

CONSPECTUS
Collaboration between academia and industry is a growing phenomenon within the chemistry community. These sectors have long held strong ties since academia traditionally trains the future scientists of the corporate world, but the recent drastic decrease of public funding is motivating the academic world to seek more private grants. This concept of industrial “sponsoring” is not new, and in the past, some companies granted substantial amounts of money per annum to various academic institutions in exchange for a prime access to all their scientific discoveries and inventions. However, academic and industrial interests were not always aligned, and therefore the investment has become increasingly difficult to justify from industry's point of view. With fluctuating macroeconomic factors, this type of unrestricted grant has become more rare and has been largely replaced by smaller and more focused partnerships. In our view, forging a partnership with industry can be a golden opportunity for both parties and can represent a true symbiosis. This type of project-specific collaboration is engendered by industry's desire to access very specific academic expertise that is required for the development of new technologies at the forefront of science. Since financial pressures do not allow companies to spend the time to acquire this expertise and even less to explore fundamental research, partnering with an academic laboratory whose research is related to the problem gives them a viable alternative. From an academic standpoint, it represents the perfect occasion to apply “pure science” research concepts to solve problems that benefit humanity. Moreover, it offers a unique opportunity for students to face challenges from the “real world” at an early stage of their career. Although not every problem in industry can be solved by research developments in academia, we argue that there is significant scientific overlap between these two seemingly disparate groups, thereby presenting an opportunity for a symbiosis. This type of partnership is challenging but can be a win-win situation if both parties agree on some general guidelines.
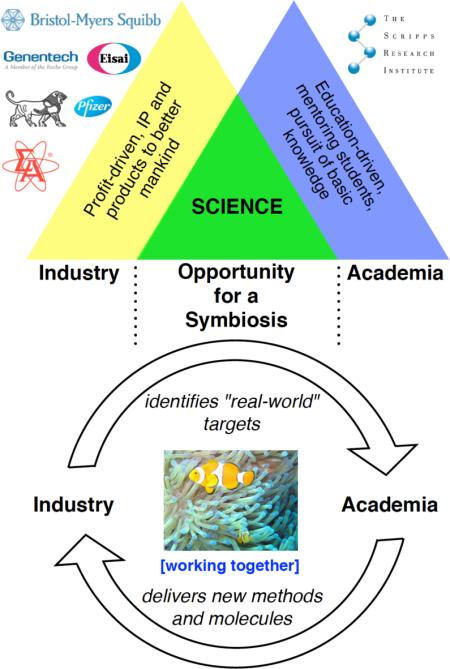
Keywords: Academia, industry, collaboration, symbiosis, organic chemistry
INTRODUCTION
Collaboration between academia and industry has recently been a topic of interest and discussion across various fields of science in journal publications1 and newsletters.2 Its importance is widely recognized and even promoted by public funding agencies such as the National Institutes of Health,3 the National Science Foundation,4 and Marie Curie Actions in Europe.5 However, the concept of collaboration between academia and industry is not new, and the recognition that there is a difficulty to translate academic research findings into commercial products of public interest is also not new. This recent resurgence of academia–industry collaborations is spurred by financial pressures, as both parties reach out to each other in order to render their practice of research and development more time- and cost-efficient. On the one hand, academia, which traditionally studies “pure science,” is leaning more toward “applied science” because public funding agencies can no longer justify spending taxpayers’ money for basic science endeavors alone. On the other hand, industry, which traditionally studies “applied science,” is turning more toward “pure science” to generate creative solutions for the exploration of new chemical space amid a very crowded intellectual property (IP) landscape. In previous models of academia–industry collaborations, industry paid for all of academia's findings and then selected the technologies that they deemed most useful for their purposes (i.e., blanket agreements and first rights deals). Now, it is becoming increasingly popular for industry to pay for the academic technology that is developed specifically for their intended purpose (i.e., sponsor-funded programs).
Some recent examples of partnerships from other groups include the collaboration of the Buchwald group and Merck for the development of palladium pre-catalysts,6 the Molander group and Janssen Pharmaceuticals for the development of alkyltrifluoroborate reagents,7 the Beller group and Evonik Industries for the development of alkane dehydrogenation catalysts,8 the Chirik group and Merck for the development of cobalt catalysts for asymmetric hydrogenation,9 and the Hawker group and Dow Chemical for the fabrication of polymer brush nanostructures via light-mediated iridium catalysis.10 Our laboratory has particularly benefitted from academia–industry collaborations, from which over a dozen fruitful projects have materialized. In this Account, we describe a series of vignettes to identify numerous elements that support a symbiotic project. Furthermore, we recount how our own research program has been shaped and influenced by our industrial colleagues.
DISCUSSION
Genentech
In 2009, a collaboration was forged with Genentech to study the bioactivity of haouamine A (2; Figure 1).11 Despite some initial reports of interesting in vitro cytotoxicity, an in-depth biological study of 2 could not be undertaken by Genentech, since isolation from marine tunicates does not afford enough natural sample. Although our laboratory had previously completed the total synthesis of haouamine A (2),12 the synthetic route was not amenable to large-scale synthesis and therefore, prompted by Rami Hannoush at Genentech, the sequence was revised. Since we strive to achieve an “ideal” synthesis whenever possible,13 the development of a scalable synthesis of this complex heptacyclic alkaloid was an academic goal that became aligned with an industrial purpose. Thus, our laboratory embarked on a third-generation synthesis of 2, which began by the preparation of over 15 grams of intermediate 1 in 8 steps.11 Then, a point-to-planar chirality transfer strategy was designed for the end-game, enabling the preparation of ample quantities of 2 and its atropisomer 3. With this newly available supply of haouamine A (2), Genentech was able to show that both 2 and 3 are highly active against PC3 human prostate cancer cells (IC50 = 29 ± 2 μM for 2 and IC50 = 32 ± 3 μM for 3) and that the cyclophane moiety is required for bioactivity.
