation (iTRAQ) [122,123] or tandem mass tag (TMT) labeling [73,124,125], and other methods, including spiking in peptides for absolute quantitation. Finally, it is possible to employ label-free methods for quantitation, which do not rely on tags, including integrating signal methods and MS spectral counting [126–131].
Figure 3. Proteome analysis.
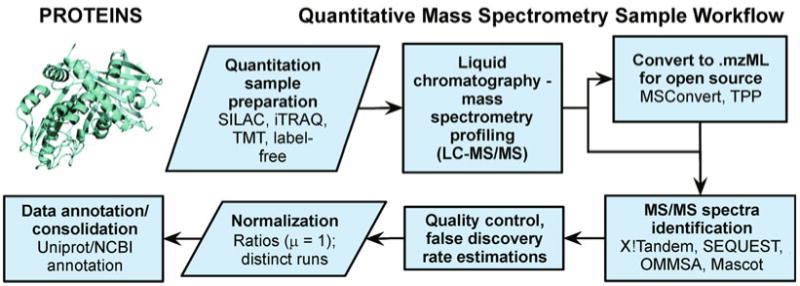
In quantitative proteomics using mass spectrometry typical approaches employ trypsin digestion coupled with tagging methods — non label-free methods include use of isotopic labeling (SILAC) or isobaric tagging (iTRAQ, TMT). One typical bottom-up-approach setup uses a combination of high affinity liquid chromatography coupled with two rounds of mass spectrometry (LC-MS/MS) to fractionate peptides for identification and obtain their mass spectra. Raw files may be analyzed using vendor software or converted to open formats (such as .mzXML, .mzData or the current standard .mzML [247–249], e.g., using MSConvert [250]). The mass spectra can be mapped to known protein using a protein library, or less frequently de novo assembled, using an array of programs (e.g., X!Tandem [251], SEQUEST [252], Mascot [253], Open Mass Spectrometry Search Algorithm (OMSSA) [254], Proteome Discoverer by Thermo Scientific, or MassHunter Workstation by Agilent). Quality control includes estimation of false discovery rates (FDR), often using a reverse database search [105,255,256]. Quantitation can be carried out to estimate relative levels of proteins in different samples (employing standardization and normalization of average sample ratios to a unit mean). Finally annotation is made using databases such as UniProt or NCBI. Some of the analysis can be performed using suites and programs, such as PEAKS [257], the Trans-Proteomic Pipeline (TPP) [258–261], multiple tools from ProteoWizard [250], OpenMS [262–264] or vendor complete solutions Proteome Discoverer and MassHunter Workstation mentioned above. Multiple other programs for mass spectrometry are available (e.g., see http://www.msutils.org).
In comparison to whole transcriptome profiling, the numbers of proteins identified in proteome profiling tend to be less in comparison, particularly since low peptide levels cannot be amplified (cf. polymerase chain reaction methods for sequencing methods). Additionally, the current bottom-up (shotgun) proteomics methodology uses digestion with endopeptidases such as trypsin to obtain peptides of small enough mass to be identified by MS/MS, resulting in many fragments that cannot be identified in MS, which may possibly be alleviated by top down approaches that do not employ a digestion step [132–136]. However, proteomics provides insights that are missing from transcriptomic analysis, especially given the low correlations between protein and transcriptome differential gene expressions [73,137–142].
Multiple proteomes have been quantitatively profiled, including characterization of ovarian cancer [143], an integrated approach that combines transcriptome and proteome information in a human cancer cell line by Nagaraj et al. [144], integrative gastric cancer characterization and effects of post-translational modifications [145], and looking for biomarkers in other cancers [146,147].
In addition to developments in proteomics, MS has encouraged the study of small molecules. The behavior of small molecules in cells though difficult to track provides insight into many common disorders. The set of all cellular small molecules is collectively called the metabolome. Metabolic processes are vital in biological pathways and a systems analysis of molecular cell complexity might lead to biomarker discovery, and possibly disease risk assessment, diagnosis and treatment [148]. Similar to proteomics, metabolomics can employ mass spectrometry to identify compounds [149] (Figure 4) and cataloguing is under way, with thousands of metabolites identified by structure, mass and occasionally associated biological processes [150–161]. The identification of compounds can be based on MS/MS application and use of known compound spectra, or via use of standards against which mass spectra are compared. The profiling of metabolic components on an individualized basis can provide insights into pharmacogenomics and personalized medications, in addition to potential biomarkers, for example cholesterol levels and coronary artery [162,163]. The metabolomics of cancer has been extensively studied [164–166] and Type 2 Diabetes has been investigated [167], and in vivo interactions with proteins are being evaluated [168].
Other omics
Genomes, transcriptomes and metabolomes have received widespread attention and currently offer the most quantitative data, provided by robust and comprehensive omics technologies, both in terms of experimental, as well as computational methodology. However multiple other omics are available, and these numbers are increasing, with a few notable technologies mentioned below:
Autoantibodyomes
In addition to profiling of proteins directly, the reactivity of proteins to autoantibodies may be profiled on a large scale. Spotted protein arrays [169–173] have been implemented to study for example effects in cancer [174], immune response [175] and recently diabetes [176]. Another approach is the Nucleic Acid Programmable Protein Array (NAPPA) constructed by spotting plasmid DNA to effectively express and code the proteins on the array and used for immunoprofiling [177,178]. Furthermore functional peptide arrays have also been constructed [179,180]. Complementary technologies such as bead-based immunoassays are also being actively developed, such as the Luminex xMAP assay [181].
Microbiomes
Omics profiling could also include mapping of the personal microbiome, the complete set of microbes in an individual (e.g., found mainly on the skin or in the gut, conjunctiva, saliva and mucosa) using possibly a combined omics approach to look at genetic makeup and metabolic components [182–187]. The human microbiota (http://www.human-microbiome.org) have been associated to obesity [188] and diabetes [189,190] and have also been suspected to play an active role in the development of immunity [191]. The dynamic monitoring of microbiome-related changes can help identify the specific microbiota involved in disease responses, elucidate microbiome-host interactions and how the individual variability in components impacts developmental and metabolic processes.
Methylomes
In addition to genomics, epigenomic information, such probing the methylome, i.e., identifying all genomic sites of cytosine methylation [192,193], might provide information about differentiation and regulation of gene expression. Methylation analysis and data interpretation can be challenging [194,195] but methods are improving as more data becomes available. Methylome analysis has now been carried out in blood components [196], stem cells [197] and ovarian cancer [61], and it might prove invaluable in assessing epigenomic effects on individual development and health.
