

Abstract
The qualitative dimension of gene expression data and its heterogeneous nature in cancerous specimens can be accounted for by phylogenetic modeling that incorporates the directionality of altered gene expressions, complex patterns of expressions among a group of specimens, and data-based rather than specimen-based gene linkage. Our phylogenetic modeling approach is a double algorithmic technique that includes polarity assessment that brings out the qualitative value of the data, followed by maximum parsimony analysis that is most suitable for the data heterogeneity of cancer gene expression. We demonstrate that polarity assessment of expression values into derived and ancestral states, via outgroup comparison, reduces experimental noise; reveals dichotomously expressed asynchronous genes; and allows data pooling as well as comparability of intra- and interplatforms. Parsimony phylogenetic analysis of the polarized values produces a multidimensional classification of specimens into clades that reveal shared derived gene expressions (the synapomorphies); provides better assessment of ontogenic pathways and phyletic relatedness of specimens; efficiently utilizes dichotomously expressed genes; produces highly predictive class recognition; illustrates gene linkage and multiple developmental pathways; provides higher concordance between gene lists; and projects the direction of change among specimens. Further implication of this phylogenetic approach is that it may transform microarray into diagnostic, prognostic, and predictive tool.
Introduction
Gene microarray has been employed in studying comparative gene expression in cancer, genetic disorders, infections, drug response and interactions, as well as other biological processes (Quackenbush, 2006), and its data used to generate cancer taxonomy (Bittner et al., 2000; Golub, et al., 1999; Lossos and Morgensztern, 2006), diagnosis, prognosis (Beer, et al., 2002), subtyping/class discovery (Alizadeh, et al., 2000; Beer et al., 2002), and biomarker detection (Lossos and Morgensztern, 2006). However, after more than a decade since its introduction and subsequent wide usage, microarray gene expression is still facing a number of problems that are limiting its usefulness and potential (Harrison et al., 2007; Millenaar et al., 2006; Wang, et al., 2005). There are the problems of reproducibility of measurements between runs, instruments, or laboratories; the inability to perform intra- and interplatform comparability, pooling, and insufficient concordance of gene lists. Furthermore, there is the lack of an optimal bioinformatic tool to model the heterogeneity of gene expression of cancerous specimens, and due to the multiphasic nature of cancer, statistically significant gene expressions are not necessarily biologically meaningful during all phases of cancer. Current analytical paradigms such as phenetic clustering and maximum likelihood (including Bayesian) have not resolved these issues (Abu-Asab et al., 2008), and there is a total lack of an analytical paradigm that can transform microarray data into a multi-dimensional bioinformatic tool useful for a clinical setting.
Cancer incipience, progression, and maintenance are all evolutionary processes at the cellular and tissue levels; they mirror similar evolutionary processes at the population levels in that they all involve genetic modifications within an individual, selective pressure, and clonal propagation. Tumors derived from the same primary tumor become diverse and contain heterogeneous patterns of gene expression after a brief time of divergence. Data heterogeneity points out the existence of several phenomena: high genomic diversity in diseased specimens, high mutation rate, and possibly multiple pathways of disease development. To efficiently and accurately model these phenomena, biologically compatible methods of analysis should be used.
In an attempt to resolve some of the above listed problems through biologically compatible methodology and broaden the bioinformatic potential of the microarray technology, we introduce a parsimony phylogenetic approach for microarray data analysis that is based on outgroup comparison (a.k.a. polarity assessment) and maximum parsimony. This approach is a double-algorithmic procedure where the data values are first polarized into derived or ancestral depending on whether they fall within the range of the outgroup, which is usually composed of normal healthy specimens, then the polarized data is processed with a maximum parsimony algorithm. Maximum parsimony produces a phylo-genetic classification of the specimens that recognizes monophyletic classes (clades) that are delimited by shared derived gene expressions (the synapomorphies); it achieves that by finding the phylogenetic tree with the minimum steps to construct.
Biologically meaningful modeling and interpretation of the data, and better correlation with clinical characteristics, diagnosis, and outcomes are highly desired criteria in an analytical tool (Allison et al., 2006; Beer, et al., 2002; Bittner, et al., 2000; Golub, et al., 1999). Clustering specimens into unidimensional classification of discernable entities on the basis of overall quantitative gene expression similarities has some serious drawbacks (Allison et al., 2006; Lyons-Weiler et al., 2004), and appears to be incongruent with the nature of disease development (Abu-Asab, et al., 2006; 2008). In this report, we are demonstrating that the use of parsimony phylogenetic analysis of microarray data resolves the issues of gene-ranking discrepancies, improves interplatform concordance, makes possible intra- and interplatform comparability, eliminates biases in the gene linkage criteria, and casts gene expression profiles into a biologically relevant and predictive model of class discovery.
A superior classification is one that summarizes maximum knowledge about its specimens, reflects their true ontogenic relationships to one another, and offers predictivity (Farris, 1979; Golub, et al., 1999). The latter would especially be significant when the classification is applied in a clinical setting for diagnosis, prognosis, or posttreatment evaluation. We are utilizing parsimony phylogenetics because of its inherent ability to produce a robust classification of relationships—class discovery; and its forecasting power to reveal the characters of a specimen when its place in the classification is established—class prediction (Albert, 2005b). Parsimony models the heterogeneity of cancerous microarray data without any a priori assumptions (Goloboff and Pol, 2005; Siddall, 1998; Stefankovic and Vigoda, 2007). Additionally, a phylogenetic approach elucidates the direction of change among specimens that leads to their molecular and cellular diversity: the presence of one or more developmental pathway (Abu-Asab et al., 2008), and novel expressions that are involved in the progression and maintenance of the disease.
A strict parsimony phylogenetic analysis uses only shared derived values, synapomorphies, to delimit a natural group of specimens within a clade (Wiley and Siegel-Causey, 1991). Shared derived values of a gene among several specimens constitute a synapomorphy; therefore, only a synapomorphy is indicative of their relatedness. Because synapomorphies define clades at various grouping levels, a parsimonious phylogenetic classification reflects hierarchical shared developmental pathways among a group of specimens and may reveal the presence of subclasses with each having its own uniquely derived gene expression synapomorphies. In biological and clinical senses, class discovery and prediction should be based on shared derived gene expressions (i.e., synapomorphies). For example, a cancer class (a clade in phylogenetic terminology) is delimited by one or more synapomorphies, and a cancerous specimen will be placed in a class only if it shares the same synapomorphies with the members of the clade.
In this study, we are describing a double-algorithmic analytical method of microarray gene-expression data based on polarity assessment algorithm, UNIPAL (Abu-Asab, et al., 2006) where the polarized values can be used by a parsimony algorithm, MIX (Felsenstein, 1989) to produce a phylogenetic classification of specimens. This approach brings in a systematic solution to class discovery through phylogenetic classification whereby every class is delimited by shared derived gene expressions—i.e., synapomorphies-delimited clades. Because such a classification reflects the shared aberrations of gene expressions of the specimens, we expect it to have a biological and clinical relevance, and to advance targeted treatments of disease.
Materials and Methods
Gene expression datasets
In order to demonstrate the applicability of parsimony phylogenetics to microarray gene expression data, and test the results of interplatform concordance and comparability, we downloaded three publicly available datasets of gene expression comparative studies, GDS484 (Hoffman et al., 2004), GDS533 (Quade et al., 2004), and GDS1210 (Hippo et al., 2002), from NCBI’s Gene Expression Omnibus (http://www. ncbi.nlm.nih.gov/geo/). The GDS484 was conducted on GPL96 (Affymetrix GeneChip Human Genome U133 Array Set HG-U133A), and the other two studies on GPL80 (Affymetrix GeneChip Human Full Length Array HuGeneFL). The GDS484 was comprised of normal myometrium (n = 5) and uterine leiomyomas (n = 5) obtained from fibroid afflicted patients. The GDS533 study encompassed normal myometrium (n = 4), benign uterine leiomyoma (n = 7), as well as malignant uterine (n = 9) and extrauterine (n = 4) leiomyosarcoma specimens. The GDS1210 study included expression profiling of 22 primary advanced gastric cancer tissues and 8 normal specimens.
Polarity assessment and parsimony analysis
Polarity assessment through outgroup comparison does not use comparison of means and folds but rather it converts the continuous values into discontinuous ones through the assessment of each gene’s values against that of the normals’ range and produces a matrix of polarized values (0s and 1s). Our polarity assessment program, UNIPAL, compares independently each gene’s value of experimental specimens against its corresponding range within the outgroup, and scores each as either derived (1) or ancestral (0), so the matrix of gene expression values is transformed into a matrix of polarized scores (0s and 1s).
We used all the expression data points of all specimens in the analysis without any a priori selection of only a specific cluster of data. For polarity assessment (apomorphic [or derived] versus plesiomorphic [or ancestral]), data was polarized with our customized algorithm (UNIPAL) that recognized derived values of each gene when compared with the outgroups (Abu-Asab et al., 2006). Outgroups here were composed of normal healthy specimens only. Ideally, the outgroup should be large enough to encompass the maximum variation within normal healthy population. UNIPAL is freely available for noncommercial use from the authors.
The phylogenetic analysis was carried out with MIX, the maximum parsimony program of PHYLIP ver. 3.57c (Felsenstein, 1989), to produce separate parsimony phylogenetic analyses for each dataset, and the inclusive matrix of the two sets (GDS533 and GDS1210), which included all their specimens. MIX was run in randomized and nonrandomized inputs, and no significant differences were observed between the two options.
Phylogenetic trees were drawn using TreeView (Page, 1996).
Interplatform concordance and comparability
To test interplatform concordance when analyzed parsimoniously, we compared the synapomorphies of the two uterine leiomyoma datasets, GDS484 and GDS533, and recorded the percentage of concordance.
To test interplatform comparability (i.e., whether datasets can be pooled together for a parsimony analysis), we combined the polarized matrices of the two identical platform datasets, GDS533 and GDS1210, processed the combined matrix by MIX, and compared the result to their separate cladograms.
Results
The implications of a parsimonious analysis of the gene expression data are realized at several aspects: the recognition and utilization of partially asynchronous genes and dichotomously expressed asynchronous genes; the implications of outgroup selection and its effect on significant gene listing, better interplatform concordance and comparability, as well as the practical usefulness of the multidimensional cladograms.
Dichotomously expressed asynchronous (DEA) genes
Our analysis identified a specific punctuated pattern of gene expression that seemed to occur only in a set of specimens where a gene’s expression values were around the normals’ distribution (over and underexpressed), but did not overlap with it (Table 1–Table 7). This pattern has been only recognized once in the literature but was not named (Lyons-Weiler et al., 2004); we termed this phenomenon dichotomous asynchronicity to reflect its two-tailed distribution and deviation from the normal expression range.
TABLE 1.