Figure 1.

Development of a scalable route to haouamine A (2) for bioactivity testing at Genentech.
LEO Pharma
In 2011, we embarked on an ambitious multi-year partnership with LEO Pharma to develop practical syntheses of complex natural products for the treatment of skin diseases.14 The overall objective has been to apply in-house chemistry methodologies and synthesis strategies to prepare novel synthetic intermediates and analogs for biological evaluation against LEO Pharma targets. In particular, LEO Pharma has been pursuing complex natural terpenes of biological significance, which fortuitously has been our own area of expertise over the last decade.15 The cornerstone of our association with LEO Pharma was the diterpene ingenol (6; Figure 2).16 An esterified version of 6, ingenol mebutate (or ingenol-3-angelate; 7), belongs to the LEO Pharma portfolio and is commercialized under the trade name Picato™ for the treatment of actinic keratosis. Biological studies showed that the C3 hydroxyl group has to be functionalized to ensure potency.17 However, in solution, ingenol mebutate (7) undergoes rapid hydrolysis or transesterification due to the hydroxy triad (C3, C4 and C5), which limits its current use to epidermal application as a gel. The development of a total synthesis of ingenol (6) and various analogs could thus enable a better understanding of the structure-activity relationship of ingenanes, and also enable the potential identification of an orally stable derivative that cannot be accessed from 6. From an academic perspective, ingenol (6) presents a great synthetic challenge due to its highly strained in,out-[4.4.1]bicycloundecane core. While the previous syntheses showcase incredibly elegant transformations, all routes are over 35 steps, thus calling for the design of a shorter and more scalable route. LEO Pharma only had experience in working with contract research organizations at the time, so they took a big leap of faith when they decided to partner with an academic group. Indeed, they had no guarantees that we could deliver a concise and practical synthesis of ingenol (6) in a reasonable timescale. As it turned out, a two-phase terpene synthesis logic18 facilitated our task of constructing the terpene core, and we were able to synthesize our ‘cyclase phase endpoint,’ tigliane intermediate 5, in 7 steps and in gram-scale from (+)-carene (4).16 Successful implementation of a biomimetic pinacol shift and regioselective oxidations delivered ingenol (6) in 7 more steps. This scalable total synthesis has since enabled the preparation of various analogs that will allow for a better understanding of the mechanism of action of this ‘first-in-class’ drug. Through biweekly teleconferences and frequent email exchanges, the team at LEO pharma led by Jakob Felding has continuously supported and guided the synthesis strategies, and further provided critical advice for the types of structures that would be beneficial for analog synthesis. Notably, the Scripps–LEO route to ingenol (6) and related analogs are the subject of two separate patent filings.
Figure 2.

Inception of a symbiotic relationship with LEO Pharma through ingenol synthesis.
While the quest for ingenol was the linchpin of our partnership with LEO Pharma, other synergetic interests appeared along the way and transformed it into a lasting relationship. The synthesis of natural products and related analogs in the bioactive sesquiterpene19 and meroterpene20,21 families has allowed us to showcase several synthesis strategies in complex molecule settings all the while providing LEO Pharma a library of unique structural frameworks for medicinal chemistry endeavors (Figure 3).
Figure 3.

Divergent synthesis of bioactive terpene families.
Keeping with the theme of complex terpene synthesis, we reported the synthesis of ouabagenin (16), a polyhydroxylated steroid (Figure 4).22 Ouabagenin (16) belongs to the class of cardenolides, and its parent glycoside, ouabain, is used as a treatment for congestive heart failure but suffers from a very narrow therapeutic index. The preparation of analogs could solve this longstanding industrial problem, however, the only previous synthesis of ouabain required a lengthy 41 steps.23 Consequently, academic and industrial interests met in this daunting task: a scalable synthesis of a molecule as complex as ouabagenin (16) was needed. We decided on a quasi-biomimetic approach starting with the inexpensive adrenosterone and exploiting the oxidation at C11 to install oxidation at C19 and then, C5 and C1. This “redox relay” strategy enabled the preparation of over half a gram of protected ouabageninone (14) in 14 steps. This intermediate is currently serving as a platform to access various analogs such as C14-fluorinated 15, derivatives of which are being tested by LEO Pharma. The importance of the C19-hydroxyl group for the bioactivity of ouabain and other steroidal compounds is also under current investigation and is the subject of a separate patent filing. Finally, to complete our initial objective, ouabagenin (16) was prepared in 6 steps from 14.
Figure 4.

Synthesis of a polyhydroxylated cardenolide, ouabagenin (16), and analogs thereof.
Bristol-Myers Squibb
In 2012, The Scripps Research Institute (TSRI) and Bristol-Myers Squibb (BMS) entered a long term multi-laboratory partnership centered on projects of mutual interest.24 This sponsor-funded program enables the application of TSRI's scientific know-how to the development of therapeutics against diseases of interest at BMS. Since both medicinal and process chemistry groups were involved from BMS, the types of projects that we undertook are varied in nature. Natural product synthesis and modification, as well as method development are all part of this collaboration portfolio. What follows are examples of completed projects that span many areas of interest in medicinal and process chemistry.