Personalized Medicine
The developments of the many different omics technologies outlined above have given us tremendous insight into the human genome and associations to diseases, especially with the rise of the personal genome. The NHGRI recognizing the importance of these developments and the directions necessary to enhance health care, outlined in 2011 a vision for the future of personalized medicine [198] encompassing five domains of development that included understanding the structure of genomes, their biology, improving our understanding of the biology of disease, advancing medicine and improving the effectiveness of healthcare. The aims had been set to a shift towards personalized medicine within two decades, but the availability of the technology and constant decreasing costs have made pilot investigations of personalized medicine a current possibility [73]. Genetic variation has proven adequate for understanding group differences in disorders, but a truly personalized implementation needs to consider an individual. Clinicians are already considering molecular markers in their evaluation of patients, and particularly cancer [199–203]. The typical clinical diagnosis involves the observation of symptoms traditionally confirmed utilizing a small set of molecular markers. In diseases that share a common set of symptoms, some rare, such diagnosis is often complicated and prolonged, especially for heterogeneous disorders that need additional information to enable classification and subsequent specific treatments. Genetic and environmental factors create additional variability in disease severity, progression and treatment responses. Thus, traditional assays together with the aforementioned current omics technologies, that allow monitoring of thousands of molecular components, will facilitate and accelerate differential diagnostics and sub-classification through utilizing a more complete set of disease markers. A personalized approach will result in better targeting of diseases, introduce higher precision through measurement of larger sets of molecular components and ideally implemented at an early age to assess disease risk and have a preventative rather than retrospective treatment focus.
A personal approach is by its nature an n = 1 study, which helps eliminate variation between individuals that are treated as a group, but still requires some verification and establishment of a baseline for comparison. As such, the profiling of healthy physiological states in a longitudinal approach may provide such a basis, if multiple time points with similar physiological state makeup are sampled. Multiple omics can supply multiple supporting datasets at each time point, with each complementary technology providing additional supporting information for a baseline establishment. This introduces the concept of complete omics monitoring of individuals over time, making personalized medicine a more dynamic proposition. The dynamic changes of molecular components may be associated to the individual's changing physiological states, and mapped onto pathways to identify the onset and progression of disease, including possible preventive measures. In our suggested implementation, termed integrative Personal Omics Profiling (iPOP) which we followed in the study discussed below [73] we integrate the omics components discussed above in a longitudinal approach with three essential steps (Figure 5):
Figure 5. iPOP for personalized medicine.
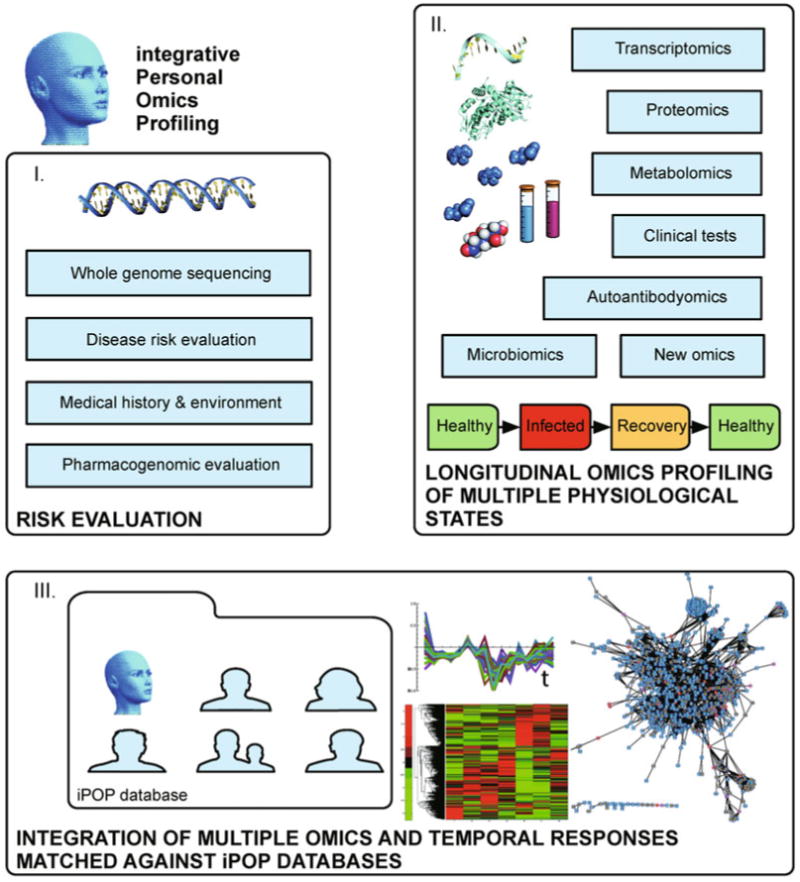
The framework described in the text employs multi-omics analyses (see above and Figures 1–4) that may be implemented for individuals. In step I) Risk estimation for disease is carried out using a whole genome sequencing to perform variant analysis coupled to medical history, environmental considerations and pharmacogenomics evaluations. In step II) Dynamic profiling of multiple omics using an array of technologies follows multiple omics longitudinally in a subject as they progress through their different physiological states, including healthy, disease, and recovery states. Thus thousands of molecular components are collected over time for III) Data integration and biological impact assessment, using temporal patterns to obtain matched omics information, correlate and classify responses, compare against pathway databases and visualize components, e.g., current pathway tools include DAVID [206,272], KEGG [151], Reactome [157–161], Ingenuity Pathway Analysis (IPA); networks can be visualized using Cytoscape [207], various R packages through Bioconductor [234], Matlab by MathWorks and several others. The future iPOP implementations may be gathered into a curated database of iPOP-disease associations that may help in categorizing an omics dynamic response to a catalogued physiological state and disease onset, with potential diagnostic capabilities.
I) Risk estimation
As discussed above the personal and common genomic variants determined in an individual genome can be associated to disease [76], with pharmacogenomic evaluation to determine possible drug response. An early age whole genome sequencing, possibly at birth, can provide a list of possible increased risk disorders and lead to taking preventive measures. This may be done in combination with a complete medical and family history, as for example implemented in the PGP project, and in conjunction with classical clinical risk factor profiling.
II) Dynamic profiling of multiple omics
Starting with a healthy or ‘steady state’ baseline, by monitoring changes in the molecular components over multiple time points, drastic or gradual changes in physiological states might be assessed and the dynamic onset of disease profiled, and possibly prevented. Such profiling may be done on blood components, which are easily obtainable currently in the clinic. The individual blood components are excellent reflectors of generalized physiological state of an individual, as the blood circulates and receives inputs from multiple tissues throughout the body. The components may be processed to track multiple omics, such as transcriptome, proteome, metabolome and autoantibody-ome, etc., which as mentioned offer complementary information, especially given the modest correlation observed between transcriptomic and proteomic components [137–142]. A recent study of profiles of tumors changing over time also employed an integrative approach on genomic and transcriptomic components [204]. Implementing this monitoring on healthy individuals will allow the monitoring of disease onset and physiological changes from various healthy, disease and recovery states, and following thousands of molecular component levels and responses at corresponding physiological states.
III) Data integration and biological impact assessment
The multiple omics data can be analyzed individually to characterize their temporal response profile. This may be done using standard statistical time-series analysis, extensively used in all quantitative disciplines, such as physics, economics and finance, as discussed by Bar-Joseph et al. [205]. The dynamic signature of the signals for each molecular component can be studied for autocorrelation, periodicity or spikey behavior, corresponding to causal changes or abnormal physiological state conditions resulting from the onset of disease, infections, or environmental effects. The different classes of temporal response can be checked for biological pathway and gene ontology enrichment [151,157–161,206–210], and corresponding disease associations in comparison to a database of other longitudinal profiles (coupled to complete electronic records of omic and medical histories). Such a database is a necessary and powerful resource towards the realization of personalized medicine based on omics data profiling.