SYNAPOMORPHIES DEFINING A CLADE OF LEIOMYOMA AND LEIOMYOSARCOMA SPECIMENS IN COMPARISON TO NORMAL SPECIMENS (GDS533)
| A. Overexpressed synapomorphic genes: | ||
| D00596 | TYMS thymidylate synthetase | OE[1, 2] |
| B. Underexpressed synapomorphic genes: | ||
| L19871 | ATF3 activating transcription factor 3 | UE[1, 2] |
| U62015 | CYR61 cysteine-rich, angiogenic inducer, 61 | UE[1, 2] |
| X68277 | DUSP1 dual specificity phosphatase 1 | UE[1, 2] |
| V01512 | FOS v-fos FBJ murine osteosarcoma viral oncogene homolog | UE[1], NS [2] |
| L49169 | FOSB FBJ murine osteosarcoma viral oncogene homolog B | NS[1], UE[2] |
| J04111 | JUN v-jun sarcoma virus 17 oncogene homolog (avian) | UE[1, 2] |
| Y00503 | KRT19 keratin 19 | UE[1], NS [2] |
| U24488 | TNXB tenascin XB | UE[1], UE,OE[2] |
| C. Dichotomously-expressed synapomorphic genes: | ||
| M31994 | ALDH1A1 aldehyde dehydrogenase 1 family, member A1 | UE[1], NS[2] |
| X05409 | ALDH2 aldehyde dehydrogenase 2 family (mitochondrial) | NS[1, 2] |
| D25304 | ARHGEF6 Rac/Cdc42 guanine nucleotide exchange factor (GEF) 6 | NS[1, 2] |
| K03430 | C1QB complement component 1, q subcomponent, B chain | NS[1], OE[2] |
| U60521 | CASP9 caspase 9, apoptosis-related cysteine peptidase | NS[1, 2] |
| M73720 | CPA3 carboxypeptidase A3 (mast cell) | NS[1, 2] |
| HG2663-HT2759_at | Cpg-Enriched DNA, Clone S19 (HG3995-HT4265) | NS[1, 2] |
| M14676 | FYN oncogene related to SRC, FGR, YES | NS[1, 2] |
| M34677 | F8A1 coagulation factor VIII-associated (intronic transcript) 1 | OE,UE[2] |
| U60061 | FEZ2 fasciculation and elongation protein zeta 2 (zygin II) | NS[1, 2] |
| U86529 | GSTZ1 glutathione transferase zeta 1 (maleylacetoacetate isomerase) | NS[1, 2] |
| HG358-HT358_at | Homeotic Protein 7, Notch Group (HG358-HT358) | NS[2] |
| AB002365 | KIAA0367 BCH motif-containing molecule at the carboxyl terminal region 1 | NS[1], OE,UE[2] |
| U37283 | MFAP5 microfibrillar associated protein 5 | NS[1, 2] |
| HG406-HT406_at | MFI2 antigen p97 (melanoma associated) identified by monoclonal antibodies 133.2 and 96.5 | NS[1, 2] |
| M55593 | MMP2 matrix metallopeptidase 2 (gelatinase A, 72kDa gelatinase, 72kDa type IV collagenase) | NS[1], OE,UE[2] |
| M76732 | MSX1 msh homeobox homolog 1 | NS[1, 2] |
| L48513 | PON2 paraoxonase 2 | NS[1, 2] |
| U77594 | RARRES2 retinoic acid receptor responder (tazarotene induced) 2 | NS[1, 2] |
| M11433 | RBP1 retinol binding protein 1 | NS[1, 2] |
| L03411 | RDBP RD RNA binding protein | NS[1], OE[2] |
| Z29083 | TPBG trophoblast glycoprotein | NS[1, 2] |
| S73591 | TXNIP thioredoxin interacting protein | NS[1, 2] |
Synapomorphies include: OE gene, 8 UE genes, and 23 DE genes. Last column reports the status of the synapomorphies as described by [1] Hoffman et al. (2004) and [2] Quade et al. (2004) in their significant genes’ lists. DE = dichotomously-expressed; NS = not significant; OE = overexpressed; UN = underexpressed.
TABLE 7.
INTERPLATFORM CONCORDANCE
| GDS533 | GDS484 |
|---|---|
| A. Overexpressed synapomorphic genes: | |
| a. Identical Synapomorphies | |
| DDB2 | DDB2 |
| FUT8 | FUT8 |
| MEST | MEST |
| TMSL8 | TMSL8 |
| TYMS | TYMS |
| b. Homologous synapomorphies | |
| CACNB3 | CACNA1C |
| COL5A2 | COL4A5 |
| KIAA0367 | KIAA0101 |
| PRKAR1B | PRKACB |
| B. Underexpressed synapomorphic genes: | |
| a. Identical synapomorphies | |
| ALDH1A1 | ALDH1A1 |
| ALDH2 | ALDH2 |
| ATF3 | ATF3 |
| CEBPD | CEBPD |
| CXADR | CXADR |
| CYR61 | CYR61 |
| DUSP1 | DUSP1 |
| FOS | FOS |
| HRASLS3 | HRASLS3 |
| IER2 | IER2 |
| JUN | JUN |
| KRT19 | KRT19 |
| RARRES2 | RARRES2 |
| TACSTD2 | TACSTD2 |
| TNXB | TNXB |
| VIL2 | VIL2 |
| b. Homologous synapomorphies | |
| CASP9 | CASP4 |
| CYBA | CYB5R1 |
| FOSB | FOS |
| JUNB | JUN |
| PPP4C | PPP1R10 |
| SLC20A1 | SLC18A2 |
| THBS1 | THBD |
| WDR43 | WDR37 |
| C. Dichotomously expressed synapomorphic genes: | |
| a. Identical synapomorphies | |
| CTSB | CTSB |
| b. Homologous synapomorphies | |
| ARL4D | ARL4C |
| FOXO1A | FOXJ3 |
| GNB3 | GNB1L |
| ITGA6 | ITGA2B |
| ITGA9 | ITGA2B |
| KCNK1 | KCNJ5 |
| MFAP5 | MFAP4 |
| PSMC3 | PSMC2 |
| SELP | SELL |
| TXNIP | TXNDC13 |
| ZNF43 | ZNF259P |
A list of overlapping identical (22) and homologous (23) synapomorphic genes in leiomyoma specimens of GDS484 and GDS533. These include: 9 OE, 24 UE, and 12 DE.
Although t-statistic and fold-change may dismiss these asynchronous genes from the list of differentially expressed genes, or misrepresent their significance (Lyons-Weiler et al., 2004), an outgroup polarity assessment will assess each value as derived and let the parsimony algorithm reveal its significance in relation to the rest of the genes. A parsimony phylogenetic algorithm uses the polarized values of all genes to produce the most parsimonious classification, the one with the lowest number of reversals and parallelisms (i.e., minimizes multiple origins of expression states in hypothesizing the relationships among the specimens) (Albert, 2005a; Felsenstein, 2004).
Through polarity assessment a large number of DEA genes were recognized. All these genes had their expression values above and below that of the normal specimens’ range, that is, derived in relation to outgroups thus pointing out the heterogeneity that exists among specimens. DEA genes were found in all the three datasets studied here (Table 1–Table 7), and were included within all the analyses.
Most parsimonious cladograms
Parsimony analysis produced one most parsimonious cladogram (having the least number of steps in constructing a classification of specimens) for the uterine GDS533 dataset (Fig. 1). The topology of the tree showed one large inclusive clade that encompassed all of the leiomyomas and leiomyosarcomas delimited by 32 synapomorphies (Table 1), a terminal clade with nine sarcoma specimens, middle sarcoma clade with four specimens, five small basal leiomyoma clades in tandem arrangement, followed by four basal normal clades.
FIG. 1.
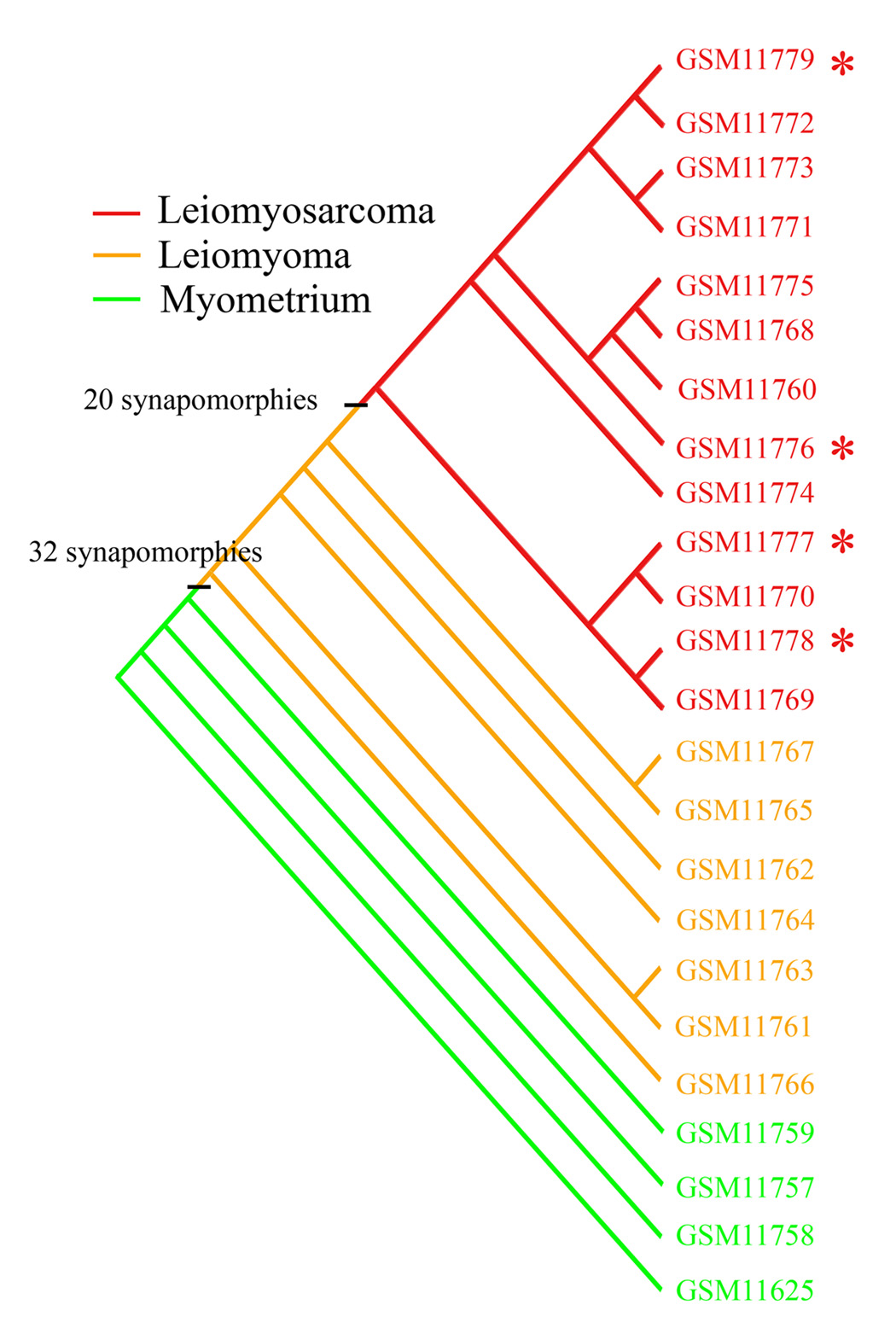
A cladogram of a parsimony phylogenetic analysis of microarray gene-expression data representing normal myometrium (n = 4), leiomyoma (n = 7), leiomyosarcoma (n = 9), and extrauterine leiomyosarcoma (n = 4) specimens. The leiomyomas and leiomyosarcomas form a clade defined by 32 synapomorphies (Table 1). The leiomyosarcoma specimens form a terminal clade that is circumscribed by 20 synapomorphies (Table 3). Asterisk (*) denotes extrauterine leiomyosarcomas.