Betulin (17) and betulinic acid (18), two pentacyclic triterpenes of the lupane family, were at the heart of the first joint effort between our laboratory and BMS medicinal chemistry (Figure 5).25 These two natural products that only differ by the oxidation at C28 present very interesting in vitro bioactivities. Betulinic acid (18) especially shows promising potency as an anti-inflammatory, anticancer, and anti-HIV agent. Unfortunately, any potential therapeutic application would be hampered by their scarce solubility in aqueous media. One solution to render 17 and 18 more soluble in water is to adorn their skeleton with heteroatoms. This problem appeared to us as an unparalleled opportunity to test some C–H oxidation methodologies on highly complex substrates. A dual chemical and enzymatic approach afforded eight oxidized betulin derivatives (some of which are natural products themselves), which were used for solubility assays. These assays were conducted by BMS and revealed a drastic increase of solubility for some compounds when compared to 17 or 18. This study demonstrates the power of C–H oxidation in the field of drug discovery as a tool to improve not only the potency of a pharmaceutical lead, but also its physicochemical properties. Furthermore, this work would not have been possible without a constant exchange of data and molecules between our laboratory and several groups at BMS, and Alicia Regueiro-Ren was instrumental in orchestrating this multifaceted research. Unpublished analogs arising from the developed C–H oxidation reactions will be patented by BMS in the near future.
Figure 5.

Beginning of a symbiotic relationship with BMS: performing C–H oxidation reactions on the biologically active framework of betulin (17) and betulinic acid (18) to enhance solubility profiles.
Academia–industry partnerships are not limited to medicinal chemistry research and can be beneficial at any stage of drug development. Thus, we joined forces with the process department at BMS to develop a new regioselective bromination of heteroarenes (Figure 6).26 A plethora of bioactive compounds possess C2-substituted heteroarenes, which are traditionally synthesized by cross-coupling reactions. Hence, C2 halogenation of heteroarenes is often a requisite step in drug synthesis. While chlorination methodology is abundant, regioselective bromination reactions proceeding under mild conditions are scarce. Building upon previous BMS findings made by Martin Eastgate and Ke Chen,27 it was found that Ts2O and nBu4NBr results in a mild bromination that can be applied to a wide array of fused pyridine N-oxides (21). This new reaction is currently being applied to prepare kilogram quantities of a drug candidate at BMS.
Figure 6.

Extending BMS process chemistry findings to fill a methodology gap in heteroarene bromination.
Due to the cornucopia of nitrogenous heterocycles in drug discovery, functionalization of heteroarenes is of primordial importance for the pharmaceutical industry. Radical C–H functionalization of heterocycles has been a longstanding area of research in our laboratory that resulted in collaborations with various pharmaceutical groups (vide infra). Among these desired transformations, an innate C–H amination of heteroarenes was highly sought. We aimed to develop a stable reagent that could generate an N-centered radical, which adds into arenes to make aminated or imidated products under mild conditions (Figure 7).28 After much exploration and optimization, N-succinimidyl perester (NSP) was found to generate an imidyl radical in situ, which then adds to (hetero)arenes to give imidated products. An essential component of the development of this work, which spanned over a year and a half, was the critical insight and careful suggestions from Martin Eastgate at BMS. On many occasions, this project was very close to being abandoned but it was the patience and optimism from our BMS colleague that paved the way to fruition.
Figure 7.
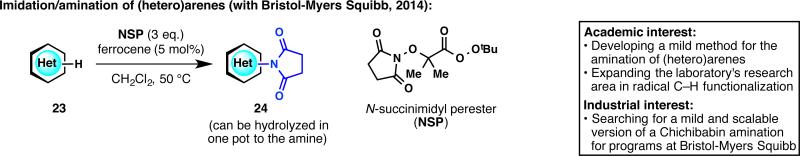
Radical C–H amination of (hetero)arenes and new reagent development.
Continuing with the theme of heteroarene functionalization, a chlorination method was then sought. Although chlorination of (hetero)aromatic compounds is a very well-known process, the “go-to” reagent, N-chlorosuccinimide (NCS), is not always sufficient in terms of reactivity. Pharmaceutical companies are therefore interested in alternative (hetero)arene chlorination strategies. For example, BMS had a few scaffolds for which NCS was sluggish but for which SO2Cl2 or Cl2 was excessively reactive. Based on a curious finding during the synthesis of pyrrole-imidazole alkaloids,29 a guanidine-based chlorinating reagent was invented (Figure 8).30 This reagent was named CBMG, or more affectionately as Palau'chlor (which is based on the name of the iconic pyrrole-imidazole natural product, palau'amine). Aided by the process chemistry expertise of BMS, we were able to assess the stability of this reagent and safety of its use in large-scale procedures. Thanks to the distribution capacity of Sigma-Aldrich (vide infra), Palau'chlor is already being used in drug discovery programs at BMS and in many other pharmaceutical companies.
Figure 8.
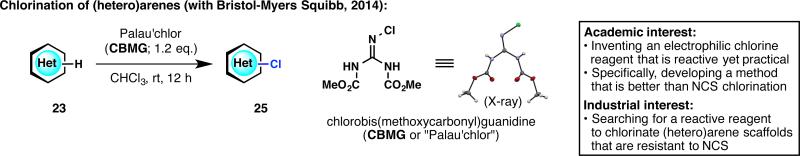
Revisiting the old problem of aromatic chlorination with a new chlorinating agent, Palau'chlor.
Pfizer
Pfizer is a prime example depicting a paradigm shift in academia–industry collaborations that has been witnessed over the last decade. From 2006 to 2011, TSRI and Pfizer had been in a 5-year research collaboration wherein Pfizer had the first right to partner with TSRI on every project (a “first-rights deal”) in exchange for a lump-sum payment.31 After this collaboration ended, Pfizer re-entered into a more focused partnership with selected individual laboratories, including our group. In this symbiosis, we gained access to compounds in confidential projects at Pfizer for internal scientific use. In return, unpublished results were shared with Pfizer scientists for incorporation into their research programs. Numerous projects have been completed together resulting in many joint publications, which will be discussed below.