Example implementation of personalized medicine: iPOP
To show the feasibility and practical applicability of iPOP we profiled a healthy individual, 54, over a period of initially 14 (now 33) months [73]. This initial time series covered healthy states, and two viral states, including a human rhinovirus (HRV) infection at the initiation of the study and a respiratory syncytial virus (RSV) infection 289 days later. The iPOP used blood samples to extract omic components from peripheral blood mononuclear cells (PBMCs) and serum, which were analyzed to obtain a complete DNA, RNA, protein, metabolite and autoantibody profile. Initially a complete medical exam was performed with standard clinical tests before time-point profiling began. In a first step, WGS with two platforms was carried out (Complete Genomics and Illumina, at 150- and 120-fold coverage respectively) and WES with three platforms (Nimblegen, Illumina and Agilent) and helped identify a large number of variants (> 3 × 106 SNPs; > 2 × 105 indels; > 2000 SVs). Using multiple platforms allowed us to determine high-confidence and novel variants (using HugeSeq [211]). Evaluation of genetic disease risks based on variants was carried out, both by looking for known disease associations using dbSNP and the Online Mendelian Inheritance in Man (OMIM, http://omim.org/) database and using the RiskO-Gram algorithm [76] which integrates information from multiple alleles to assess risk against a similarly matched data cohort. This revealed significantly increased risk for various disorders, including open angle glaucoma, dyslipidemia, coronary artery disease, basal cell carcinoma, type 2 diabetes (T2D), age related macular degeneration and psoriasis. This encouraged the subject to follow up on these disorders, and also start monitoring glucose and glycated hemoglobin (HbA1c) levels, which surprisingly increased beyond normal levels following the RSV infection, and the subject was diagnosed by his physician for T2D 369 days into the study. Related to T2D, pharmacogenomic considerations revealed a possibly favorable (glucose lowering) response to diabetic drugs rosiglitazone and metformin, should treatment become necessary. Furthermore, the autoantibodyome profiling of the subject (Invitrogen ProtoArrays profiling of 9483 protein reactivities to Immunoglobulin G (IgG)) revealed increased reactivity in multiple proteins, including DOK6 (related to insulin receptors), and GOSR1, BTK and ASPA, previously reported to show high reactivity by Winer et al. in insulin resistant patients [176]. The subject initiated and still maintains a strict dietary and exercise regiment supplemented with low doses of acetylsalicylic acid, which helped him control his glucose and HbA1c levels, which after a considerable time period (∼months) have now returned to normal levels.
In addition a range of omics were profiled over time for up to 20 different timepoints over the span of the study including high coverage transcriptome (RNA-Seq of PBMCs, 2.67 billion reads mapped to 19714 isoforms corresponding to 12659 genes), proteome (MS of PBMCs, identifying a total of 6280 proteins; 3731 consistently across most timepoints), metabolome (MS of serum, profiling 6862 and 4228 metabolites during periods of HRV and RSV infections respectively, with ∼20% identified based on mass and retention times alone). The dynamic transcriptome, proteome and metabolome profiles were analyzed in a novel integrated framework based on spectral analysis of the time series. This allowed the identification of temporal patterns in the combined data, corresponding to biological processes that varied with physiological state changes, including the onset of T2D seen in multiple omics components, and common signatures of HRV and RSV infections. While several gene associations to pathways were known, multiple genes showed similar patterns that had not been reported before and merit further investigation.
Other Considerations and Future Directions
The iPOP study discussed above revealed the complexities and characteristics of personal genomes, transcriptomes, proteomes and metabolomes and showed the feasibility of personalized longitudinal profiling that can provide actionable health information. Multiple omics data integration still presents a formidable challenge and merits further development. Each omics technology produces different kinds of data, including multiple formats (e.g., data files range from simple text, and extensible markup, e.g., .xml, to vendor closed-source formats). Additionally, each omics set requires its own quality control analysis, further confounded by different error and noise levels associated to the different technologies. As each of the data sets also presents different signal and noise distributions, this makes uniform normalization approaches across omics challenging, especially if considering multimodal dynamic data. Furthermore, the amounts of information per omics set can vary, e.g., ∼5000 proteins, ∼20000 transcript isoforms, ∼6000–10000 metabolites, ∼9000 autoantibody-protein reactivities and so forth. Hence, gene-centric approaches, that integrate data corresponding to, associated or interacting with the same genes, will not always work, as the different components may not match. The integration of information per component is made more difficult with multiple existing gene and protein annotations, often resulting in a many-to-many map in the gene-protein integration, and correspondingly lacking metabolite-protein/gene annotations and associations. Finally, if considering dynamic datasets, this also results in multiple instances where time points might be missing data for some of the molecular components (especially evident in mass spectrometry and shotgun proteomics, where proteins are identified through different peptides). These complications of omics data integration necessitate that each individual omics data set is analyzed independently up to normalization, and then integrated with the other information. New integrative methodology has to account for such different normalizations, missing data, and also integration that is not gene-based, but rather incorporates time-series analyses, as for example was carried out in the iPOP study [73]. Classification of changes by temporal response, and possibly interaction data leads to an interpretation of components based on shared similar dynamics and avoids some of the issues of insufficient annotations and missing information. Such an interpretation lends itself to a clinical setting where dynamic changes are associated to varying personalized physiological states, and may be adopted by the medical community.
To facilitate the wide adoption of the methods into personalized medicine, the integrated data analysis will require optimization of current computational tools to rapidly and efficiently handle as well as visualize the multiple omics data. As a first step, the amount of computation time for different analyses must be reduced from days (in the case of mapping sequence data and quantitative proteomics in current omics analyses presented above) to hours or less to have immediate relevance to active medical examinations. Secondly, better visualizations of omics data, though difficult, are also necessary, as multidimensional information is difficult to collate, present, and interpret (many efforts are addressing this, e.g., Circos plots that allow multiple sequence information to be displayed together are now widely adopted [212]). Incorporating such information with clinical data and phenotypes presents a new challenge, requiring browsers that combine temporal information with multi-dimensional omics sets. We believe network analysis [213–217] presents an excellent visualization and integration possibility, allowing the combinations of multiple levels of networks, dynamically changing, that will include cellular information, component and corresponding disease temporal progressions, as well as medical assay data in a modularized approach. The computational analyses and visualization of omics data integration also reveal the known need to manage large amounts of data [218,219], both in terms of processing power, as well as storage capacity and maintaining easy accessibility, especially for the practicing clinician — with the recent advent of cloud computing providing one possible solution. Finally, the combination of omics data with medical records presents another challenge, with privacy and ethical issues that must be considered. Such improvements and standardization of approaches will help make the analysis available in a clinical setting and an increasingly larger set of patients, while encouraging the early adaptation of the integrated approaches by the scientific community towards personalized medicine applications.
As technology improves we expect to see advancements in each omics implementation discussed above. In terms of sequencing, continual improvements in depth and read length will allow unambiguous precise sequence mapping and additionally the querying of lower gene expression, coupled to higher accuracy in variant calling. With sequencing times becoming faster (e.g., whole genome sequencing in ∼5–30 hours depending on platform at deep, ∼100 × coverage), and hardware more compact, eventually such technology will be available in the clinic, enabling the incorporation of all genomic, transcriptomic, microbiomic and autoantibodyomic profiling as parts of regular medical examinations. Correspondingly, mass spectrometry improvements (including table-top hardware now available) will improve mass accuracy, and higher sensitivity, allowing increases in the number of proteins identified and better quantitation, which can already be implemented in a clinical setting. The MS improvements in combination with better metabolite cataloguing will also improve the identification of small molecules. The protocol and methodology advancements will allow using a smaller volume of patient sample needed for iPOP (decreasing from ∼80 mL to drops of blood) making it feasible to probe the omics on more regular basis for each patient, even providing home kits to send in self-collected samples (akin to what is already implemented to some degree by companies, e.g., 23andMe, that collect saliva samples for phenotyping).
