

Abstract
We previously developed cell-based vaccines as therapeutics for metastatic cancers. The vaccines were aimed at activating type I CD4+T cells and consisted of tumor cells transfected with genes encoding syngeneic MHC class II and CD80 costimulatory molecules, and lacking the MHC II-associated invariant chain. The vaccines showed some efficacy in mice with sarcoma, melanoma, and breast cancer and activated MHC class II syngeneic T cells from breast, lung, and melanoma patients. During the course of the vaccine studies, we observed that CD80 not only costimulated naïve T cells, but also bound to PD-L1 and prevented tumor cell-expressed PD-L1 from binding to its receptor PD-1 on activated T cells. A soluble form of CD80 (CD80-Fc) had the same effect and sustained IFNγ production by both human and murine PD-1+ activated T cells in the presence of PD-L1+ human or mouse tumor cells, respectively. In vitro studies with human tumor cells indicated that CD80-Fc was more effective than antibodies to either PD-1 or PD-L1 in sustaining T cell production of IFNγ. Additionally, in vivo studies with a murine tumor demonstrated that CD80-Fc was more effective than antibodies to PD-L1 in extending survival time. Studies with human T cells blocked for CD28 and with T cells from CD28 knockout mice demonstrated that CD80-Fc simultaneously inhibited PD-L1/PD-1-mediated immune suppression and delivered costimulatory signals to activated T cells, thereby amplifying T cell activation. These results suggest that CD80-Fc may be a useful monotherapy that minimizes PD-1 pathway immune suppression while simultaneously activating tumor-reactive T cells.
Keywords: Tumor-induced immune suppression, T cell activation, Checkpoint blockade molecules, PD-1 pathway, Cancer immunotherapy, PIVAC 14
Introduction
Cancer immunotherapy was recognized by Science Magazine in 2013 as the “Breakthrough of the Year” [1], and monoclonal antibodies (mAbs) to PD-L1 (Programmed Death Ligand 1; also known as B7 homolog 1 (B7-H1) or CD274) and PD-1 (Programmed Death 1 or CD279) were described as “Drugs of the Year” because of their ability to neutralize PD-L1/PD-1-mediated immune suppression [2].
PD-L1 is a major obstacle to antitumor immunity because it (i) tolerizes/anergizes tumor-reactive T cells by binding to its receptor PD-1 [3, 4]; (ii) renders tumor cells resistant to CD8+ T cell and FasL-mediated lysis [5]; and (iii) tolerizes T cells by reverse signalling through T cell-expressed CD80 [6]. Many malignant cells express PD-L1, either constitutively or after induction by IFNγ [3, 5, 7, 8]. The role of PD-1 as a significant obstacle to antitumor immunity is supported by studies demonstrating that antibody blocking of PD-1 improves T cell activation and reduces tumor progression [7, 9–11] and that antibody blocking of PD-L1 reverse signalling through CD80 prevents T cell anergy [6]. These effects are at least partially due to blocking of PD-L1 on dendritic cells (DC) since the expression of PD-L1 by activated DC limits effector cell differentiation and the generation of CD8+ T cell memory [12]. The critical role of PD-L1 in human cancer was unequivocally established by recent clinical trials in which 19–30 % of patients with certain advanced cancers treated with antibodies to PD-L1 or PD-1 had partial or complete remissions [13, 14]. On-going studies combining mAb therapy to PD-L1 or PD-1 with immune activating strategies have demonstrated that optimal efficacy is achieved if PD-L1/PD-1 suppression is blocked while concurrently providing immune activation signals such as with cancer vaccines [15, 16]. Based on these findings, we are developing a therapeutic strategy that combines in a single reagent the ability to inhibit PD-1 pathway immune suppression while concomitantly delivering activating signals to tumor-reactive T cells. Our approach makes use of the fact that in addition to binding to the receptor PD-1, PD-L1 also binds to the costimulatory molecule CD80 (also known as B7.1) [17, 18]. This unique binding capacity has led us to hypothesize that a soluble version of CD80 has the potential to facilitate antitumor immunity via three independent mechanisms: (i) CD80 could inhibit the PD-1 suppressive pathway and maintain the activity of activated PD-1+ T cells by binding to PD-L1 and thereby preventing the binding of PD-L1+ tumor cells to PD-1+ T cells; (ii) by binding to PD-L1+ cells, CD80 would also prevent the suppression caused by PD-L1–CD80 reverse signalling into CD80+ T cells; and (iii) as a dimer, CD80 could simultaneously bind to PD-L1 and CD28. This dual binding could bridge PD-L1+ target tumor cells and tumor-reactive CD4+ and CD8+ T cells and concomitantly prevent PD-1 pathway immune suppression while delivering activating costimulatory signals through CD28. These mechanisms are shown schematically in Fig. 1. This article will review the in vitro and in vivo data from murine and human systems that support our hypothesis and identify a soluble form of CD80 as a potential novel cancer immunotherapeutic reagent.