In 2011, our laboratory reported a method for heteroarene trifluoromethylation in which CF3–SO2Na (Langlois reagent) was used under oxidative conditions to react with heteroarene C–H bonds.32 Building on this work, we surmised that heteroarene difluoromethylation could be of interest in medicinal chemistry for isostere-based drug design (Figure 9).33 Although we had originally aimed to make the analogous CF2H–SO2Na, the synthesis of this reagent was not possible and instead, it was found that zinc difluoromethanesulfinate (DFMS) could be prepared as a crystalline compound that reacts readily with heteroarene C–H bonds at room temperature. Michael Collins at Pfizer has been our industrial colleague ever since the initiation of this joint work, providing us the valuable perspective of a practicing medicinal chemist. Aided by Pfizer La Jolla's close relationship with our laboratory, both figuratively and geographically, our groups have been able to meet in person every few weeks to exchange ideas as well as chemical compounds. Finally, as described at the end of this Account, DFMS catalyzed the beginning of our relationship with Sigma-Aldrich, which spurred an institution-wide partnership in 2013.
Figure 9.
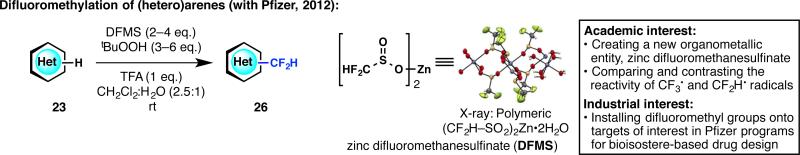
Establishment of a symbiotic relationship with Pfizer that led to the creation of a mild, rapid and practical way to difluoromethylate (hetero)arenes.
Based on the rapid widespread use of DFMS in both academia and industry, a series of zinc bis(alkanesulfinate) reagents were designed (Figure 10).34,35 Zinc trifluoromethanesulfinate (TFMS) was found to be superior to the sodium version in both reactivity and efficiency. Other zinc bis(alkanesulfinate) reagents that contain trifluoroethyl, monofluoromethyl, isopropyl, tri(ethylene glycol), monochloromethyl, (methoxycarbonyl)methyl, cyclohexyl, and perfluorohexyl groups were also synthesized. This expansion of the types of (fluoro)alkyl groups that can be appended onto heteroarenes not only provided us a better understanding of sulfinate radical chemistry but also served to diversify numerous small molecules for Pfizer's drug discovery programs.
Figure 10.
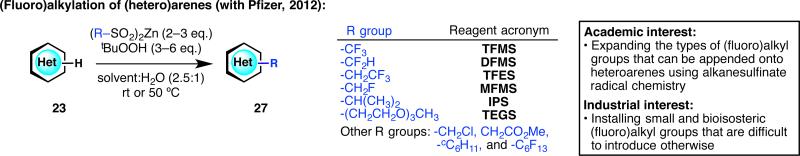
Expansion of the scope of the (fluoro)alkylating agent to generate a suite of diversifying reagents for medicinal chemistry.
In 2013, we had read with great interest a review titled “Profound Methyl Effects in Drug Discovery and a Call for New C–H Methylation Reactions.”36 Although heteroarene methylation was already an objective of ours, this review gave us renewed motivation to expand the sulfinate radical method for methylation (Figure 11).37 Meanwhile, Pfizer chemists (now consisting of Michael Collins, Aaron Burns and Martha Ornelas) wanted to respond to the “call for new C–H methylation reactions” but in a way that guarantees ease of product purification, knowing that it is notoriously difficult to separate the product from starting material in methylation reactions. To this end, we jointly invented a two-step methylation method, wherein a new reagent, zinc (phenylsulfonyl)methanesulfinate (PSMS), allowed for the appendage of a −CH2SO2Ph group onto heteroarenes. The formed intermediate is much more polar than the heteroarene starting material (and often crystalline) and therefore the purification process is extremely simple even in the event of incomplete conversion. This intermediate can then be desulfonylated under three orthogonal reaction conditions, allowing for functional group compatibility when necessary. This strategy has been field-tested using building blocks in a current Pfizer program and continues to be used for heteroarene functionalization and divergent synthesis.
Figure 11.
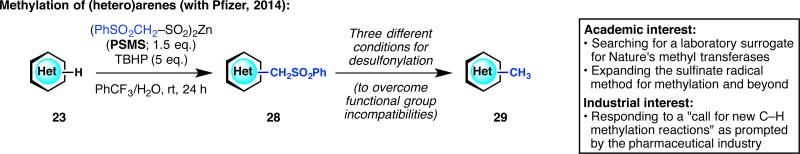
Responding to the pharmaceutical industry's “call for new C–H methylation reactions.”
Another article in 2013, titled “Metabolically Stable tert-Butyl Replacement,” made it clear to the chemistry community that trifluoromethylcyclopropylation is a much-needed reaction for bioisostere-based drug discovery.38 Both our group and Pfizer aimed to find a simple way to introduce this trifluoromethylcyclopropyl unit, and the logical flow of our research program identified the requirement of synthesizing trifluoromethylcyclopropylsulfinate (Figure 12).39 At this point, we had been making an assortment of alkanesulfinate reagents from alkanesulfonyl chlorides,33–35 alkyl halides,34,37 and difluoromethyl 2-pyridyl sulfone,40 but none of these methods could be applied to trifluoromethylcyclopropanecarboxylic acid (30), the only source of commercially available trifluoromethylcyclopropane. A solution to this challenge was found by exploiting the Barton decarboxylation reaction, where an interrupted version of this classic transformation was the key to success.39 While the academic focus was primarily to convert inexpensive carboxylic acids into valuable alkanesulfinate reagents, our industrial collaborators were able to pinpoint scaffolds that would make a great impact in medicinal chemistry. The utility of the resulting reagents, including sodium trifluoromethylcyclopropylsulfinate (TFCS-Na), has been demonstrated at Pfizer and is expected to be widespread in other pharmaceutical companies.