The technological and methodological advancements will allow for effective iPOP implementations with multiple patients, but it will still take some time to evaluate what constitutes actionable information and which components will be most informative. Once these relevant components are identified monitoring technologies can be further developed to help possible clinical implementations. This will certainly be alleviated by multiple iPOP studies providing the necessary aggregated information. However, clinical and psychological concerns need to be addressed and the possible impact to patient health being of paramount importance, in a medical process in which the patient is actively participating [220]. Such active participation requires the training of the public and health professionals to an understanding of genomic information, and how this omics knowledge impacts their health, and their families. Genetic counseling is a necessity, and the number of trained genetic counselors is steadily increasing. Informed consent will be necessary, but this requires an understanding of basic genomic terms that are not apparent to non-experts. To facilitate this, probably school curriculum adjustments will be needed to enable early education of the public.
The emergence of quantitative Personal Omics, including genomes transcriptomes, proteomes, metabolomes and other omics allows us to now combine them to yield personalized actionable health care information. Such research is at the forefront of medical science, and may help the characterization of disorders and the implementation of precise personal medicine aimed towards prevention rather than treatment. Careful forward planning, coupled to the continuing interest and participation of the public, government agencies and researchers, assures that the development of personalized omics will proceed beyond possible hurtles into a novel approach for the 21st century health care implementations.
Acknowledgments
We would like to thank the Stanford Genetics Department and the NIH for support through grant P50HG02357. GIM would also like to thank the NIH for support through training grant T32HG000044. We also thank Drs. Rui Chen, Jennifer Li Pook Than and Hogune Im for useful discussions.
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 7.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder M, Du J, Gerstein M. Personal genome sequencing: current approaches and challenges. Genes Dev. 2010;24:423–431. doi: 10.1101/gad.1864110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mardis ER. A decade's perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- 10.Tucker T, Marra M, Friedman JM. Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet. 2009;85:142–154. doi: 10.1016/j.ajhg.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ronaghi M, Uhlén M, Nyrén P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363, 365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 12.Ronaghi M, Karamohamed S, Pettersson B, Uhlén M, Nyrén P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem. 1996;242:84–89. doi: 10.1006/abio.1996.0432. [DOI] [PubMed] [Google Scholar]
- 13.Nyrén P. The history of pyrosequencing. Methods Mol Biol. 2007;373:1–14. doi: 10.1385/1-59745-377-3:1. [DOI] [PubMed] [Google Scholar]
- 14.Nutter RC. New frontiers in plant functional genomics using next generation sequencing technologies. In: Kahl G, Meksem K, editors. The Handbook of Plant Functional Genomics: Concepts and Protocels. Chapter 21. Wiley-VCH Verlag GmbH & Co. KGaA; 2008. pp. 431–446. [Google Scholar]
- 15.Dai M, Thompson RC, Maher C, Contreras-Galindo R, Kaplan MH, Markovitz DM, Omenn G, Meng F. NGSQC: cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics. 2010;11:S7. doi: 10.1186/1471-2164-11-S4-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandey V, Nutter RC, Prediger E. Applied biosystems SOLiD™ system: ligation-based sequencing. In: Janitz M, editor. Next Generation Genome Sequencing: Towards Personalized Medicine. Chapter 3. Wiley-VCH Verlag GmbH & Co. KGaA; 2008. pp. 29–42. [Google Scholar]
- 17.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 18.Braslavsky I, Hebert B, Kartalov E, Quake SR. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci USA. 2003;100:3960–3964. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korlach J, Bjornson KP, Chaudhuri BP, Cicero RL, Flusberg BA, Gray JJ, Holden D, Saxena R, Wegener J, Turner SW. Real-time DNA sequencing from single polymerase molecules. Methods Enzymol. 2010;472:431–455. doi: 10.1016/S0076-6879(10)72001-2. [DOI] [PubMed] [Google Scholar]
- 20.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 21.Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Genet. 2010;19:R227–R240. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- 22.Hayden E. Nanopore genome sequencer makes its debut. Nature. 2012 doi: 10.1038/nature.2012.10051. [DOI] [Google Scholar]
- 23.Bainbridge MN, Wang M, Burgess DL, Kovar C, Rodesch MJ, D'Ascenzo M, Kitzman J, Wu YQ, Newsham I, Richmond TA, et al. Whole exome capture in solution with 3 Gbp of data. Genome Biol. 2010;11:R62. doi: 10.1186/gb-2010-11-6-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, Butte AJ, Snyder M. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The International HapMap Consortium. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altshuler D, Clark AG. Genetics. Harvesting medical information from the human family tree. Science. 2005;307:1052–1053. doi: 10.1126/science.1109682. [DOI] [PubMed] [Google Scholar]
- 29.Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC, Szpiech ZA, Degnan JH, Wang K, Guerreiro R, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 30.Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kidd JM, Gravel S, Byrnes J, Moreno-Estrada A, Musharoff S, Bryc K, Degenhardt JD, Brisbin A, Sheth V, Chen R, et al. Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation. Am J Hum Genet. 2012;91:660–671. doi: 10.1016/j.ajhg.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galanter JM, Fernandez-Lopez JC, Gignoux CR, Barnholtz-Sloan J, Fernandez-Rozadilla C, Via M, Hidalgo-Miranda A, Contreras AV, Figueroa LU, Raska P, et al. Development of a panel of genome-wide ancestry informative markers to study admixture throughout the Americas. PLoS Genet. 2012;8:e1002554. doi: 10.1371/journal.pgen.1002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bryc K, Auton A, Nelson MR, Oksenberg JR, Hauser SL, Williams S, Froment A, Bodo JM, Wambebe C, Tishkoff SA, et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad Sci USA. 2010;107:786–791. doi: 10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haraksingh RR, Abyzov A, Gerstein M, Urban AE, Snyder M. Genome-wide mapping of copy number variation in humans: comparative analysis of high resolution array platforms. PLoS ONE. 2011;6:e27859. doi: 10.1371/journal.pone.0027859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang Z, Snyder M, Gerstein MB. PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009;10:R23. doi: 10.1186/gb-2009-10-2-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinlan AR, Hall IM. Characterizing complex structural variation in germline and somatic genomes. Trends Genet. 2012;28:43–53. doi: 10.1016/j.tig.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The ENCODE Project Consortium. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ecker JR, Bickmore WA, Barroso I, Pritchard JK, Gilad Y, Segal E. Genomics: ENCODE explained. Nature. 2012;489:52–55. doi: 10.1038/489052a. [DOI] [PubMed] [Google Scholar]
- 45.Birney E. The making of ENCODE: lessons for big-data projects. Nature. 2012;489:49–51. doi: 10.1038/489049a. [DOI] [PubMed] [Google Scholar]