Fig. 1.

A soluble recombinant molecule consisting of the extracellular domains of CD80 linked to the Fc domain of Ig (soluble CD80 or CD80-Fc) has the potential to maintain and facilitate T cell activation through three independent mechanisms. (i) CD80-Fc could block the PD-1 suppressive pathway by binding to PD-L1 on tumor cells or activated T cells and preventing binding to PD-1 on activated T cells; (ii) CD80-Fc could activate tumor-reactive T cells by binding to PD-L1 on tumor cells and simultaneously costimulating through CD28 on T cells; (iii) CD80-Fc could prevent the tolerance induced by reverse signalling of PD-L1 through CD80 on T cells by binding to PD-L1 on tumor cells. TcR = T cell receptor; pMHC = peptide-MHC complex; APC = antigen-presenting cell; ? = CD80-Fc probably does not bind to CTLA4 although theoretically it should bind
Membrane-bound human or mouse CD80 prevents PD-L1–PD-1 interactions and maintains IFNγ production by activated PD-1+ T cells in the presence of PD-L1+ human or mouse tumor cells
We initially observed that tumor cells transfected with membrane-bound CD80 plus MHC class II molecules syngeneic to the recipient were therapeutic vaccines for mice with up to 21-day established, large (up to 6 mm diameter) vascularized, subcutaneous tumors with extensive stroma [19], as well as for mice with established, spontaneous metastatic disease [20]. At the time we attributed the efficacy of CD80 to its serving as a costimulatory molecule. In vitro studies confirmed that the genetically modified tumor cells were both antigen-presenting cells (APC) for tumor peptides [21, 22] and a source of tumor peptide/MHC I and II complexes for “cross-dressing” dendritic cells [23, 24]. When we realized that most of the mouse tumors were strongly PD-L1+, or were induced by IFNγ to express PD-L1, we examined the possibility that CD80 was also maintaining T cell activation by preventing PD-L1–PD-1 interactions. We tested this hypothesis by expressing membrane-bound CD80 on seven different PD-L1+ human tumor cell lines including lung adenocarcinoma, mammary carcinoma, and cutaneous and uveal melanomas. For all PD-L1+ tumors, co-expression of membrane-bound CD80 prevented the binding of PD-1 as assessed by flow cytometry [25]. The functional relevance of preventing PD-L1–PD-1 interactions was shown by coculture experiments in which activated human T cells were incubated with human PD-L1+ tumor cells. Whereas PD-L1+ tumor cells suppressed IFNγ production, CD80+PD-L1+ tumor cells prevented the suppression and maintained IFNγ production by activated PD-1+ human T cells. IFNγ production by both CD4+ and CD8+ T cells was sustained. Expression of membrane-bound CD80 on two PD-L1+ murine tumor cell lines similarly prevented PD-1 binding and maintained IFNγ production by PD-1+ murine T cells [26], further suggesting that CD80 has potential as a therapeutic agent to prevent PD-L1–PD-1-mediated immune suppression [25].