Figure 12.

Responding to the pharmaceutical industry's need for a convenient access to trifluoromethylcyclopropanated (hetero)arenes.
Our industrial partners can often identify “real-world” problems better than we can, owing to their years of experience in multidisciplinary research targeting diseases. A fitting example is the challenge associated in preventing or reducing aldehyde oxidase (AO)-mediated metabolism. Building upon previous work by Pfizer,41 our Pfizer colleagues had hoped to find a practical method for the chemical prediction and prevention of AO metabolism by blocking specific sites of a lead compound. Since electron-deficient heteroarenes are often substrates toward AO metabolism, which is similar to the trend for the sulfinate reagent DFMS, we conjectured that DFMS could be a potential candidate for AO prediction (Figure 13).42 As a result, DFMS suitably reacts with heteroaromatic scaffolds that are vulnerable to AO metabolism, and can often block the exact site of AO metabolism. Furthermore, efficient reaction at room temperature (within 2 h), and simple recognition of a difluoromethylated product peak at M+50 by LCMS analysis allow for a convenient “litmus test” for routine use in drug discovery.
Figure 13.
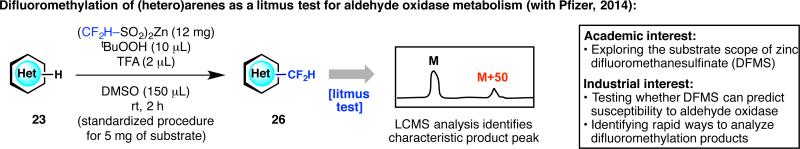
Merging interests with Pfizer's aldehyde oxidase (AO) metabolism program: the utilization of zinc difluoromethanesulfinate (DFMS) as a litmus test.
Morphotek/Eisai
Another symbiotic relationship can be witnessed in a project developed with Morphotek (a subsidiary of Eisai). A major commitment of Morphotek is to develop antibody-drug conjugate (ADC) platforms for oncology, and they were interested in expanding the scope of drugs that can be attached to monoclonal antibodies. Their objective was thus in line with our goal of expanding the scope of substrates that can be reacted with difluoroalkyl-containing sulfinate reagents.40 With the sulfinate radical chemistry, we were confident that we would be able to tag traditionally “untaggable” substrates that only contain C–H bonds. To this end, (difluoroalkylazido)sulfinate (DAAS-Na), a reagent containing a difluoromethanesulfinate unit as well as an azido terminus, was developed (Figure 14).43 Bioactive natural products as well as synthetic drugs were tested for reaction with DAAS-Na, and upon successful tagging, the azide unit was engaged in a “click” reaction with an antibody containing an alkyne-capped linker. The resulting ADC demonstrates great potential for labeling and bioconjugation of pharmaceutically relevant scaffolds that do not present conventional functional groups.
Figure 14.

Initiating a symbiosis with Eisai/Morphotek to achieve “native chemical tagging.”
Sigma-Aldrich
One final industry partner that has been a significant presence throughout all the projects described above is Sigma-Aldrich, a leading supplier of chemical reagents and products. Since Sigma-Aldrich is not a pharmaceutical company, the academia–industry relationship in this example differs from the rest. Ever since we became engaged in methodology development, we have believed that the best way to disseminate our scientific findings for widespread use is to commercialize the newly invented reagents through a purveyor of chemical reagents with a global presence. In particular, our colleagues in industry, whose stringent time constraints preclude them from making most reagents in-house, only incorporate new methodology into their research programs if these new reagents are readily accessible. From the Sigma-Aldrich standpoint, if they are the first supplier to add these new reagents to their portfolio, it will expand their market share. This would concomitantly expedite cutting-edge research, which would ultimately lead to increased sales. The institution-wide partnership between TSRI and Sigma-Aldrich began in 2013, after the invention of the first sulfinate reagents. Two reagents in particular, DFMS and TFMS, spawned this partnership due to their immediate sales following publication, which surpassed the $100,000 mark in less than 18 months. Following this model, TSRI and Sigma-Aldrich began a symbiotic relationship with the main objective of reducing the delay between the invention of novel reagents and their commercialization.44 In order to achieve this goal, unpublished work that can lead to the development of a new reagent is communicated with Sigma-Aldrich on a biweekly basis, and in return, Sigma-Aldrich provides the necessary chemicals that constitute these new reagents and expedites the commercialization process. As a result, dozens of reagents developed in our laboratory have been made commercially available, with a selected set depicted in Figure 15.28,30,33,34,37,39,40,43,45,46
Figure 15.

Instituting an unprecedented symbiosis of an academic group with a reagent company to generate a series of new reagents for widespread benefit.
CONCLUSION
As exemplified throughout the vignettes of this review, true symbiotic relationships between academia and industry can be achieved and be beneficial for both parties. They led us to develop many new reagents and new reactions, as well as a number of total syntheses and derivatizations of complex molecules. On the industrial side, the results of these partnerships have impacted every step of the drug discovery process, from the synthesis of a few milligrams of potential therapeutic leads to the improvement of kilogram-scale preparations of drug candidates. Commercialization of our new reagents through Sigma-Aldrich will hopefully serve both the academic and industrial worlds. Moreover, establishing partnerships with various companies has shaped general expectations for the roles of both parties in such a relationship (Table 1). This type of symbiosis generally starts from industry's identification of “real-world” problems meeting academia's capacity to deliver out-of-the-box solutions. Academia needs to provide specialized scientific education to the students, which can be complemented by industry's multidisciplinary expertise. Transparent communication between both groups is required through progress reports and (telephone) conferences, and an agreement must be reached regarding intellectual properties and deadlines. Finally, both sides must be driven by the will of discovering new science. While we recognize that not all projects are suited for a symbiotic partnership, we believe that many can be tailored such that mutual success can be achieved. We hope that this kind of symbiotic relationship will complement the existing models of funding and that the present Account will stimulate the launch of more fruitful partnerships between academia and industry.