- 46.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, Uziel G, Lamantea E, Invernizzi F, Poulton J, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- 50.Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 51.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M, Gronka S, Wuu J, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bilgüvar K, Oztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoğlu D, Tüysüz B, Cağlayan AO, Gökben S, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467:207–210. doi: 10.1038/nature09327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, et al. Exome sequencing, ANGPTL3 mutations, and familialcombined hypolipidemia. N Engl J Med. 2010;363:2220–2227. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. Exome sequencing of head and neck squamous cell carcinomareveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873–885. doi: 10.1016/j.cell.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 61.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Küntzer J, Maisel D, Lenhof HP, Klostermann S, Burtscher H. The Roche Cancer Genome Database 2.0. BMC Med Genomics. 2011;4:43. doi: 10.1186/1755-8794-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, Escaramis G, Jares P, Beà S, González-Díaz M, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yost SE, Smith EN, Schwab RB, Bao L, Jung H, Wang X, Voest E, Pierce JP, Messer K, Parker BA, et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 2012;40:e107. doi: 10.1093/nar/gks299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Natrajan R, Mackay A, Lambros MB, Weigelt B, Wilkerson PM, Manie E, Grigoriadis A, A'hern R, van der Groep P, Kozarewa I, et al. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol. 2012;227:29–41. doi: 10.1002/path.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Link DC, Schuettpelz LG, Shen D, Wang J, Walter MJ, Kulkarni S, Payton JE, Ivanovich J, Goodfellow PJ, Le Beau M, et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA. 2011;305:1568–1576. doi: 10.1001/jama.2011.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dewey FE, Chen R, Cordero SP, Ormond KE, Caleshu C, Karczewski KJ, Whirl-Carrillo M, Wheeler MT, Dudley JT, Byrnes JK, et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011;7:e1002280. doi: 10.1371/journal.pgen.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, Reid JG, Fink JK, Morgan MB, Gingras MC, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med. 2011;3:87re3. doi: 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, Dudley JT, Ormond KE, Pavlovic A, Morgan AA, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lesko LJ, Schmidt S. Individualization of drug therapy: history, present state, and opportunities for the future. Clin Pharmacol Ther. 2012;92:458–466. doi: 10.1038/clpt.2012.113. [DOI] [PubMed] [Google Scholar]
- 78.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–468. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 79.Zineh I, Johnson JA. Pharmacogenetics of chronic cardiovascular drugs: applications and implications. Expert Opin Pharmacother. 2006;7:1417–1427. doi: 10.1517/14656566.7.11.1417. [DOI] [PubMed] [Google Scholar]
- 80.Gupta S, Jain S, Brahmachari SK, Kukreti R. Pharmacogenomics: a path to predictive medicine for schizophrenia. Pharmacogenomics. 2006;7:31–47. doi: 10.2217/14622416.7.1.31. [DOI] [PubMed] [Google Scholar]
- 81.Thorn CF, Klein TE, Altman RB. Pharmacogenomics and bioinformatics: PharmGKB. Pharmacogenomics. 2010;11:501–505. doi: 10.2217/pgs.10.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark Med. 2011;5:795–806. doi: 10.2217/bmm.11.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lunshof JE, Bobe J, Aach J, Angrist M, Thakuria JV, Vorhaus DB, Hoehe MR, Church GM. Personal genomes in progress: from the human genome project to the personal genome project. Dialogues Clin Neurosci. 2010;12:47–60. doi: 10.31887/DCNS.2010.12.1/jlunshof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ball MP, Thakuria JV, Zaranek AW, Clegg T, Rosenbaum AM, Wu X, Angrist M, Bhak J, Bobe J, Callow MJ, et al. A public resource facilitating clinical use of genomes. Proc Natl Acad Sci USA. 2012;109:11920–11927. doi: 10.1073/pnas.1201904109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Church GM. The personal genome project. Mol Syst Biol. 2005;1:2005.0030. doi: 10.1038/msb4100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jones B. Genomics: personal genome project. Nat Rev Genet. 2012;13:599. doi: 10.1038/nrg3309. [DOI] [PubMed] [Google Scholar]
- 87.Clark TA, Sugnet CW, Ares M., Jr Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science. 2002;296:907–910. doi: 10.1126/science.1069415. [DOI] [PubMed] [Google Scholar]
- 88.Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–1154. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 89.Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–2246. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 90.Yamada K, Lim J, Dale JM, Chen H, Shinn P, Palm CJ, Southwick AM, Wu HC, Kim C, Nguyen M, et al. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science. 2003;302:842–846. doi: 10.1126/science.1088305. [DOI] [PubMed] [Google Scholar]
- 91.David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci USA. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okoniewski MJ, Miller CJ. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinformatics. 2006;7:276. doi: 10.1186/1471-2105-7-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Royce TE, Rozowsky JS, Gerstein MB. Toward a universal microarray: prediction of gene expression through nearest-neighbor probe sequence identification. Nucleic Acids Res. 2007;35:e99. doi: 10.1093/nar/gkm549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bähler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 96.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 98.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mayr C, Bartel DP. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell. 2009;138:673–684. doi: 10.1016/j.cell.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Delahaye NF, Rusakiewicz S, Martins I, Ménard C, Roux S, Lyonnet L, Paul P, Sarabi M, Chaput N, Semeraro M, et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat Med. 2011;17:700–707. doi: 10.1038/nm.2366. [DOI] [PubMed] [Google Scholar]
- 104.Rajan P, Elliott DJ, Robson CN, Leung HY. Alternative splicing and biological heterogeneity in prostate cancer. Nat Rev Urol. 2009;6:454–460. doi: 10.1038/nrurol.2009.125. [DOI] [PubMed] [Google Scholar]
- 105.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- 107.Cravatt BF, Simon GM, Yates JR., 3rd The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 108.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 109.Aebersold R. Quantitative proteome analysis: methods and applications. J Infect Dis. 2003;187:S315–S320. doi: 10.1086/374756. [DOI] [PubMed] [Google Scholar]
- 110.Aebersold R. A mass spectrometric journey into protein and proteome research. J Am Soc Mass Spectrom. 2003;14:685–695. doi: 10.1016/S1044-0305(03)00289-7. [DOI] [PubMed] [Google Scholar]
- 111.Yates JR, 3rd, Gilchrist A, Howell KE, Bergeron JJ. Proteomics of organelles and large cellular structures. Nat Rev Mol Cell Biol. 2005;6:702–714. doi: 10.1038/nrm1711. [DOI] [PubMed] [Google Scholar]
- 112.Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. doi: 10.1146/annurev-biochem-061308-093216. [DOI] [PubMed] [Google Scholar]
- 113.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 114.Allmer J. Existing bioinformatics tools for the quantitation of post-translational modifications. Amino Acids. 2012;42:129–138. doi: 10.1007/s00726-010-0614-3. [DOI] [PubMed] [Google Scholar]