The cladogram in Figure 1 showed that the leiomyoma specimens did not form a natural group by themselves—they did not form their own clade separating them from the leiomyosarcomas, and there were no synapomorphies circumscribing them as a clade when the ingroup was composed of leiomyoma and leiomyosarcoma and the outgroup composed of the normals. However, the leiomyomas shared 146 synapomorphies distinguishing them from the normals (Table 2).
TABLE 2.
SYNAPOMORPHIES OF LEIOMYOMA SPECIMENS IN COMPARISON TO NORMAL SPECIMENS (GDS533)
| A. Overexpressed synapomorphic genes: | ||
| D16469 | ATP6AP1 ATPase, H+ transporting, lysosomal accessory protein 1 | NS[1, 2] |
| U07139 | CACNB3 calcium channel, voltage-dependent, beta 3 subunit | NS[1], OE[2] |
| M11718 | COL5A2 collagen, type V, alpha 2 | NS[1, 2] |
| U18300 | DDB2 damage-specific DNA binding protein 2, 48 kDa | NS[1, 2] |
| D38550 | E2F3 E2F transcription factor 3 | NS[1, 2] |
| M34677 | F8A1 coagulation factor VIII-associated (intronic transcript) 1 | NS[1, 2] |
| D89289 | FUT8 fucosyltransferase 8 (alpha (1,6) fucosyltransferase) | NS[1, 2] |
| D86962 | GRB10 growth factor receptor-bound protein 10 | NS[1, 2] |
| M32053 | H19, imprinted maternally expressed untranslated mRNA | NS[1, 2] |
| U07664 | HLXB9 homeobox HB9 | OE[1, 2] |
| D87452 | IHPK1 inositol hexaphosphate kinase 1 | NS[1, 2] |
| U51336 | ITPK1 inositol 1,3,4-triphosphate 5/6 kinase | NS[1, 2] |
| ABA002365 | KIAA0367 | NS[1], OE[2] |
| D78611 | MEST mesoderm specific transcript homolog (mouse) | OE[1], NS[2] |
| U19718 | MFAP2 microfibrillar-associated protein 2 | NS[1, 2] |
| M55593 | MMP2 matrix metallopeptidase 2 (gelatinase A, 72 kDa gelatinase, 72kDa type IV collagenase) | OE[1, 2] |
| U79247 | PCDH11X protocadherin 11 X-linked | NS[1, 2] |
| L24559 | POLA2 polymerase (DNA directed), alpha 2 (70 kDa subunit) | NS[1, 2] |
| M65066 | PRKAR1B protein kinase, cAMP-dependent, regulatory, type I, beta | NS[1, 2] |
| D14694 | PTDSS1 phosphatidylserine synthase 1 | NS[1, 2] |
| U24186 | RPA4 replication protein A4, 34 kDa | NS[1, 2] |
| U85658 | TFAP2C transcription factor AP-2 gamma (activating enhancer binding protein 2 gamma) | NS[1, 2] |
| D82345 | TMSL8 thymosin-like 8 | NS[1, 2] |
| D85376 | TRHR thyrotropin-releasing hormone receptor | NS[1, 2] |
| D00596 | TYMS* thymidylate synthetase | OE[1, 2] |
| B. Underexpressed synapomorphic genes: | ||
| X0330 | ADH1B alcohol dehydrogenase IB (class I), beta polypeptide | NS[1, 2] |
| M31994 | ALDH1A1* aldehyde dehydrogenaes 1 family, member A1 | UE[1], NS[2] |
| X05409 | ALDH2* aldehyde dehydrogenase 2 family (mitochondrial) | NS[1, 2] |
| L19871 | ATF3* activating transcription factor 3 | UE[1, 2] |
| U60521 | CASP9 caspase 9, apoptosis-related cysteine peptidase | NS[1, 2] |
| D49372 | CCL11 chemokine (C—C motif) ligand 11 | NS[1, 2] |
| X05323 | CD200 molecule | NS[1, 2] |
| M83667 | CEBPD CCAAT/enhancer binding protein (C/EBP), delta | NS[1, 2] |
| U90716 | CXADR coxsackie virus and adenovirus receptor | NS[1, 2] |
| M21186 | CYBA cytochrome b-245, alpha polypeptide | NS[1, 2] |
| U62015 | CYR61* cysteine-rich, angiogenic inducer, 61 | UE[1, 2] |
| Z22865 | DPT dermatopontin | NS[1, 2] |
| X56807 | DSC2 desmocollin 2 | NS[1, 2] |
| X68277 | DUSP1* dual specificity phosphatase 1 | UE[1, 2] |
| V01512 | FOS* v-fos FBJ murine osteosarcoma viral oncogene homolog | UE[1], NS[2] |
| L49169 | FOSB FBJ murine osteosarcoma viral oncogene homolog B | NS[1], UE[2] |
| L11238 | GP5 glycoprotein V (platelet) | NS[1, 2] |
| M36284 | GYPC glycophorin C (Gerbich blood group) | NS[1, 2] |
| M60750 | HIST1H2BG histone cluster 1, H2bg | NS[1, 2] |
| X79200 | Homo spaiens mRNA for SYT-SSX protein | NS[1, 2] |
| X92814 | HRASLS3 HRAS-like suppressor 3 | NS[1, 2] |
| M62831 | IER2 immediate early response 2 | NS[1, 2] |
| J04111 | JUN* v-jun sarcoma virus 17 oncogene homolog (avian) | UE[1, 2] |
| Y00503 | KRT19* keratin 19 | NS[1, 2] |
| X89430 | MECP2 methyl CpG binding protein 2 (Rett syndrome) | NS[1, 2] |
| U46499 | MGST1 microsomal glutathione S-transferase 1 | NS[1, 2] |
| M93221 | MRC1 mannose receptor, C type 1 | NS[1, 2] |
| M76732 | MSX1* msh homeobox homolog 1 (Drosophila) | NS[1, 2] |
| S71824 | NCAM1 neural cell adhesion molecule 1 | OE[1], NS[2] |
| X70218 | PPP4C protein phosphatase 4 | NS[1, 2] |
| U02680 | PTK9 protein tyrosine kinase 9 | NS[1, 2] |
| U79291 | PTPN11 protein tyrosine phosphatase, non-receptor type 11 (Noonan syndrome 1) | NS[1, 2] |
| U77594 | RARRES2* retinoic acid receptor responder (tazarotene induced) 2 | NS[1, 2] |
| M11433, X07438 | RBP1* retinol binding protein 1, cellular | NS[1, 2] |
| L20859 | SLC20A1 solute carrier family 20 (phosphate transporter), member 1 | NS[1, 2] |
| M97935 | STAT1 signal transducer and activator of transcription 1, 91kDa | NS[1, 2] |
| J04152 | TACSTD2 tumor-associated calcium signal transducer 2 | NS[1, 2] |
| X14787 | THBS1 thrombospondin 1 | NS[1, 2] |
| U24488 | TNXB* tenascin XB | UE[1, 2] |
| Z29083 | TPBG* trophoblast glycoprotein | NS[1, 2] |
| X51521 | VIL2 villin 2 (ezrin) | UE[1], NS[2] |
| D87716 | WDR43 WD repeat domain 43 | NS[1, 2] |
| C. Dichotomously-expressed synapomorphic genes: | ||
| ABCB1; ADRM1; AIM1; ALDH1A3; AMDD; ARHGEF6; ARL4D; ATP5B; Atp8a2; C1QB; CA9; CALM2; CTSB; CCRL2; CD52; CD99; CPA3; DPYD; DSG2; Emx2; FEZ2; FLNA; FOXO1A; FYN; GAPDH; GNB3; GSTZ1; H1F0; H2-ALPHA; HBG2; Ubx, Notch1; Hox5.4; HTR2C; ICA1; IGF2; INSR; ITGA6; ITGA9; KCNK1; KIAA0152; MAP1D; MATK; MBP; MDM4; MFAP5; MFI2 antigen p97; MLH1; MPZ; NDUFS1; NELL2; NNAT; NOS3; NR4A1; OASL; ODC1; OLFM1; PKN2; PON2; PRMT2; PSMC3; PTR2; RANBP2; RBMX; RDBP; RHOG; SAFB2; SCRIB; SELFP; SERPINF1; SMS; SPOCK2; ST3GAL1; THRA; TNXB; TTLL4; TXNIP; UPK2; XA; ZNF43 | ||
These comprise: 25 OE genes, 42 UE genes, and 79 DE genes.
Asterisk (*) indicates a synapomorphy for leiomyosarcoma as well. Last column reports the status of the synapomorphies as described by [1] Hoffman et al. (2004) and [2] Quade et al. (2004) in their significant genes lists.
The 13 leiomyosarcoma specimens separated into a large terminal clade that was delimited by 20 synapomorphies in comparison with an outgroup composed of leiomyoma and normal specimens (Table 3), and 29 synapomorphies derived in relation to leiomyomas only as an outgroup (Table 4). Extrauterine sarcoma specimens did not assemble together, but rather were scattered within the sarcoma clades (denoted by * on the cladogram in Fig. 1). When the leiomyomas were removed from the comparison, there were 156 synapomorphies delimiting the sarcomas (Table 5).
TABLE 3.
A CLADE OF ALL LEIOMYOSARCOMA SPECIMENS DEFINED BY 20 SYNAPOMORPHIES IN COMPARISON TO NORMAL AND LEIOMYOMA SPECIMENS
| A. Overexpressed synapomorphic genes: | ||
| X54942 | CKS2 CDC28 proteininase regulatory subunit 2 | NS |
| U68566 | HAX1 HCLS1 associated protein X-1 | NS |
| L03411 | RDBP RD RNA binding protein | OE |
| X59543 | RRM1 ribonucleotide reductase M1 polypeptide | NS |
| B. Underexpressed synapomorphic genes: | ||
| D13639 | CCND2, cyclin D2 | UE |
| D21337 | COL4A6 collagen, type IV, alpha 6 | UE |
| HG2810-HT2921_at | Csh2 chorionic somatomammotropin hormone 2 [Rattus norvegicus] | NS |
| L36033 | CXCL12 chemokine (C-X-C motif) ligand 12 (stromal cell-derived factor 1) | NS |
| HG2663-HT2759_at | EMX2 empty spiracles homolog 2 (Drosophila). Homeotic Protein Emx2 | NS |
| HG2663-HT2759_at | Homeotic Protein Emx2 | NS |
| HG2810-HT2921_at | HOXA10 homeobox A10 Expressed in the adult human endometrium | UE |
| AB002382 | LOC284394 hypothetical gene supported by NM_001331 | NS |
| U69263 | MATN2 matrilin 2 | UE |
| U85707 | Meis1, myeloid ecotropic viral integration site 1 homolog (mouse) | UE |
| Z29678 | MITF microphthalmia-associated transcription factor | UE |
| L35240 | PDLIM7 PDZ and LIM domain 7 (enigma) | NS |
| D87735 | RL14 ribosomal protein L14 | NS |
| L14076 | SFRS4 splicing factor, arginine/serine-rich 4 | UE |
| J05243 | SPTAN1 spectrin, alpha, nonerythrocytic 1 (alpha-fodrin) | NS |
| C. Dichotomously-expressed synapomorphic genes: | ||
| M33197 | GAPDH glyceraldehyde-3-phosphate dehydrogenase | NS |
Last column reports the status of the synapomorphies as described by Quade et al. (2004) in their significant genes list.