Table 1.
General expectations in a fruitful academia-industry symbiosis.
| What is to be expected from academia | What is to be expected from industry |
|---|---|
| Out-of-the-box solutions | “Real-world” problems |
| Education of students | Multidisciplinary expertise |
| Biweekly progress | Biweekly phone conference or meeting |
| Meeting deadlines | Patience when facing challenges |
| Sharing discoveries through publications | Filing patents |
| Discovering new science | Applying collaborative science |
ACKNOWLEDGMENT
The authors thank Bristol-Myers Squibb for a graduate fellowship (Q.M.), the National Institutes of Health (NIH/NIGMS GM-073949 and GM-106210), and our many industrial partners (LEO Pharma, Bristol-Myers Squibb, Pfizer, Morphotek/Eisai, and Sigma-Aldrich) for funding. The authors are grateful for the opportunity to work with John Kadow (Bristol-Myers Squibb), Luigi Grasso (Morphotek/Eisai), Chris Thomas (Sigma-Aldrich), and other industrial colleagues whose names are mentioned throughout the manuscript.
Biography
Quentin Michaudel was born in France in 1986. He received his B.Sc. (2008) and M.Sc. (2010) at the École Normale Supérieure de Lyon, which included a 6-month research internship at The Scripps Research Institute (TSRI) with Prof. Phil S. Baran. He then returned to the Baran laboratory for his Ph.D. studies, where he is working on new C–H oxidations and their applications to complex molecule synthesis.
Yoshihiro Ishihara was born in Kyoto, Japan in 1984 but was raised in Montreal, Canada. He received his B.Sc. (2005) and his M.Sc. (2007) at McGill University, after which he obtained his Ph.D. (2012) under the tutelage of Prof. Phil S. Baran at TSRI. Currently, he is a senior staff scientist in the Baran laboratory with interests ranging from academic total synthesis projects to collaborative research.
Phil S. Baran was born in New Jersey in 1977 and received his undergraduate education from New York University in 1997. After earning his Ph.D. at TSRI in 2001, he pursued postdoctoral studies at Harvard University until 2003, at which point he returned to TSRI to begin his independent career. He was promoted to the rank of Professor in 2008 and is currently the Darlene Shiley Chair in chemistry. His laboratory is dedicated to the study of fundamental organic chemistry through the auspices of natural product total synthesis.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- 1.a Gray NS. Drug discovery through industry-academic partnerships. Nat. Chem. Biol. 2006;2:649–653. doi: 10.1038/nchembio1206-649. [DOI] [PubMed] [Google Scholar]; b Nicolaou KC. Academic–industrial partnerships in drug discovery and development. Angew. Chem. Int. Ed. 2014;53:4730–4730. doi: 10.1002/anie.201402816. [DOI] [PubMed] [Google Scholar]
- 2.a Tachibana C, Science Careers Opening industry-academic partnerships. 2013 http://sciencecareers.sciencemag.org/career_magazine/previous_issues/articles/2013_04_12/science.opms.r1300132.; b Eales J, Business Weekly Academic-industry partnerships: a broad spectrum of opportunity. 2014 http://www.businessweekly.co.uk/blog/business-weekly-guest-blog/17422-academic-industry-partnerships-a-broad-spectrum-of-opportunity.; c Larkin M, Elsevierconnect Building successful partnerships between academia and industry. 2014 http://www.elsevier.com/connect/building-successful-partnerships-between-academia-and-industry.
- 3.National Institutes of Health, Department of Health and Human Services Academic-industrial partnerships for translation of in vivo imaging systems for cancer investigations (R01) http://grants.nih.gov/grants/guide/pa-files/PAR-13-169.html.
- 4.National Science Foundation Grant Opportunities for Academic Liaison with Industry (GOALI) http://www.nsf.gov/funding/pgm_summ.jsp?pims_id=13706.
- 5.European Commission Research & Innovation Innovation Industry-Academia Partnerships and Pathways (IAPP) — Marie Curie Actions. http://ec.europa.eu/research/mariecurieactions/about-mca/actions/iapp/index_en.htm.