- 115.Michalski A, Damoc E, Hauschild JP, Lange O, Wieghaus A, Makarov A, Nagaraj N, Cox J, Mann M, Horning S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol Cell Proteomics. 2011;10:M111.011015. doi: 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 117.Ong SE, Mann M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol Biol. 2007;359:37–52. doi: 10.1007/978-1-59745-255-7_3. [DOI] [PubMed] [Google Scholar]
- 118.Ong SE, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC) Nat Protoc. 2006;1:2650–2660. doi: 10.1038/nprot.2006.427. [DOI] [PubMed] [Google Scholar]
- 119.Ong SE, Kratchmarova I, Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2003;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 120.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 121.Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y, Mann M. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 2011;6:147–157. doi: 10.1038/nprot.2010.192. [DOI] [PubMed] [Google Scholar]
- 122.Choe L, D'Ascenzo M, Relkin NR, Pappin D, Ross P, Williamson B, Guertin S, Pribil P, Lee KH. 8-plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. Proteomics. 2007;7:3651–3660. doi: 10.1002/pmic.200700316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 124.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 125.Dayon L, Hainard A, Licker V, Turck N, Kuhn K, Hochstrasser DF, Burkhard PR, Sanchez JC. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal Chem. 2008;80:2921–2931. doi: 10.1021/ac702422x. [DOI] [PubMed] [Google Scholar]
- 126.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 127.Zybailov BL, Florens L, Washburn MP. Quantitative shotgun proteomics using a protease with broad specificity and normalized spectral abundance factors. Mol Biosyst. 2007;3:354–360. doi: 10.1039/b701483j. [DOI] [PubMed] [Google Scholar]
- 128.Mueller LN, Rinner O, Schmidt A, Letarte S, Bodenmiller B, Brusniak MY, Vitek O, Aebersold R, Müller M. SuperHirn — a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics. 2007;7:3470–3480. doi: 10.1002/pmic.200700057. [DOI] [PubMed] [Google Scholar]
- 129.May D, Fitzgibbon M, Liu Y, Holzman T, Eng J, Kemp CJ, Whiteaker J, Paulovich A, McIntosh M. A platform for accurate mass and time analyses of mass spectrometry data. J Proteome Res. 2007;6:2685–2694. doi: 10.1021/pr070146y. [DOI] [PubMed] [Google Scholar]
- 130.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics. 2010;7:39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 131.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 132.Kusunoki M, Tsutsumi K, Nakayama M, Kurokawa T, Nakamura T, Ogawa H, Fukuzawa Y, Morishita M, Koide T, Miyata T. Relationship between serum concentrations of saturated fatty acids and unsaturated fatty acids and the homeostasis model insulin resistance index in Japanese patients with type 2 diabetes mellitus. J Med Invest. 2007;54:243–247. doi: 10.2152/jmi.54.243. [DOI] [PubMed] [Google Scholar]
- 133.Shaffer JP. Controlling the false discovery rate with constraints: the Newman-Keuls test revisited. Biom J. 2007;49:136–143. doi: 10.1002/bimj.200610297. [DOI] [PubMed] [Google Scholar]
- 134.Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol. 2003;21:921–926. doi: 10.1038/nbt849. [DOI] [PubMed] [Google Scholar]
- 135.Ahdesmäki M, Lähdesmäki H, Pearson R, Huttunen H, Yli-Harja O. Robust detection of periodic time series measured from biological systems. BMC Bioinformatics. 2005;6:117. doi: 10.1186/1471-2105-6-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 137.Washburn MP, Koller A, Oshiro G, Ulaszek RR, Plouffe D, Deciu C, Winzeler E, Yates JR., 3rd Protein pathway and complex clustering of correlated mRNA and protein expression analyses in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2003;100:3107–3112. doi: 10.1073/pnas.0634629100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ning K, Fermin D, Nesvizhskii AI. Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. J Proteome Res. 2012;11:2261–2271. doi: 10.1021/pr201052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lundberg E, Fagerberg L, Klevebring D, Matic I, Geiger T, Cox J, Algenäs C, Lundeberg J, Mann M, Uhlen M. Defining the transcriptome and proteome in three functionally different human cell lines. Mol Syst Biol. 2010;6:450. doi: 10.1038/msb.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kislinger T, Cox B, Kannan A, Chung C, Hu P, Ignatchenko A, Scott MS, Gramolini AO, Morris Q, Hallett MT, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 141.Gry M, Rimini R, Strömberg S, Asplund A, Pontén F, Uhlén M, Nilsson P. Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics. 2009;10:365. doi: 10.1186/1471-2164-10-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Greenbaum D, Jansen R, Gerstein M. Analysis of mRNA expression and protein abundance data: an approach for the comparison of the enrichment of features in the cellular population of proteins and transcripts. Bioinformatics. 2002;18:585–596. doi: 10.1093/bioinformatics/18.4.585. [DOI] [PubMed] [Google Scholar]
- 143.Petricoin EF, III, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359:572–577. doi: 10.1016/S0140-6736(02)07746-2. [DOI] [PubMed] [Google Scholar]
- 144.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Pääbo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Guo T, Fan L, Ng WH, Zhu Y, Ho M, Wan WK, Lim KH, Ong WS, Lee SS, Huang S, et al. Multidimensional identification of tissue biomarkers of gastric cancer. J Proteome Res. 2012;11:3405–3413. doi: 10.1021/pr300212g. [DOI] [PubMed] [Google Scholar]
- 146.Woolfson A, Ellmark P, Chrisp JS, Scott MA, Christopherson RI. The application of CD antigen proteomics to pharmacogenomics. Pharmacogenomics. 2006;7:759–771. doi: 10.2217/14622416.7.5.759. [DOI] [PubMed] [Google Scholar]
- 147.Griffin NM, Schnitzer JE. Overcoming key technological challenges in using mass spectrometry for mapping cell surfaces in tissues. Mol Cell Proteomics. 2011;10:R110.000935. doi: 10.1074/mcp.R110.000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Suhre K, Gieger C. Genetic variation in metabolic phenotypes: study designs and applications. Nat Rev Genet. 2012;13:759–769. doi: 10.1038/nrg3314. [DOI] [PubMed] [Google Scholar]
- 149.Theodoridis G, Gika HG, Wilson ID. Mass spectrometry-based holistic analytical approaches for metabolite profiling in systems biology studies. Mass Spectrom Rev. 2011;30:884–906. doi: 10.1002/mas.20306. [DOI] [PubMed] [Google Scholar]
- 150.Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, et al. The human serum metabolome. PLoS ONE. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Zhou Z, Han L, Karapetyan K, Dracheva S, Shoemaker BA, et al. PubChem's BioAssay Database. Nucleic Acids Res. 2012;40:D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Caspi R, Altman T, Dreher K, Fulcher CA, Subhraveti P, Keseler IM, Kothari A, Krummenacker M, Latendresse M, Mueller LA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40:D742–D753. doi: 10.1093/nar/gkr1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tautenhahn R, Cho K, Uritboonthai W, Zhu Z, Patti GJ, Siuzdak G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat Biotechnol. 2012;30:826–828. doi: 10.1038/nbt.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sana TR, Roark JC, Li X, Waddell K, Fischer SM. Molecular formula and METLIN Personal Metabolite Database matching applied to the identification of compounds generated by LC/TOF-MS. J Biomol Tech. 2008;19:258–266. [PMC free article] [PubMed] [Google Scholar]