TABLE 4.
A CLADE OF ALL LEIOMYOSARCOMA SPECIMENS DEFINED BY 29 SYNAPOMORPHIES IN COMPARISON TO LEIOMYOMA SPECIMENS ONLY (GDS533)
| A. Overexpressed synapomorphic genes: | ||
| X54941 | CKS1B CD28 protein kinase regulatory subunit 1B | OE |
| X54942 | CKS2 CD28 protein kinase regulatory subunit 2 | NS |
| J03060 | GBAP glucosidase, beta; acid, pseudogene | NS |
| U78027 | GLA galactosidase, alpha (associated w/Fabry’s) RPL36A ribosomal protein L36a | NS |
| Y00433 | GPX1 glutathione peroxidase 1 | NS |
| U68566 | HAX1 HCLS1 associated protein X-1 | NS |
| X59543 | RRM1 ribonucleotide reductase M1 polypeptide | NS |
| U12465 | RPL35 ribosomal protein L35 | OE |
| U67674 | SLC10A2 solute carrier family 10 (sodium/bile acid cotransporter family), member 2 | NS |
| B. Underexpressed synapomorphic genes: | ||
| U87223 | CNTNAP1 contactin associated protein 1 | UE |
| D30655 | EIF4A2 eukaryotic translation initiation factor 4A, isoform 2 | UE |
| L20814 | GRIA2 glutamate receptor, ionotropic, AMPA 2 | UE |
| M10051 | INSR insulin receptor | NS |
| D79999 | LOC221181 hypothetical gene supported by NM_006437 | NS |
| D14812 | MORF4L2 mortality factor 4 like 2 | UE |
| L36151 | PIK4CA phosphatidylinositol 4-kinase, catalytic, alpha | NS |
| D42108 | PLCL1 phospholipase C-like 1 | NS |
| L13434 | RpL41 Ribosomal protein L41 | NS |
| HG921-HT3995_at | Serine/Threonine Kinase, Receptor 2-2, Alt. Splice 3 | NS |
| D31891 | SETDB1 SET domain, bifurcated 1 | UE |
| AB002318 | Talin2 | NS |
| U53209 | TRA2A transformer-2 alpha | NS |
| D87292 | TST thiosulfate sulfurtransferase (rhodanese) | NS |
| M15990 | YES1 v-yes-1 Yamaguchi sarcoma viral oncogene homolog 1 | NS |
| C. Dichotomously expressed synapomorphic genes: | ||
| U56417 | AGPAT1 1-acylglycerol-3-phosphate O-acyltransferase 1 (lysophosphatidic acid acyltransferase, alpha) | NS |
| M63167 | AKT1 v-akt murine thymoma viral oncogene homolog 1 | NS |
| L27560 | IGFBP5 insulin-like growth factor binding protein 5 | NS |
| U40223 | P2RY4 pyrimidinergic receptor P2Y, G-protein coupled, 4 | NS |
| D76444 | RNF103 ring finger protein 103 | NS |
Last column reports the status of the synapomorphies as described by Quade et al. (2004) in their significant genes list
TABLE 5.
A CLADE COMPOSED OF ALL LEIOMYOSARCOMA SPECIMENS IS DEFINED IN RELATION TO NORMAL SPECIMENS (GDS533)
| A. Overexpressed synapomorphic genes: | ||
| S78187 | CDC25B cell division cycle 25B | NS |
| U40343 | CDKN2D cyclin-dependent kinase inhibitor 2D (p19, inhibits CDK4) | NS |
| X54942 | CKS2 CDC28 protein kinase regulatory subunit 2 | NS |
| X79353 | CDI1 GDP dissociation inhibitor 1 | NS |
| X14850 | H2AFX H2A histone family, member X | NS |
| U51127 | IRF5 interferon regulatory factor 5 | NS |
| U04209 | MFAP1 microfibrillar-associated protein 1 | NS |
| U43177 | MpV17 mitochondrial inner membrane protein | NS |
| U19796 | MRPL28 mitochondrial ribosomal protein L28 | OE |
| U37690 | POLR2L polymerase (RNA) II (DNA directed) polypeptide L, 7.6 kDa | NS |
| M22960 | PPGB protective protein for beta-galactosidase (galactosialidosis) | NS |
| U09210 | SLC18A3 solute carrier family 18 (vesicular acetylcholine), member 3 | NS |
| M86752 | STIP1 stress-induced-phosphoprotein 1 (Hsp70/Hsp90-organizing protein) | OE |
| M26880 | UBC ubiquitin C | OE |
| U43177 | UCN urocortin | NS |
| B. Underexpressed synapomorphic genes: | ||
| M12963 | ADH1A alcohol dehydrogenase 1A (class I), alpha polypeptide | UE |
| HG3638-HT3849_s_at | Amyloid Beta (A4) Precursor Protein, Alt. Splice 2, A4(751) | NS |
| L28997 | ARL1 ADP-ribosylation factor-like 1 | NS |
| Z49269 | CCL14 chemokine (C—C motif) ligand 14 | UE |
| M92934 | CTGF connective tissue growth factor | UE |
| M74099 | CUTL1 cut-like 1, CCAAT displacement protein (Drosophila) | NS |
| M96859 | DPP6 dipeptidyl-peptidase 6 | UE |
| U94855 | EIF3S5 eukaryotic translation initiation factor 3, subunit 5 epsilon, 47kDa | NS |
| L25878 | EPHX1 epoxide hydrolase 1, microsomal (xenobiotic) | NS |
| U60061-U69140 | FEZ2 fasciculation and elongation protein zeta 2 (zygin II) | NS |
| X67491 | GLUDP5 glutamate dehydrogenase pseudogene 5 | NS |
| HG4334-HT4604_s_at | Glycogenin | NS |
| X53296 | IL1RN interleukin 1 receptor antagonist | NS |
| X55740 | NT53 5′-nucleotidase, ecto (CD73) | UE |
| X78136 | PCBP2 poly(rC) binding protein 2 | UE |
| Z50194 | PHLDA1 pleckstrin homology-like domain, family A, member 1 | NS |
| J02902 | PPP2R1A protein phosphatase 2 (formerly 2A), regulatory subunit A (PR 65), alpha isoform | NS |
| J03805 | PPP2CB protein phosphatase 2, catalytic subunit, beta isoform | NS |
| U25988 | PSG11 pregnancy specific beta-1-glycoprotein 11 | NS |
| M98539 | PTGDS prostaglandin D2 synthase 21kDa (brain) | UE |
| X54131 | PTPRB protein tyrosine phosphatase, receptor type, B | NS |
| M12174 | RHOB ras homolog gene family, member B | NS |
| HG1879-HT3521_at | RHOQ ras homolog gene family, member Q | NS |
| X98534 | VASP vasodilator-stimulated phosphoprotein | NS |
| X51630 | WT1 Wilms tumor 1 | UE |
| HG3426-HT3610_s_at | Zinc Finger Protein Hzf-16, Kruppel-Like, Alt. Splice 1 | NS |
| M92843 | ZFP36 zinc finger protein 36, C3H type, homolog (mouse) | UE |
| C. Dichotomously expressed synapomorphic genes: | ||
| U80226 | ABAT 4-aminobutyrate aminotransferase | NS |
| M14758 | ABCB1 ATP-binding cassette, sub-family B (MDR/TAP), member 1 | NS |
| M95178 | ACTN1 actinin, alpha 1 | NS |
| U76421 | ADARB12 adenosine deaminase, RNA-specific, B1 (RED1 homolog rat) | NS |
| U46689 | ALDH3A2 aldehyde dehydrogenase 3 family, member A2 | NS |
| L34820 | ALDH5A1 aldehyde dehydrogenase 5 family, member A1 (succinate-semialdehyde dehydrogenase) | NS |
| M84332 | ARF1 ADP-ribosylation factor 1 | NS |
| D14710 | ATP5A1 ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1, cardiac muscle | NS |
| X84213 | BAK1 BCL2-antagonist/killer 1 | NS |
| U23070 | BAMBI BMP and activin membrane-bound inhibitor homolog (Xenopus laevis) | NS |
| M33518 | BAT2 HLA-B associated transcript 2 | NS |
| X61123 | BTG1 B-cell translocation gene 1, anti-proliferative | NS |
| S60415 | CACNB2 calcium channel, voltage-dependent, beta 2 subunit | NS |
| M19878 | CALB1 calbindin 1, 28 kDa | NS |
| L76380 | CALCRL calcitonin receptor-like | NS |
| M21121 | CCL5 chemokine (C—C motif) ligand 5 | NS |
| D14664 | CD302 CD302 molecule | NS |
| X72964 | CETN2 centrin, EF-hand protein, 2 | NS |
| U66468 | CGREF1 cell growth regulator with EF-hand domain 1 | NS |
| M63379 | CLU clusterin | NS |
| X52022 | COL6A3 collagen, type VI, alpha 3 | UE |
| L25286 | COL15A1 collagen, type XV, alpha 1 | NS |
| S45630 | CRYAB crystallin, alpha B | NS |
| X95325 | CSDA cold shock domain protein A | NS |
| U03100 | CTNNA1 catenin (cadherin-associated protein), alpha 1, 102kDa | NS |
| X52142 | CTPS CTP synthase | NS |