- 6.Bruno NC, Tudge MT, Buchwald SL. Design and preparation of new palladium precatalysts for C–C and C–N cross-coupling reactions. Chem. Sci. 2013;4:916–920. doi: 10.1039/C2SC20903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Presset M, Oehlrich D, Rombouts F, Molander GA. Copper-mediated radical trifluoromethylation of unsaturated potassium organotrifluoroborates. J. Org. Chem. 2013;78:12837–12843. doi: 10.1021/jo4023233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdhury AD, Weding N, Julis J, Franke R, Jackstell R, Beller M. Towards a practical development of light-driven acceptorless alkane dehydrogenation. Angew. Chem. Int. Ed. 2014;53:6477–6481. doi: 10.1002/anie.201402287. [DOI] [PubMed] [Google Scholar]
- 9.Friedfeld MR, Shevlin M, Hoyt JM, Krska SW, Tudge MT, Chirik PJ. Cobalt precursors for high-throughput discovery of base metal asymmetric alkene Hydrogenation Catalysts. Science. 2013;342:1076–1080. doi: 10.1126/science.1243550. [DOI] [PubMed] [Google Scholar]
- 10.Poelma JE, Fors BP, Meyers GF, Kramer JW, Hawker CJ. Fabrication of complex three-dimensional polymer brush nanostructures through light-mediated living radical polymerization. Angew. Chem. Int. Ed. 2013;52:6844–6848. doi: 10.1002/anie.201301845. [DOI] [PubMed] [Google Scholar]
- 11.Burns NZ, Krylova IN, Hannoush RN, Baran PS. Scalable total synthesis and biological evaluation of haouamine A and its atropisomer. J. Am. Chem. Soc. 2009;131:9172–9173. doi: 10.1021/ja903745s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a Baran PS, Burns NZ. Total synthesis of (±)-haouamine A. J. Am. Chem. Soc. 2006;128:3908–3909. doi: 10.1021/ja0602997. [DOI] [PubMed] [Google Scholar]; b Burns NZ, Baran PS. On the origin of the haouamine alkaloids. Angew. Chem. Int. Ed. 2008;47:205–208. doi: 10.1002/anie.200704576. [DOI] [PubMed] [Google Scholar]
- 13.Gaich T, Baran PS. Aiming for the ideal synthesis. J. Org. Chem. 2010;75:4657–4673. doi: 10.1021/jo1006812. [DOI] [PubMed] [Google Scholar]
- 14.The Scripps Research Institute News & Views Baran lab collaborates with LEO Pharma. 2011 http://www.scripps.edu/newsandviews/e_20110718/etc.html.
- 15.For examples, see: Shenvi RA, Guerrero CA, Shi J, Li C-C, Baran PS. Synthesis of (+)-cortistatin A. J. Am. Chem. Soc. 2008;130:7241–7243. doi: 10.1021/ja8023466. Maimone TJ, Shi J, Ashida S, Baran PS. Total synthesis of vinigrol. J. Am. Chem. Soc. 2009;131:17066–17067. doi: 10.1021/ja908194b.
- 16.a Jørgensen L, McKerrall SJ, Kuttruff CA, Ungeheuer F, Felding J, Baran PS. 14-step synthesis of (+)-ingenol from (+)-3-carene. Science. 2013;341:878–882. doi: 10.1126/science.1241606. [DOI] [PubMed] [Google Scholar]; b McKerrall SJ, Jørgensen L, Kuttruff CA, Ungeheuer F, Baran PS. Development of a concise synthesis of (+)-ingenol. J. Am. Chem. Soc. 2014;136:5799–5810. doi: 10.1021/ja501881p. [DOI] [PubMed] [Google Scholar]
- 17.Liang X, Grue-Sørensen G, Månsson K, Vedsø P, Soor A, Stahlhut M, Bertelsen M, Engell KM, Högberg T. Syntheses, biological evaluation and SAR of ingenol mebutate analogues for treatment of actinic keratosis and non-melanoma skin cancer. Bioorg. Med. Chem. Lett. 2013;23:5624–5629. doi: 10.1016/j.bmcl.2013.08.038. [DOI] [PubMed] [Google Scholar]
- 18.Chen K, Baran PS. Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature. 2009;459:824–828. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 19.Foo K, Usui I, Götz DCG, Werner EW, Holte D, Baran PS. Scalable, enantioselective synthesis of germacrenes and related sesquiterpenes inspired by terpene cyclase phase logic. Angew. Chem. Int. Ed. 2012;51:11491–11495. doi: 10.1002/anie.201206904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon DD, Lockner JW, Zhou Q, Baran PS. Scalable, divergent synthesis of meroterpenoids via “borono-sclareolide”. J. Am. Chem. Soc. 2012;134:8432–8435. doi: 10.1021/ja303937y. [DOI] [PubMed] [Google Scholar]
- 21.Rosen BR, Simke LR, Thuy-Boun PS, Dixon DD, Yu J-Q, Baran PS. C–H functionalization logic enables synthesis of (+)-hongoquercin A and related compounds. Angew. Chem. Int. Ed. 2013;52:7317–7320. doi: 10.1002/anie.201303838. [DOI] [PubMed] [Google Scholar]
- 22.Renata H, Zhou Q, Baran PS. Strategic redox relay enables a scalable synthesis of ouabagenin, a bioactive cardenolide. Science. 2013;339:59–63. doi: 10.1126/science.1230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang H, Sridhar Reddy M, Phoenix S, Deslongchamps P. Total synthesis of ouabagenin and ouabain. Angew. Chem. Int. Ed. 2008;47:1272–1275. doi: 10.1002/anie.200704959. [DOI] [PubMed] [Google Scholar]
- 24.The Scripps Research Institute News & Views Scripps Research and Bristol-Myers Squibb enter into five-year collaboration. 2014 http://www.scripps.edu/newsandviews/e_20120618/bms.html.