- 156.Smith CA, O'Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G. METLIN: a metabolite mass spectral database. Ther Drug Monit. 2005;27:747–751. doi: 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 157.Vastrik I, D'Eustachio P, Schmidt E, Gopinath G, Croft D, de Bono B, Gillespie M, Jassal B, Lewis S, Matthews L, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39. doi: 10.1186/gb-2007-8-3-r39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37:D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Matthews L, D'Eustachio P, Gillespie M, Croft D, de Bono B, Gopinath G, Jassal B, Lewis S, Schmidt E, Vastrik I, et al. An introduction to the reactome knowledgebase of human biological pathways and processes. Bioinformatics Primer, NCI/Nature Pathway Interaction Database 2007 [Google Scholar]
- 160.Joshi-Tope G, Gillespie M, Vastrik I, D'Eustachio P, Schmidt E, de Bono B, Jassal B, Gopinath GR, Wu GR, Matthews L, et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Croft D, O'Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dumont J, Huybrechts I, Spinneker A, Gottrand F, Grammatikaki E, Bevilacqua N, Vyncke K, Widhalm K, Kafatos A, Molnar D, et al. FADS1 genetic variability interacts with dietary α-linolenic acid intake to affect serum non-HDL-cholesterol concentrations in European adolescents. J Nutr. 2011;141:1247–1253. doi: 10.3945/jn.111.140392. [DOI] [PubMed] [Google Scholar]
- 163.Lu Y, Feskens EJ, Dollé ME, Imholz S, Verschuren WM, Müller M, Boer JM. Dietary n-3 and n-6 polyunsaturated fatty acid intake interacts with FADS1 genetic variation to affect total and HDL-cholesterol concentrations in the Doetinchem Cohort Study. Am J Clin Nutr. 2010;92:258–265. doi: 10.3945/ajcn.2009.29130. [DOI] [PubMed] [Google Scholar]
- 164.Serkova NJ, Glunde K. Metabolomics of cancer. Methods Mol Biol. 2009;520:273–295. doi: 10.1007/978-1-60327-811-9_20. [DOI] [PubMed] [Google Scholar]
- 165.Griffin JL, Shockcor JP. Metabolic profiles of cancer cells. Nat Rev Cancer. 2004;4:551–561. doi: 10.1038/nrc1390. [DOI] [PubMed] [Google Scholar]
- 166.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15:606–614. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li X, Gianoulis TA, Yip KY, Gerstein M, Snyder M. Extensive in vivo metabolite-protein interactions revealed by large-scale systematic analyses. Cell. 2010;143:639–650. doi: 10.1016/j.cell.2010.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 170.Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2:RESEARCH0004. doi: 10.1186/gb-2001-2-2-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Robinson WH, Steinman L, Utz PJ. Protein arrays for autoantibody profiling and fine-specificity mapping. Proteomics. 2003;3:2077–2084. doi: 10.1002/pmic.200300583. [DOI] [PubMed] [Google Scholar]
- 172.Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, Dean EJ, Fournel S, Fong D, Genovese MC, de Vegvar HE, et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med. 2002;8:295–301. doi: 10.1038/nm0302-295. [DOI] [PubMed] [Google Scholar]
- 173.Sharon D, Chen R, Snyder M. Systems biology approaches to disease marker discovery. Dis Markers. 2010;28:209–224. doi: 10.3233/DMA-2010-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hudson ME, Pozdnyakova I, Haines K, Mor G, Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. Proc Natl Acad Sci USA. 2007;104:17494–17499. doi: 10.1073/pnas.0708572104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhu H, Hu S, Jona G, Zhu X, Kreiswirth N, Willey BM, Mazzulli T, Liu G, Song Q, Chen P, et al. Severe acute respiratory syndrome diagnostics using a coronavirus protein microarray. Proc Natl Acad Sci USA. 2006;103:4011–4016. doi: 10.1073/pnas.0510921103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. 2011;17:610–617. doi: 10.1038/nm.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Miersch S, LaBaer J. Nucleic Acid programmable protein arrays: versatile tools for array-based functional protein studies. Curr Protoc Protein Sci. 2011;Chapter 27 doi: 10.1002/0471140864.ps2702s64. Unit27.2. [DOI] [PubMed] [Google Scholar]
- 178.Sibani S, LaBaer J. Immunoprofiling using NAPPA protein microarrays. Methods Mol Biol. 2011;723:149–161. doi: 10.1007/978-1-61779-043-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Andresen H, Bier FF. Peptide microarrays for serum antibody diagnostics. Methods Mol Biol. 2009;509:123–134. doi: 10.1007/978-1-59745-372-1_8. [DOI] [PubMed] [Google Scholar]
- 180.Andresen H, Grötzinger C, Zarse K, Kreuzer OJ, Ehrentreich-Förster E, Bier FF. Functional peptide microarrays for specific and sensitive antibody diagnostics. Proteomics. 2006;6:1376–1384. doi: 10.1002/pmic.200500343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wong SJ, Demarest VL, Boyle RH, Wang T, Ledizet M, Kar K, Kramer LD, Fikrig E, Koski RA. Detection of human anti-flavivirus antibodies with a West Nile virus recombinant antigen microsphere immunoassay. J Clin Microbiol. 2004;42:65–72. doi: 10.1128/JCM.42.1.65-72.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Weinstock GM. Genomic approaches to studying the human microbiota. Nature. 2012;489:250–256. doi: 10.1038/nature11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Grice EA, Segre JA. The human microbiome: our second genome. Annu Rev Genomics Hum Genet. 2012;13:151–170. doi: 10.1146/annurev-genom-090711-163814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kuczynski J, Lauber CL, Walters WA, Parfrey LW, Clemente JC, Gevers D, Knight R. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13:47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sonnenburg JL, Fischbach MA. Community health care: therapeutic opportunities in the human microbiome. Sci Transl Med. 2011;3:78ps12. doi: 10.1126/scitranslmed.3001626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 189.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 191.Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pelizzola M, Ecker JR. The DNA methylome. FEBS Lett. 2011;585:1994–2000. doi: 10.1016/j.febslet.2010.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 194.Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 195.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 196.Li Y, Zhu J, Tian G, Li N, Li Q, Ye M, Zheng H, Yu J, Wu H, Sun J, et al. The DNA methylome of human peripheral blood mononuclear cells. PLoS Biol. 2010;8:e1000533. doi: 10.1371/journal.pbio.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O'Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Green ED, Guyer MS National Human Genome Research Institute. Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470:204–213. doi: 10.1038/nature09764. [DOI] [PubMed] [Google Scholar]
- 199.Moch H, Blank PR, Dietel M, Elmberger G, Kerr KM, Palacios J, Penault-Llorca F, Rossi G, Szucs TD. Personalized cancer medicine and the future of pathology. Virchows Arch. 2012;460:3–8. doi: 10.1007/s00428-011-1179-6. [DOI] [PubMed] [Google Scholar]
- 200.Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul SA, Naing A, Janku F, Luthra R, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center Initiative. Clin Cancer Res. 2012;18:6373–6383. doi: 10.1158/1078-0432.CCR-12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Parkinson DR, Johnson BE, Sledge GW. Making personalized cancer medicine a reality: challenges and opportunities in the development of biomarkers and companion diagnostics. Clin Cancer Res. 2012;18:619–624. doi: 10.1158/1078-0432.CCR-11-2017. [DOI] [PubMed] [Google Scholar]