| D38549 | CYFIP1 cytoplasmic FMR1 interacting protein 1 | NS |
| X64229 | DEK DEK oncogene (DNA binding) | NS |
| M63391 | DES desmin | UE |
| Z34918 | EIF4G3 eukaryotic translation initiation factor 4 gamma, 3 | NS |
| U97018 | EML1 echinoderm microtubule associated protein like 1 | NS |
| U12255 | FCGRT Fc fragment of IgG, receptor, transporter, alpha | NS |
| U36922 | FOXO1A forkhead box O1A (rhabdomyosarcoma) | NS |
| U91903 | FRZB frizzled-related protein | NS |
| M33197 | GAPDH glyceraldehyde-3-phosphate dehydrogenase | NS |
| U09587 | GARS glycyl-tRNA synthetase | NS |
| U66075 | GATA6 GATA binding protein 6 | NS |
| D13988 | GDI2 GDP dissociation inhibitor 2 | NS |
| U31176 | GFER growth factor, augmenter of liver regeneration (ERV1 homolog, S. cerevisiae) | NS |
| U28811 | GLG1 golgi apparatus protein 1 | NS |
| U66578 | GPR23 G protein-coupled receptor 23 | NS |
| L40027 | GSK3A glycogen synthase kinase 3 alpha | NS |
| U77948 | GTF2I general transcription factor II, i | UE |
| Z29481 | HAAO 3-hydroxyanthranilate 3,4-dioxygenase | NS |
| D16480 | HADHA hydroxyacyl-Coenzyme A dehydrogenase/3-ketoacyl-Coenzyme A thiolase/enoyl-Coenzyme A hydratase (trifunctional protein), alpha subunit | NS |
| U50079 | HDAC1 histone deacetylase 1 | NS |
| U50078 | HERC1 hect (homologous to the E6-AP (UBE3A) carboxyl terminus) domain and RCC1 (CHC1)-like domain (RLD) 1 | NS |
| M95623 | HMBS hydroxymethylbilane synthase | NS |
| X79536 | HNRPA1 heterogeneous nuclear ribonucleoprotein A1 | NS |
| L15189 | HSPA9B heat shock 70 kDa protein 9B (mortalin-2) | NS |
| U05875 | IFNGR2 interferon gamma receptor 2 (interferon gamma transducer 1) | NS |
| X57025 | IGF1 insulin-like growth factor 1 (somatomedin C) | UE |
| HG3543- | IGF2 insulin-like growth factor 2 (somatomedin A) | NS |
| U40282 | ILK integrin-linked kinase | NS |
| X74295 | ITGA7 integrin, alpha 7 | NS |
| X57206 | ITPKB inositol 1,4,5-trisphosphate 3-kinase B | NS |
| AB002365 | KIAA0367 | UE |
| J00124 | KRT14 keratin 14 (epidermolysis bullosa simplex, Dowling-Meara, Koebner) | NS |
| X05153 | LALBA lactalbumin, alpha- | NS |
| X02152 | LDHA lactate dehydrogenase A | NS |
| HG3527-HT3721_f_at | LHB luteinizing hormone beta polypeptide | NS |
| X86018 | LRRC41 leucine rich repeat containing 41 | NS |
| L38486 | MFAP4 microfibrillar-associated protein 4 | NS |
| D87742 | MIA3 melanoma inhibitory activity family, member 3 | NS |
| M69066 | MSN moesin | NS |
| AB003177 | mRNA for proteasome subunit p27 | NS |
| U47742 | MYST3 MYST histone acetyltransferase (monocytic leukemia) 3 | NS |
| M30269 | NID1 nidogen 1 | NS |
| U80669 | NKX3-1 NK3 transcription factor related, locus 1 (Drosophila) | NS |
| M10901 | NR3C1 nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) | NS |
| M16801 | NR3C2 nuclear receptor subfamily 3, group C, member 2 | NS |
| U52969 | PCP4 Purkinje cell protein 4 | UE |
| J03278 | PDGFRB platelet-derived growth factor receptor, beta polypeptide | NS |
| D37965 | PDGFRL platelet-derived growth factor receptor-like | NS |
| Z49835 | PDIA3 protein disulfide isomerase family A, member 3 | NS |
| U78524 | PIAS1 protein inhibitor of activated STAT, 1 | NS |
| U60644 | PLD3 phospholipase D family, member 3 | NS |
| D11428 | PMP22 peripheral myelin protein 22 | NS |
| U79294 | PPAP2B phosphatidic acid phosphatase type 2B | NS |
| S71018 | PPIC peptidylprolyl isomerase C (cyclophilin C) | NS |
| X07767 | PRKACA protein kinase, cAMP-dependent, catalytic, alpha | NS |
| X83416 | PRNP prion protein (p27–30) | NS |
| M555671 | PROZ protein Z, vitamin K-dependent plasma glycoprotein | NS |
| U72066 | RBBP8 retinoblstoma binding protein 8 | NS |
| L25081 | RHOC ras homolog gene family, member C | NS |
| U40369 | SAT1 spermidine/spermine N1-acetyltransferase 1 | NS |
| M97287 | SATB1 special AT-rich sequence binding protein 1 (binds to nuclear matrix/scaffold-associating DNAs) | NS |
| U83463 | SDCBP syndecan binding protein (syntenin) | NS |
| U28369 | SEMA3B sema domain, immunoglobulin domain (Ig), short basic domain, secreted, (semaphorin) 3B | NS |
| HG3925-ht4195_at | SFTPA2 surfactant, pulmonary-associated protein A2 | NS |
| L31801 | SLC16A1 solute carrier family 16, member 1 (monocarboxylic acid transporter 1) | NS |
| M91463 | SLC2A4 solute carrier family 2 (facilitated glucose transporter), member 4 | NS |
| U66617 | SMARCD1 SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily d, member 1 | NS |
| U50383 | SMYD5 SMYD family member 5 | NS |
| D43636 | SNRK SNF related kinase | NS |
| D87465 | SPOCK2 sparc/osteonectin, cwcv and kazal-like domains proteoglycan (testican) 2 | NS |
| M61199 | SSFA2 sperm specific antigen 2 | NS |
| U15131 | ST5 suppression of tumorigenicity 5 | NS |
| U95006 | STRA13 stimulated by retinoic acid 13 homolog (mouse) | NS |
| M74719 | TCF4 transcription factor 4 | NS |
| X14253 | TDGF1 teratocarcinoma-derived growth factor 1 | NS |
| U52830 | TERT telomerase reverse transcriptase | NS |
| U12471 | THBS1 thrombospondin 1 | NS |
| U16296 | TIAM1 T-cell lymphoma invasion and metastasis 1 | NS |
| L01042 | TMF1 TATA element modulatory factor 1 | NS |
| U03397 | TNFRSF9 tumor necrosis factor receptor subfamily, member 9 | NS |
| X05276 | TMP4 tropomyosin 4 | UE |
| HG4683-HT5108_s_at | TRAF2 TNF receptor-associated factor 2 | NS |
| U64444 | UFD1L ubiquitin fusion degradation 1 like (yeast) | NS |
| U39318 | UBE2D3 ubiquitin-conjugating enzyme E2D 3 (UBC4/5 homolog, yeast) | NS |
| X59739 | ZFX zinc-finger protein, X-linked | NS |
Last column reports the status of the synapomorphies as described by Quade et al. (2004) in their significant genes list
The various combinations of comparisons (several outgroup and ingroup compositions) illustrate the effect of outgroup and ingroup selections on the results (Table 1–Table 5). These comparisons also show the similarities and differences between two diseases that arise within the same tissue, as well as the relationship between the leiomyoma and leiomyosarcoma; and the possibility of the latter arising within leiomyoma.
For the gastric dataset, GDS1210, parsimony analysis produced one most parsimonious cladogram (Fig. 2). The cladogram topology showed two terminal clades with six and five specimens, respectively, and a tandem arrangement of six small clades with the largest having three specimens. The inclusive gastric cancer clade was circumscribed by 34 synapomorphies (Table 6). In a list by list comparison, our 34 identified synapomorphies for the gastric cancer overlapped only with one common gene (CST4) from the gene list of the authors of the study (Hippo et al., 2002).
FIG. 2.
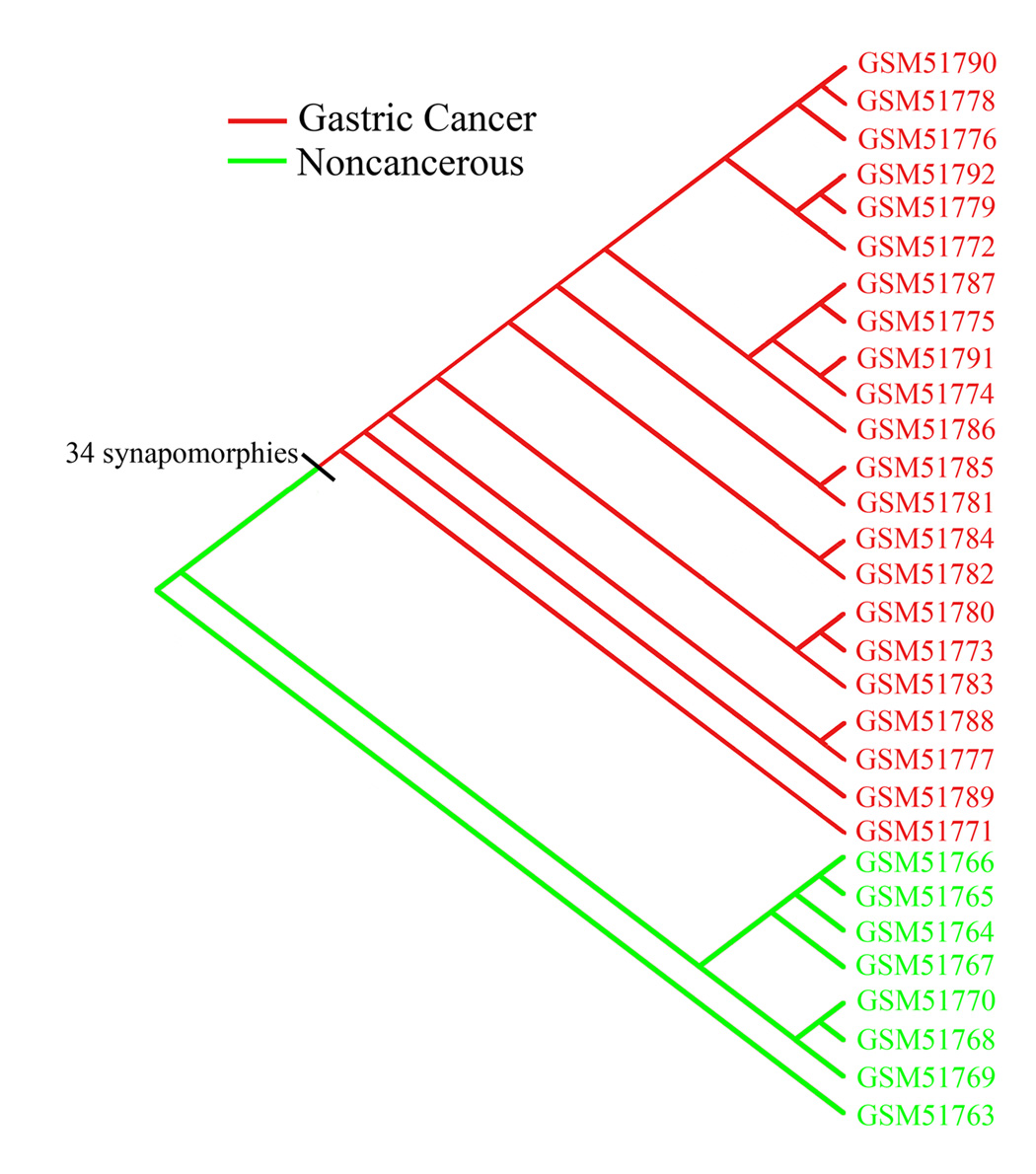
A cladogram of a parsimony phylogenetic analysis of gastric cancer and noncancerous specimens. It shows a clade delineated by 34 synapomorphies (Table 6) encompassing all cancer specimens.
TABLE 6.