- 25.Michaudel Q, Journot G, Regueiro-Ren A, Goswami A, Guo Z, Tully TP, Zou L, Ramabhadran RO, Houk KN, Baran PS. Improving physical properties via C– H oxidation: chemical and enzymatic approaches. Angew. Chem. Int. Ed. 2014;53:12091–12096. doi: 10.1002/anie.201407016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wengryniuk SE, Weickgenannt A, Reiher C, Strotman NA, Chen K, Eastgate MD, Baran PS. Regioselective bromination of fused heterocyclic N-oxides. Org. Lett. 2013;15:792–795. doi: 10.1021/ol3034675. [DOI] [PubMed] [Google Scholar]
- 27.Eastgate MD, Bultman MS, Chen K, Fanfair DD, Fox RJ, La Cruz TE, Mudryk BM, Risatti CA, Simpson JH, Soumeillant MC, Tripp JC, Xiao Y. Methods for the preparation of HIV attachment inhibitor piperazine prodrug compound. PCT Int. Appl. WO2013119625 A1. 2013 [Google Scholar]
- 28.Foo K, Sella E, Thomé I, Eastgate MD, Baran PS. A mild, ferrocene-catalyzed C–H imidation of (hetero)arenes. J. Am. Chem. Soc. 2014;136:5279–5282. doi: 10.1021/ja501879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez RA, Barrios Steed D, Kawamata Y, Su S, Smith PA, Steed TC, Romesberg FE, Baran PS. Axinellamines as broad-spectrum antibacterial agents: scalable synthesis and biology. J. Am. Chem. Soc. 2014;136:15403–15413. doi: 10.1021/ja508632y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez RA, Pan C-M, Yabe Y, Kawamata Y, Eastgate MD, Baran PS. Palau’chlor: a practical and reactive chlorinating reagent. J. Am. Chem. Soc. 2014;136:6908–6911. doi: 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Scripps Research Institute News Release The Scripps Research Institute enters major five-year $100 million collaboration with Pfizer. 2014 http://www.scripps.edu/news/press/2006/113006.html.
- 32.Ji Y, Brueckl T, Baxter RD, Fujiwara Y, Seiple IB, Su S, Blackmond DG, Baran PS. Innate C–H trifluoromethylation of heterocycles. Proc. Natl Acad. Sci. 2011;108:14411–14415. doi: 10.1073/pnas.1109059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujiwara Y, Dixon JA, Rodriguez RA, Baxter RD, Dixon DD, Collins MR, Blackmond DG, Baran PS. A new reagent for direct difluoromethylation. J. Am. Chem. Soc. 2012;134:1494–1497. doi: 10.1021/ja211422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujiwara Y, Dixon JA, O'Hara F, Funder ED, Dixon DD, Rodriguez RA, Baxter RD, Herlé B, Sach N, Collins MR, Ishihara Y, Baran PS. Practical and innate carbon–hydrogen functionalization of heterocycles. Nature. 2012;492:95–99. doi: 10.1038/nature11680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O'Hara F, Baxter RD, O'Brien AG, Collins MR, Dixon JA, Fujiwara Y, Ishihara Y, Baran PS. Preparation and purification of zinc sulfinate reagents for drug discovery. Nat. Prot. 2013;8:1042–1047. doi: 10.1038/nprot.2013.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schönherr H, Cernak T. Profound methyl effects in drug discovery and a call for new C–H methylation reactions. Angew. Chem. Int. Ed. 2013;52:12256–12267. doi: 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- 37.Gui J, Zhou Q, Pan C-M, Yabe Y, Burns AC, Collins MR, Ornelas MA, Ishihara Y, Baran PS. C–H methylation of heteroarenes inspired by radical SAM methyl transferase. J. Am. Chem. Soc. 2014;136:4853–4856. doi: 10.1021/ja5007838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes-Seeman D, Jain M, Bell L, Ferreira S, Cohen SL, Chen X-H, Amin J, Snodgrass B, Hatsis P. Metabolically stable tert-butyl replacement. ACS Med. Chem. Lett. 2013;4:514–516. doi: 10.1021/ml400045j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gianatassio R, Kawamura S, Eprile CL, Foo K, Ge J, Burns AC, Collins MR, Baran PS. Simple sulfinate synthesis enables C–H trifluoromethylcyclopropanation. Angew. Chem. Int. Ed. 2014;53:9851–9855. doi: 10.1002/anie.201406622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Q, Ruffoni A, Gianatassio R, Fujiwara Y, Sella E, Shabat D, Baran PS. Direct synthesis of fluorinated heteroarylether bioisosteres. Angew. Chem. Int. Ed. 2013;52:3949–3952. doi: 10.1002/anie.201300763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linton A, Kang P, Ornelas M, Kephart S, Hu Q, Pairish M, Jiang Y, Guo C. Systematic structure modifications of imidazo[1,2-a]pyrimidine to reduce metabolism mediated by aldehyde oxidase (AO). J. Med. Chem. 2011;54:7705–7712. doi: 10.1021/jm2010942. [DOI] [PubMed] [Google Scholar]
- 42.O'Hara F, Burns AC, Collins MR, Dalvie D, Ornelas MA, Vaz ADN, Fujiwara Y, Baran PS. A simple litmus test for aldehyde oxidase metabolism of heteroarenes. J. Med. Chem. 2014;57:1616–1620. doi: 10.1021/jm4017976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Q, Gui J, Pan C-M, Albone E, Cheng X, Suh EM, Grasso L, Ishihara Y, Baran PS. Bioconjugation by native chemical tagging of C–H bonds. J. Am. Chem. Soc. 2013;135:12994–12997. doi: 10.1021/ja407739y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The Scripps Research Institute, News Release Sigma-Aldrich® and The Scripps Research Institute partner to accelerate the commercialization of research reagents. http://www.scripps.edu/news/press/2013/20130718sigma.html. 2013 http://www.scripps.edu/news/press/2013/20130718sigma.html
- 45.Voica A-F, Mendoza A, Gutekunst WR, Fraga JO, Baran PS. Guided desaturation of unactivated aliphatics. Nat. Chem. 2012;4:629–635. doi: 10.1038/nchem.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilde NC, Isomura M, Mendoza A, Baran PS. Two-phase synthesis of (−)-taxuyunnanine D. J. Am. Chem. Soc. 2014;136:4909–4912. doi: 10.1021/ja501782r. [DOI] [PMC free article] [PubMed] [Google Scholar]