- 202.Modugno F, Edwards RP. Ovarian cancer: prevention, detection, and treatment of the disease and its recurrence. Molecular mechanisms and personalized medicine meeting report. Int J Gynecol Cancer. 2012;22:S45–S57. doi: 10.1097/IGC.0b013e31826bd1f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cho SH, Jeon J, Kim SI. Personalized medicine in breast cancer: a systematic review. J Breast Cancer. 2012;15:265–272. doi: 10.4048/jbc.2012.15.3.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Bar-Joseph Z, Gitter A, Simon I. Studying and modelling dynamic biological processes using time-series gene expression data. Nat Rev Genet. 2012;13:552–564. doi: 10.1038/nrg3244. [DOI] [PubMed] [Google Scholar]
- 206.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 207.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 210.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lam HY, Pan C, Clark MJ, Lacroute P, Chen R, Haraksingh R, O'Huallachain M, Gerstein MB, Kidd JM, Bustamante CD, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30:226–229. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Dorogovtsev SN, Goltsev AV, Mendes JFF. Critical phenomena in complex networks. Rev Mod Phys. 2008;80:1275–1335. [Google Scholar]
- 214.Albert R, Barabasi AL. Statistical mechanics of complex networks. Rev Mod Phys. 2002;74:47–97. [Google Scholar]
- 215.Alon U. Biological networks: the tinkerer as an engineer. Science. 2003;301:1866–1867. doi: 10.1126/science.1089072. [DOI] [PubMed] [Google Scholar]
- 216.Costa LF, Rodrigues FA, Cristino AS. Complex networks: the key to systems biology. Genet Mol Biol. 2008;31:591–601. [Google Scholar]
- 217.Levy ED, Pereira-Leal JB. Evolution and dynamics of protein interactions and networks. Curr Opin Struct Biol. 2008;18:349–357. doi: 10.1016/j.sbi.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 218.Schadt EE, Linderman MD, Sorenson J, Lee L, Nolan GP. Cloud and heterogeneous computing solutions exist today for the emerging big data problems in biology. Nat Rev Genet. 2011;12:224. doi: 10.1038/nrg2857-c2. [DOI] [PubMed] [Google Scholar]
- 219.Trelles O, Prins P, Snir M, Jansen RC. Big data, but are we ready? Nat Rev Genet. 2011;12:224. doi: 10.1038/nrg2857-c1. [DOI] [PubMed] [Google Scholar]
- 220.Biesecker LG. Opportunities and challenges for the integration of massively parallel genomic sequencing into clinical practice: lessons from the ClinSeq project. Genet Med. 2012;14:393–398. doi: 10.1038/gim.2011.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 222.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers. 2010;14:533–537. doi: 10.1089/gtmb.2010.0036. [DOI] [PubMed] [Google Scholar]
- 229.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wang LY, Abyzov A, Korbel JO, Snyder M, Gerstein M. MSB: a mean-shift-based approach for the analysis of structural variation in the genome. Genome Res. 2009;19:106–117. doi: 10.1101/gr.080069.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lam HY, Mu XJ, Stütz AM, Tanzer A, Cayting PD, Snyder M, Kim PM, Korbel JO, Gerstein MB. Nucleotide-resolution analysis of structural variants using BreakSeq and a breakpoint library. Nat Biotechnol. 2010;28:47–55. doi: 10.1038/nbt.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010;Chapter 11 doi: 10.1002/0471250953.bi1107s32. Unit 11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 241.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 243.Kuehn H, Liberzon A, Reich M, Mesirov JP. Using GenePattern for gene expression analysis. Curr Protoc Bioinformatics. 2008;Chapter 7 doi: 10.1002/0471250953.bi0712s22. Unit 7.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–510. doi: 10.1038/nbt.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Li JW, Schmieder R, Ward RM, Delenick J, Olivares EC, Mittelman D. SEQanswers: an open access community for collaboratively decoding genomes. Bioinformatics. 2012;28:1272–1273. doi: 10.1093/bioinformatics/bts128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Martens L, Chambers M, Sturm M, Kessner D, Levander F, Shofstahl J, Tang WH, Rompp A, Neumann S, Pizarro AD, et al. mzML — a community standard for mass spectrometry data. Mol Cell Proteomics. 2011;10:R110.000133. doi: 10.1074/mcp.R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Deutsch EW. Mass spectrometer output file format mzML. Methods Mol Biol. 2010;604:319–331. doi: 10.1007/978-1-60761-444-9_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Deutsch E. mzML: a single, unifying data format for mass spectrometer output. Proteomics. 2008;8:2776–2777. doi: 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- 250.Kessner D, Chambers M, Burke R, Agus D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 252.Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 253.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 254.Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. J Proteome Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 255.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 256.Elias JE, Gibbons FD, King OD, Roth FP, Gygi SP. Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nat Biotechnol. 2004;22:214–219. doi: 10.1038/nbt930. [DOI] [PubMed] [Google Scholar]
- 257.Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie GA, Ma B. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics. 2012;11:M111.010587. doi: 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pedrioli PG. Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol Biol. 2010;604:213–238. doi: 10.1007/978-1-60761-444-9_15. [DOI] [PubMed] [Google Scholar]
- 259.Keller A, Shteynberg D. Software pipeline and data analysis for MS/MS proteomics: the trans-proteomic pipeline. Methods Mol Biol. 2011;694:169–189. doi: 10.1007/978-1-60761-977-2_12. [DOI] [PubMed] [Google Scholar]
- 260.Deutsch EW, Shteynberg D, Lam H, Sun Z, Eng JK, Carapito C, von Haller PD, Tasman N, Mendoza L, Farrah T, et al. Trans-Proteomic Pipeline supports and improves analysis of electron transfer dissociation data sets. Proteomics. 2010;10:1190–1195. doi: 10.1002/pmic.200900567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, et al. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sturm M, Bertsch A, Gröpl C, Hildebrandt A, Hussong R, Lange E, Pfeifer N, Schulz-Trieglaff O, Zerck A, Reinert K, et al. OpenMS — an open-source software framework for mass spectrometry. BMC Bioinformatics. 2008;9:163. doi: 10.1186/1471-2105-9-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kohlbacher O, Reinert K, Gröpl C, Lange E, Pfeifer N, Schulz-Trieglaff O, Sturm M. TOPP — the OpenMS proteomics pipeline. Bioinformatics. 2007;23:e191–e197. doi: 10.1093/bioinformatics/btl299. [DOI] [PubMed] [Google Scholar]
- 264.Bertsch A, Gröpl C, Reinert K, Kohlbacher O. OpenMS and TOPP: open source software for LC-MS data analysis. Methods Mol Biol. 2011;696:353–367. doi: 10.1007/978-1-60761-987-1_23. [DOI] [PubMed] [Google Scholar]
- 265.Tautenhahn R, Patti GJ, Kalisiak E, Miyamoto T, Schmidt M, Lo FY, McBee J, Baliga NS, Siuzdak G. metaXCMS: second-order analysis of untargeted metabolomics data. Anal Chem. 2011;83:696–700. doi: 10.1021/ac102980g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 268.Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Katajamaa M, Miettinen J, Oresic M. MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics. 2006;22:634–636. doi: 10.1093/bioinformatics/btk039. [DOI] [PubMed] [Google Scholar]
- 270.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008;36:D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Caspi R, Altman T, Dale JM, Dreher K, Fulcher CA, Gilham F, Kaipa P, Karthikeyan AS, Kothari A, Krummenacker M, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2010;38:D473–D479. doi: 10.1093/nar/gkp875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2008;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]