A LIST OF 34 SYNAPOMORPHIES DEFINING A CLADE COMPOSED OF ALL GASTRIC CANCER SPECIMENS (GDS1210)
| A. Overexpressed synapomorphic genes: | ||
| X81817 | BAP31 mRNA | No |
| D50914 | BOP1 block of proliferation 1 | No |
| X54667 | CST4: cystatin S MGC71923 | Yes |
| L17131 | HMGA1 high mobility group AT-hook 1 | No |
| D63874 | HMGB1 high-mobility group box 1 | No |
| D26600 | PSMB4 proteasome (prosome, macropain) subunit, beta type, 4 | No |
| U36759 | PTCRA pre-T-cell antigen receptor alpha PT-ALPHA, PTA | No |
| X89750 | TGIF TGFB-induced factor (TALE family homeobox) | No |
| B. Underexpressed synapomorphic genes: | ||
| X76342 | ADH7 alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide ADH-4 | No |
| M63962 | ATP4A ATPase, H+/K+ exchanging, alpha polypeptide ATP6A | No |
| M75110 | ATP4B ATPase, H+/K+ exchanging, beta polypeptide ATP6B | No |
| J05401 | CKMT2 creatine kinase, mitochondrial 2 (sarcomeric) | No |
| L38025 | DNTFR ciliary neurotrophic factor receptor | No |
| M61855 | CYP2C9: cytochrome P450, family 2, subfamily C, polypeptide 9 CPC9 | No |
| D63479 | DGKD: diacylglycerol kinase, delta 130kDa DGKdelta, KIAA0145, dgkd-2 | No |
| X99101 | ESR2 estrogen receptor 2 (ER beta) | No |
| U21931 | FBP1 fructose-1, 6-bisphosphatase 1 | No |
| HG3432-HT3618_at | Fibroblast Growth Factor Receptor K-Sam, Alt. Splice 1 | No |
| M31328 | GNB3 guanine nucleotide binding protein (G protein), beta polypeptide 3 | No |
| D42047 | GPD1L glycerol-3-phosphate dehydrogenase 1-like | No |
| M62628 | Human alpha-1 Ig germline C-region membrane-coding region, 3′end | No |
| D29675 | Human inducible nitric oxide synthase gene, promoter and exon 1 | No |
| M63154 | Human intrinsic factor mRNA | No |
| Z29074 | KRT9 keratin 9 (epidermolytic palmoplantar keratoderma) EPPK, K9 | No |
| X05997 | LIPF lipase, gastric | No |
| U50136 | LTC4S leukotriene C4 synthase MGC33147 | No |
| X76223 | MAL: mal, T-cell differentiation protein | No |
| U19948 | PDIA2 protein disulfide isomerase family A, member 2 | No |
| L07592 | PPARD peroxisome proliferative activated receptor, delta | No |
| U57094 | RAB27A, member RAS oncogene family | No |
| AC002077 | SLC38A3 solute carrier family 38, member 3 | No |
| Z29574 | TNFRSF17 tumor necrosis factor recepter superfamily, member 17 | No |
| C. Dichotomously expressed synapomorphic genes: | ||
| D00408 | CYP3A7 cytochrome P450, family 3, subfamily A, polypeptide 7 CP37, P450-HFLA | No |
| U29092 | SELENBP1 selenium binding protein 1 | No |
Synapomorphies include: 8 OE genes, 24 UE genes, and 2 DE genes in comparison with the normal specimens. Last column reports the status of the synapomorphies as described by Hippo et al. (2002). Yes = listed; No = not listed.
Interplatform concordance
Testing of interplatform concordance was carried out by comparing the two lists of synapomorphies obtained from two leiomyoma studies, GDS484 and GDS533 (comparison results are summarized in Table 7 and Table 8). Out of the ~22,000 genes in the GDS484 dataset, our analysis produced a total of 1485 synapomorphic genes circumscribing the leiomyoma specimens. Although the leiomyomas of the GDS533 were delimited by 146 synapomorphies out of ~7,000 gene probes, a comparison between the two sets of leiomyomas’ synapomorphies produced 45 shared ones between the two (Table 7 and Table 8 ), a 31% concordance in synapomorphies despite the sizable difference in the number of probes between the two datasets, which is still better than the 12% concordance between the statistically produced gene lists of the two published studies (Hoffman et al., 2004; Quade et al., 2004).
TABLE 8.
SUMMARY OF CONCORDANCE RESULTS BETWEEN GDS484 AND GDS533
| GDS533 (Fibroids and Leiomyosarcomas) | ||||
|---|---|---|---|---|
| Quad et al. Gene List | Abu-Asab et al. Synapomorphies for Fibroids (146) | Abu-Asab et al. Synapomorphies for Fibroids and Leiomyosarcoma (32) | ||
| GDS484 (Fibroids) | Hoffman et al. Gene List | 12% | 18% | 19.3% |
| Abu-Asab et al. Synapomorphies for Fibroids (1485) | 20% | 31% | 48% | |
| GDS533 Quad et al. Gene List | N/A | 16.5% | 45% | |
The comparisons were carried out in various combinations: statistical versus statistical, phylogenetic versus statistical, and phylogenetic versus phylogenetic. N/A: not applicable (number of synapomorphies).
Furthermore, 48% concordance resulted when comparing the 32 synapomorphies of the leiomyomas and leiomyosarcomas clade (GDS533; Table 1) with the 1485 synapomorphies of the leiomyomas of GDS484 (Table 7); the clades’ synapomorphies overlapped as follows: 1/1 OE, 7/8 UE (except FOSB), and 8/23 DE, an 89% concordance within the OE and UE and 35% within the DE. Additionally, there was 45% concordance between the 32 synapomorphies of the leiomyomas and leiomyosarcomas clade and the gene list of Quad et al. (2004) (Table 8).
However, a lower concordance was obtained when comparing the phylogenetic synapomorphies against statistically generated gene lists. The synapomorphies of leiomyomas (GDS533; Table 2) showed 18% concordance (4/25 OE, 8/42 UE) with the 78 significant genes of Hoffman et al. (2004; GDS484, gene list produced by fold change), and 16.5% (5/25 OE, 6/42 UE) with the 146 genes of Quad et al. (2004; GDS533, gene list produced by an F statistic). This was higher than the concordance between the two gene lists of the published uterine studies, 12% (3/25 OE, 5/42 UE). The two studies had no mention of DE genes.
Data pooling and interplatform comparability
Data pooling and interplatform comparability was carried out on the combined polarized matrices of the gastric (GDS1210) and uterine (GDS533) datasets. Their inclusive parsimony analysis produced one most parsimonious cladogram (Fig. 3). Its topology showed a total separation of the gastric cancer from the uterine leiomyoma and sarcoma specimens into two large clades. However, the two types of cancers shared 16 synapomorphies that delimited a clade composed of all the gastric and uterine specimens (Table 9).
FIG. 3.
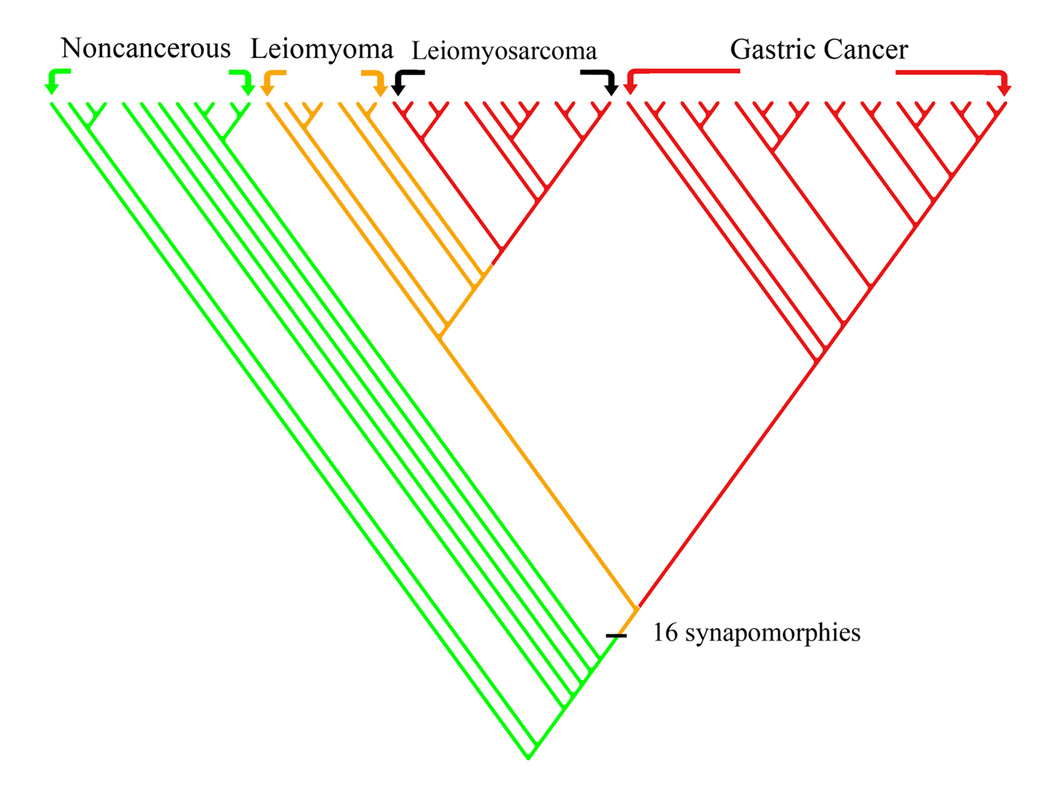
A cladogram representing a comparability analysis of the gastric (GDS1210) and uterine (GDS533) datasets. The polarized matrices of the two datasets were pooled together and processed by the parsimony phylogenetic algorithm, MIX. Each of the cancers (gastric and leiomyosarcoma) forms its own clade, and the inclusive clade encompassing the two cancers and leiomyomas is delimited by a set of synapomorphies (Table 9).
TABLE 9.
INTERPLATFORM COMPARABILITY
| ID | Gene |
|---|---|
| U52522 | ARFIP2 ADP-ribosylation factor interacting protein 2 (arfaptin 2) |
| U51478 | ATP1B3 ATPase, NaK transporting, beta 3 polypeptide |
| X66839 | CA9 carbonic anhydrase IX |
| M60974 | GADD45A growth arrest and DNA-damage-inducible, alpha |
| X01677-M33197 | GAPDH glyceraldehyde-3-phosphate dehydrogenase [two readings] |
| X14850 | H2AFX H2A histone family, member X |
| U52830 | Homo sapiens Cri-du-chat region mRNA, clone CSC8 |
| U25138 | KCNMB1 potassium large conductance calcium-activated channel, subfamily M, beta member 1 |
| D21063 | MCM2 minichromosome maintenance deficient 2 |
| L38486 | MFAP4 microfibrillar-associated protein 4 |
| D87463 | PHYHIP phytanoyl-CoA 2-hydroxylase interacting protein |
| X02419 | PLAU plasminogen activator, urokinase |
| L48513 | PON2 paraoxonase 2 |
| U29091 | SELENBP1 selenium binding protein 1 |
| Z19083 | TPBG trophoblast glycoprotein |
| M25077 | TROVE2 TROVE domain family, member 2 |
A list of 16 synapomorphies defining a clade composed of all gastric cancer (GDS1210) as well as uterine sarcoma and leiomyom specimens (GDS533).
The resulting inclusive cladogram (Fig. 3) showed an almost total agreement with the single type cladograms (Fig. 1 and Fig 2), indicating a successful pooling of datasets. However, there was a slight variation in the topology of minor branches between the cladogram of Figure 2 and the inclusive one of Figure 3. These slight differences are most likely due to the increased number of normal specimens that were used in outgroup of the inclusive cladogram. Outgroup size used here was by no means ideal; the larger the membership of the outgroup the more stable the topology of the generated cladogram (Graybeal, 1998).
Discussion
Microarray data analysis aims to identify differentially expressed genes, and subsequently characterize genetic patterns, classify specimens accordingly, and point out potential biomarkers. However, most of the problems that are currently associated with microarray analysis arise from using only the quantitative aspect of the data (the absolute continuous data values of gene expression) to carry out parametric statistical analysis. Such a statistical analysis forecasts gene linkage on the basis of quantitative correlation and not expression pattern, and lacks the power to recognize and utilize specific gene expression patterns such as dichotomous-expression and partial asynchronicities (Abu-Asab et al., 2008; Allison et al., 2006). This results in discrepancies that affect which genes are considered differentially expressed by the two main ranking criteria for generating gene lists, the t-test and fold change (Guo 2006). Our phylogenetic analysis supports a qualitative approach where the directionality of expression is the first step to designate the expression value as significant, followed by parsimony search to plot a classification of specimens with the smallest number of steps that explains the data’s distribution pattern.
This parsimonious analysis produced higher interplatform concordance than the gene lists generated with t-test and fold change (Table 7 and Table 8), and allowed the pooling and comparability of two independent experiments. Such results confer reliability to a qualitative parsimonious approach to analyzing gene expression data. Table 10 summarizes the major characteristics of a parsimony phylogenetic approach.
TABLE 10.
SUMMARY OF THE CHARACTERISTICS OF A PARSIMONIOUS PHYLOGENETIC ANALYSIS THROUGH POLARITY ASSESSMENT OF GENE-EXPRESSION VALUES FOLLOWED BY A MAXIMUM PARSIMONY ANALYSIS
| • | Offers a qualitative assessment of microarray gene expression data by sorting expression values into derived or ancestral. |
| • | Identifies synapomorphies (shared derived expression states) and uses them to delineate clades (class discovery). Synapomorphies are also the potential biomarkers. |
| • | Searches for the most parsimonious classification of specimens; the one with minimum number of steps, reversals, and parallels. |
| • | Efficiently models the heterogeneous expression profiles of the diseased specimens. Those with fast mutation rate such as cancer. |
| • | Incorporates gene expressions that violate normal distribution in a set of specimens—e.g., dichotomously expressed genes. |
| • | Reduces the sensitivity to experimental noise. |
| • | Permits pooling of multiple experiments. |
| • | Allows intra and intercomparability of data. |
| • | Produces higher concordance between gene lists than statistical methods (F & t-statistics and fold change). |
| • | Offers a nonparametric data-based, not specimen-based, gene listing and gene linkage. |
In addition to its evident scientific applications, a phylogenetic analysis as outlined here has clinical implications as well. Indeed, the results of the three microarray gene expression datasets show the clinical potential for such parsimonious analysis; they produced a total distinction of the sarcoma from the fibroid tissues (the leiomyomas), and these two classes from gastric cancer. It also identified a number of synapomorphies for gastric and uterine cancers, thus defining each as a separate disease entity with its unique shared derived expressions (see also Meza-Zepeda et al. 2006, for further support of this point). Furthermore, the combined analysis revealed the shared alterations of gene expression that are shared between the uterine and gastric cancers (Table 9). This conclusion is supported by the presence of these synapomorphic gene expressions as significant ones in other types of cancers as well: bladder, breast, colorectal, ovarian, pancreatic, prostate, and renal (Guzińska-Ustymowicz et al., 2008; Dong et al., 2007; Huang et al., 2006; Pilarsky et al., 2004).
Advantages of polarity assessment
There are several reasons for our preference of a combination of polarity assessment via outgroup comparison and parsimony over other methods for the analysis of gene expression microarray data, an approach that is also supported by other authors (Allison et al., 2006; Kolaczkowski and Thornton, 2004). Parsimony phylogenetic analysis requires polarity assessment for each data value to determine its novelty—whether it represents a change from the normal state (Abu-Asab et al., 2008). We advocate that qualitative, and not only quantitative, similarity is a better measure of common ontogenetic steps among specimens, and that a correlation of genes based on similar quantitative expression is not necessarily indicative of ontogenic relationships among genes.
Polarity assessment does not set an arbitrary stringency on gene selection, especially where the distribution pattern is gene specific within a set of specimens (e.g., DE and partially asynchronous genes), while other methods are not optimal for its assessment (Huang and Qu, 2006; Lyons-Weiler et al., 2004). Fold change and F and t-statistics may dismiss from the gene list those genes with dichotomous expressions, although they are indicative of a unique expression type and may account for some phenomena such as transitional clades located between diseased and normal clades, and multiple developmental pathways in some disease types (Abu et al., 2006; Lyons-Weiler et al., 2004). The gene lists of Table 1C–Table 7C show a large number of DE asynchronous genes that were mostly not considered significant by other methods (Hippo et al., 2002; Hoffman et al., 2004; Quade et al., 2004), or their dichotomous mode was not noticed by the authors.
Because polarity assessment transforms the quantitative data into a qualitative matrix, it reduces the data noise. The absolute quantitative nature of the microarray data restricts their use and interpretation due to their range of inconsistencies between runs, platforms, and laboratories. By polarizing each data set with its own set of outgroup specimens, the inconsistencies of the experiment are eliminated since the polarization process is a comparison between equals—data values generated at the same time. The benefit here translates into the ability to pool a large number of experiments, carry out intra- and interplatform comparabilities, and a better gene list concordance between experiments. However, as discussed below, polarity assessment is sensitive to the choice and size of the outgroup specimens.
Selection and size of the outgroup
When conducting a polarity assessment, outgroup’s selection and its effective size are very significant factors in correctly identifying synapomorphies, and therefore, optimally delimiting the classes of diseased specimens. The composition of the outgroup specimens affects the outcome of the analysis as demonstrated by the different combinations of outgroups that we used to conduct polarity assessment for the leiomyosarcomas and leiomyomas (Table 1–Table 5). We selected these various arrangements of outgroups to demonstrate that their compositions produced slightly different but still biologically meaningful results. It is important to note that the outgroup should be composed of only healthy specimens when the goal is to find out the genes involved in disease inception, progression, and maintenance. As Table 1–Table 5 show, variations of out/ingroup composition lead to variations in identifying synapomorphies. Also, Figure 3 shows that the different types of cancers will separate into their respective clades when the outgroup is composed of normal specimens; a process that can be utilized for diagnosis.
In our combined analysis (Fig. 3), the increase in outgroup size did not affect the major topology of the cladogram, but rather the internal branching of some clades (normal and gastric cancer) when compared with their single analysis (Fig. 1–Fig 2). Because increasing the number of genes in the study does not have the same effect as enlarging outgroup size (Graybeal, 1998), it is our conclusion that a successful analysis requires a good number of normal specimens to be used as the outgroup. For microarray experiments to be meaningful and provide high predictivity, the smallest number of normal specimens that encompasses the maximum variation per population should be established and used in the analysis.
Inferring gene linkage through parsimony phylogenetic analysis
Whereas gene linkage of a clustering dendrogram is based on quantitative correlations between differentially expressed genes, in a parsimony cladogram it is based on the distribution of derived and ancestral gene expression states of all genes of all the specimens; that is, it is a map of expression states—both ancestral and derived. It reflects the classification that has the lowest number of steps as well as parallels and reversals to explain the distribution of expression states among specimens.
Gene linkage here is based on the location of genes on the cladogram. The synapomorphies below a node on the cladogram are the linked genes that are shared among the specimens above that node. Because a parsimonious cladogram is hierarchical, every one of its nodes has its synapomorphy( ies). This characteristic of a cladogram presents it as a map of linked genetic alterations that produce the diversity/relatedness of its specimens and may also permit the tracing of shared ontogenic pathways that are responsible for disease initiation and progression.
Improved interplatform concordance and comparability
Improved interplatform concordance is a criterion that will confer robustness and significance on microarray as a valid experimental and clinical platform. Our tests of concordance by comparing the lists of synapomorphies generated by polarity assessment of two experiments produced better results than those of fold change and F-statistic, and better than between the latter two (Table 8). When comparing the synapomorphies of a clade composed of leiomyomas and leiomyosarcomas (GDS533) with the synapomorphies of leiomyomas (GDS484), we obtained a high concordance of 89% within over- and underexpressed and 35% within dichotomously expressed genes. The concordance between the two studies could have been higher if the number of probes of the GDS533 was closer to GDS484–7,000 versus 22,000 (Hoffman et al., 2004; Quade et al., 2004). Furthermore, even a comparison of the synapomorphies of two leiomyoma groups [GDS484 (1485 synapomorphies) and GDS533 (146 synapomorphies; Table 2)] produced 31% concordance between the two groups of leiomyoma (45/146; Table 7). Nevertheless, this was a higher percentage than was produced by statistical methods (12%).
Interplatform comparability has been difficult to carry out on microarray data because of data inconsistencies between runs, experiments, and laboratories; however, with polarity assessment, which converts the quantitative values of gene expression of every experiment into a qualitative matrix, it is possible to combine several matrices and carry out intra-and interplatform comparisons in a parsimonious phylogenetic sense. A phylogenetic interplatform comparability of microarray data can be carried out if each dataset can be polarized separately with its own outgroup to produce its polarized matrix. Furthermore, when their probes are identical, two or more polarized sets can be pooled together and analyzed as Figure 3 shows. We have successfully pooled and analyzed two separately polarized datasets (GDS533 and 1210) of gastric cancer as well as uterine leiomyoma and leiomyosarcoma, where the two datasets were prepared separately but on an identical gene chip platform, GPL80.
Implications on disease definition, profiling, diagnosis, and prognosis
Although it is assumed that each disease has its own unique developmental pathway(s) (Adsay et al., 2002; Chung, 2000; Hayashi et al., 2004), thus far, the omics data has not been used to prove this premise. Our analysis of two independently generated datasets that represent uterine (GDS533) and gastric (GDS1210) cancers confirms that each of these two types of cancer is a natural class of specimens (a clade) that is circumscribed by its own set of synapomorphies. If this can be extended to other types of cancer, then each cancer can be considered a natural clade with its unique gene expression identifiers—the synapomorphies.
There are several implications to this conclusion; the most obvious is its effect on the definition of biomarkers. If the specimens of a type of cancer form a clade, then any suggested biomarker has to be selected from the clades’ synapomorphies; otherwise, it will not be a universal diagnostic test for all the specimens of this cancer. Some of the currently applied immunohistomarkers are not universal synapomorphies. For example, the memberships of all four clades of the gastric cancers (Fig. 2) did not correlate well with the specimens’ immunoreactivity to antibodies against p53, E-cadherin, and β-catenin, and a published two-way clustering did not correlate any better (Hippo et al., 2002). The discordance between omics biomarkers and most of the currently used immunohistological markers is a problem that can be better addressed in a phylogenetic sense. The discordance between microarray and immunohistochemical (IHC) biomarkers is due to the differing natures of these two types of markers. Although gene-expression biomarkers are based on the averaging of the expression values of the number of the cells used in the study (i.e., the cell homogenate), the IHC is based on the pathologist reading of the percentage of positive cells in the stained section. The IHC result is given as a percentage figure that could start at 10%, and is used to associate the neoplasm with the normal tissue of origin. For an accurate and meaningful interpretation of the gene expression analyses, a comparison between the two types of biomarkers should be avoided, and the phylogenetic concept that is suggested here should be adopted.
A second implication is that a phylogenetic classification can be a clinical tool to carry out early detection, diagnosis, grading, prognosis, and posttreatment evaluation; these tasks can be realized through a parsimony analysis where the place of a specimen within the cladogram (i.e., the classification) will indicate its pathologic status. Alternatively, the health status of a specimen can be probed by using the synapomorphies as the biomarkers of the disease, that is, through class prediction by assigning the specimen to a clade. Because the cladogram also indicates the direction of change in gene expression among the specimens, it places those specimens with the advanced number of derived gene expression patterns at the terminal end of the cladogram, and places the specimens with the least number of gene expression changes at the lower end of the cladogram, and it may be developed for use in grading, prognosis, targeted treatment, and posttreatment assessment.
Additionally, the phylogenetic classification is a dynamic and seamless tool that will incorporate a novel specimen by placing it in the proximity of its sister groups, depending on the number of synapomorphies it shares with other members of a clade, without any radical alteration to the topology of the cladogram.
Resolving standing questions through parsimony phylogenetics: an example
Our analysis of uterine fibroids and sarcomas illustrates how a parsimony phylogenetic analysis may confront some of the unresolved issues in bioinformatics and medicine. For example, one of the persistent questions in pathology is the relationship between leiomyoma and leiomyosarcoma (Quade et al., 2004). It has been reported that approximately 1% of leiomyosarcoma may have arisen in preexisting leiomyoma (Lee et al., 2005). By analyzing data of normal uterus, leiomyoma, and leiomyosarcoma, we demonstrated that the latter two share a number of synapomorphies and form together an inclusive clade (Table 1 and Fig. 1 and Fig 3), and that leiomyosarcoma has an additional number of synapomorphies distinguishing them from leiomyoma (Table 3). Although the leiomyoma specimens, when analyzed alone, without the leiomyosarcoma, appear to have a large number of synapomorphies (Table 2), these synapomorphies are not unique to leiomyoma. Leiomyoma as a group does not form a clade within a comprehensive ingroup that includes the leiomyosarcoma; there is not even one gene expression that is unique to the group itself in this context. Because it shares with the leiomyosarcoma its synapomorphies, leiomyoma may be considered an incipient form of leiomyosarcoma.
Conclusion
The application of phylogenetic analysis through polarity assessment and parsimony to several gene expression microarray datasets provides the basis for a new paradigm to analyzing and interpreting microarray data (Table 10). It offers an alternative to F and t-statistics and fold change methods of generating differentially expressed gene listing and statistical gene linkage, brings out a higher interplatform concordance, resolves interplatform comparability problems, defines biomarkers as synapomorphies, circumscribes disease types as clades defined by synapomorphies, and possibly transforms microarray into diagnostic, prognostic, and posttreatment evaluation tool.
Acknowledgments
Competing interests: The authors have filed for US patent for their analytical method.
References
- Abu-Asab M, Chaouchi M, Amri H. Phyloproteomics: What phylogenetic analysis reveals about serum proteomics. J Proteome Res. 2006;5:2236–2240. doi: 10.1021/pr0504485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Asab M, Chaouchi M, Amri H. Evolutionary medicine: a meaningful connection between omics, disease, and treatment. Proteomic Clin Appl. 2008;2:122–134. doi: 10.1002/prca.200780047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adsay NV, Merati K, Andea A, Sarkar F, Hruban RH, Wilentz RE, et al. The dichotomy in the preinvasive neoplasia to invasive carcinoma sequence in the pancreas: differential expression of MUC1 and MUC2 supports the existence of two separate pathways of carcinogenesis. Mod Pathol. 2002;15:1087–1095. doi: 10.1097/01.MP.0000028647.98725.8B. [DOI] [PubMed] [Google Scholar]
- Albert VA. Parsimony and phylogenetics in the genomic age. In: Albert VA, editor. Parsimony, Phylogeny, and Genomics. Oxford, NY: Oxford University Press; 2005a. [Google Scholar]
- Albert VA. Parsimony, phylogeny, and genomics. Oxford, NY: Oxford University Press; 2005b. [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large Bcell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, et al. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]
- Chung DC. The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis. Gastroenterology. 2000;119:854–865. doi: 10.1053/gast.2000.16507. [DOI] [PubMed] [Google Scholar]
- Dong M, Dong Q, Zhang H, Zhou J, Tian Y, Dong Y. Expression of Gadd45a and p53 proteins in human pancreatic cancer: potential effects on clinical outcomes. J Surg Oncol. 2007;95:332–336. doi: 10.1002/jso.20684. [DOI] [PubMed] [Google Scholar]
- Farris JS. The information content of the phylogenetic system. Syst Zool. 1979;28:483–519. [Google Scholar]
- Felsenstein J. PHYLIP: phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Felsenstein J. Inferring phylogenies. Sunderland, MA: Sinauer Associates; 2004. [Google Scholar]
- Goloboff PA, Pol D. Parsimony and Bayesian phylogenetics. In: Abert VA, editor. Parsimony, Phylogeny, and Genomics. Oxford, NY: Oxford University Press; 2005. [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Graybeal A. Is it better to add taxa or characters to a difficult phylogenetic problem? Syst Biol. 1998;47:9–17. doi: 10.1080/106351598260996. [DOI] [PubMed] [Google Scholar]
- Guo L, Lobenhofer EK, Wang C, Shippy R, Harris SC, Zhang L, et al. Rat toxicogenomic study reveals analytical consistency across microarray platforms. Nat Biotechnol. 2006;24:1162–1169. doi: 10.1038/nbt1238. [DOI] [PubMed] [Google Scholar]
- Guzińska-Ustymowicz K, Stepień E, Kemona A. MCM-2, Ki-67 and PCNA protein expressions in pT3G2 colorectal cancer indicated lymph node involvement. Anticancer Res. 2008;28:451–457. [PubMed] [Google Scholar]
- Harrison AP, Johnston CE, Orengo CA. Establishing a major cause of discrepancy in the calibration of Affymetrix GeneChips. BMC Bioinformatics. 2007;8:195. doi: 10.1186/1471-2105-8-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Yamashita J, Watanabe T. Molecular genetic analysis of deep-seated glioblastomas. Cancer Genet Cytogenet. 2004;153:64–68. doi: 10.1016/j.cancergencyto.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Hippo Y, Taniguchi H, Tsutsumi S, Machida N, Chong JM, Fukayama M, et al. Global gene expression analysis of gastric cancer by oligonucleotide microarrays. Cancer Res. 2002;62:233–240. [PubMed] [Google Scholar]
- Hoffman PJ, Milliken DB, Gregg LC, Davis RR, Gregg JP. Molecular characterization of uterine fibroids and its implication for underlying mechanisms of pathogenesis. Fertil Steril. 2004;82:639–649. doi: 10.1016/j.fertnstert.2004.01.047. [DOI] [PubMed] [Google Scholar]
- Huang KC, Park DC, Ng SK, Lee JY, Ni X, Ng WC, et al. Selenium binding protein 1 in ovarian cancer. Int J Cancer. 2006;118:2433–2440. doi: 10.1002/ijc.21671. [DOI] [PubMed] [Google Scholar]
- Huang S, Qu Y. The loss in power when the test of differential expression is performed under a wrong scale. J Comput Biol. 2006;13:786–797. doi: 10.1089/cmb.2006.13.786. [DOI] [PubMed] [Google Scholar]
- Kolaczkowski B, Thornton JW. Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous. Nature. 2004;431:980–984. doi: 10.1038/nature02917. [DOI] [PubMed] [Google Scholar]
- Lee EJ, Kong G, Lee SH, Rho SB, Park CS, Kim BG, et al. Profiling of differentially expressed genes in human uterine leiomyomas. Int J Gynecol Cancer. 2005;15:146–154. doi: 10.1111/j.1048-891x.2005.15016.x. [DOI] [PubMed] [Google Scholar]
- Lossos IS, Morgensztern D. Prognostic biomarkers in diffuse large B-cell lymphoma. J Clin Oncol. 2006;24:995–1007. doi: 10.1200/JCO.2005.02.4786. [DOI] [PubMed] [Google Scholar]
- Lyons-Weiler J, Patel S, Becich MJ, Godfrey TE. Tests for finding complex patterns of differential expression in cancers: towards individualized medicine. BMC Bioinformatics. 2004;5:110. doi: 10.1186/1471-2105-5-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza-Zepeda LA, Kresse SH, Barragan-Polania AH, Bjerkehagen B, Ohnstad HO, Namløs HM, et al. Array comparative genomic hybridization reveals distinct DNA copy number differences between gastrointestinal stromal tumors and leiomyosarcomas. Cancer Res. 2006;66:8984–8993. doi: 10.1158/0008-5472.CAN-06-1972. [DOI] [PubMed] [Google Scholar]
- Millenaar FF, Okyere J, May ST, van Zanten M, Voesenek LA, Peeters AJ. How to decide? Different methods of calculating gene expression from short oligonucleotide array data will give different results. BMC Bioinformatics. 2006;7:137. doi: 10.1186/1471-2105-7-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesse RM, Stearns SC. The great opportunity: evolutionary applications to medicine and public health. Evol Appl. 2008;1:28–48. doi: 10.1111/j.1752-4571.2007.00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Pilarsky C, Wenzig M, Specht T, Saeger HD, Grützmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004;6:744–750. doi: 10.1593/neo.04277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Microarray analysis and tumor classification. New Engl J Med. 2006;354:2463–2472. doi: 10.1056/NEJMra042342. [DOI] [PubMed] [Google Scholar]
- Quade BJ, Wang TY, Sornberger K, Dal Cin P, Mutter GL, Morton CC. Molecular pathogenesis of uterine smooth muscle tumors from transcriptional profiling. Genes, Chromosomes and Cancer. 2004;40:97–108. doi: 10.1002/gcc.20018. [DOI] [PubMed] [Google Scholar]
- Siddall ME. Success of parsimony in the four-taxon case: long-branch repulsion by likelihood in the Farris zone. Cladistics. 1998;14:209–220. doi: 10.1111/j.1096-0031.1998.tb00334.x. [DOI] [PubMed] [Google Scholar]
- Stefankovic D, Vigoda E. Phylogeny of mixture models: robustness of maximum likelihood and non-identifiable distributions. J Comput Biol. 2007;14:156–189. doi: 10.1089/cmb.2006.0126. [DOI] [PubMed] [Google Scholar]
- Wang H, He X, Band M, Wilson C, Liu L. A study of inter-lab and inter-platform agreement of DNA microarray data. BMC Genomics. 2005;6:71. doi: 10.1186/1471-2164-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley EO, Siegel-Causey D. The Compleat Cladist: A Primer of Phylogenetic Procedures. Lawrence, KN: Museum of Natural History, Dyche Hall, University of Kansas; 1991. [Google Scholar]