A soluble form of CD80 similarly maintains IFNγ production by activated PD-1+ human or mouse T cells in the presence of PD-L1+ tumor cells
The studies with membrane-bound CD80 provided proof-of-principle for CD80 as a potential therapeutic; however, therapy with a membrane-bound molecule is not feasible. We therefore set out to determine whether a soluble form of CD80 was similarly effective. Membrane-bound CD80 consists of an amino-terminal Ig-like variable domain (IgV) followed by an Ig-like constant domain (IgC), a transmembrane region, and a cytoplasmic domain. PD-L1 has a similar structure (Fig. 2a). Mutation studies have established that CD80 and PD-1 bind to a similar location within the IgV domain of PD-L1 [17, 18] (Fig. 2b, c). To construct a soluble form of CD80 that binds to PD-L1, we generated recombinant fusion proteins consisting of the IgV plus IgC domains of human or mouse CD80 fused to the Fc domain of either human IgG1 or murine IgG2a, respectively (CD80-Fc). To determine whether CD80-Fc was as effective as membrane-bound CD80, phytohemag-glutinin (PHA)-activated human peripheral blood mononu-clear cells (PBMC) were cocultured with PD-L1+ human C8161 cells ± human CD80-Fc or irrelevant fusion protein TROY-Fc (toxicity and JNK inducer or TNFRSF19). PD-L1+ human tumor cells suppressed the activated CD4+ and CD8+ T cells, and inclusion of CD80-Fc reversed the suppression [26]. Experiments with three additional PD-L1+ human tumors (MCF10 mammary carcinoma, H292 squamous cell carcinoma, and H358 bronchioloal-veolar adenocarcinoma) showed similar suppression and rescue of activation in the presence of CD80-Fc [25]. The activated human T cells expressed high levels of both PD-1 and PD-L1, resulting in susceptibility to PD-1 suppression. To ascertain that CD80-Fc rescues T cell activation by preventing PD-1 suppression, we compared the ability of human PD-1-Fc vs. CD80-Fc to prevent PD-1 suppression [26]. Mouse PD-1 served as a negative control. Both human CD80 and human PD-1, but not mouse PD-1, restored IFNγ production in the presence of PD-L1+ human tumor cells, with CD80 being more effective.
Fig. 2.
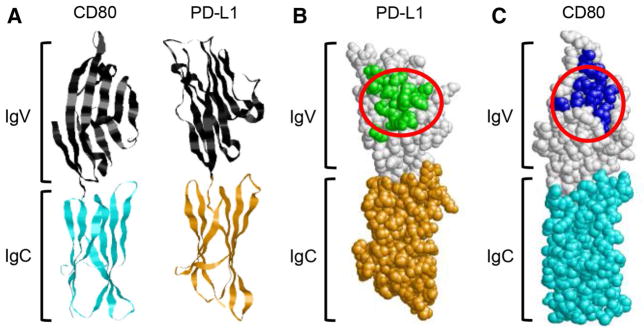
CD80, PD-1, PD-L1, and CTLA-4 share common binding sites. a Ribbon structures showing the amino-proximal IgV-like and the amino-distal IgC-like domains of CD80 and PD-L1 (structures are from RCSB Protein Databank (http://www.rcsb.org/pdb/home/home.do); accession numbers 1I8L and 3BIK, respectively). b PD-L1 space-fill structure showing the predicted binding sites for PD-1 (green residues) and CD80 (area circled in red). c CD80 space-fill structure showing the predicted binding sites for CTLA-4 (blue residues) and PD-L1 (area circled in red). Predicted binding sites are based on [17, 48, 49], and identification of amino acid residues that are within 3.9Å of the binding partner
Murine CD80 similarly prevented the binding of murine PD-1 to murine PD-L1+ tumor cells and maintained T cell production of IFNγ in the presence of PD-L1+ murine tumor cells. The 4T1 mammary carcinoma constitutively expresses PD-L1, and low levels (100 units) of IFNγ increase PD-L1 expression. PD-L1+ 4T1 cells bound soluble PD-1, and inclusion of CD80-Fc prevented the binding of PD-1. Addition of the mAb (43H12), which disrupts murine PD-L1–CD80 interactions [6], confirmed that CD80 prevented PD-1 binding because CD80 is physically associated with PD-L1. Co-expression of membrane-bound CD80 by PD-L1+ 4T1 or B16 MELF10 cells, or inclusion of CD80-Fc also prevented suppression of antigen-activated transgenic murine T cells [26]. These results demonstrated that CD80 masks PD-L1 and maintains T cell activation in the presence of multiple human and mouse PD-L1+ tumors.
CD80-Fc is more effective than mAbs to PD-L1 or PD-1 for maintaining activated T cells
MAbs to PD-L1 and PD-1 have significant efficacy in clinical trials as either monotherapies [13, 14, 27, 28], or in combination with other checkpoint blockers or cancer vaccines [29, 30]. To preliminarily determine how soluble CD80 compares to existing mAbs, we tested human CD80-Fc and five antihuman PD-L1 (29E.2A3, MIH1, 5H1, MIH3, and 130021) and five antihuman PD-1 (PD1.3.1.3, EH12.1, EH12.2H7, J116, and MIH4) mAbs in vitro for their ability to sustain T cell production of IFNγ in the presence of six different PD-L1+ human tumor cell lines (non-small cell lung cancers H292 and H358, mammary carcinoma MCF10, uveal melanoma MEL202, and cutaneous melanoma C8161). Cocultures of activated T cells plus PD-L1+ tumor cells containing CD80-Fc or CD80-transfected tumor cells consistently produced more IFNγ than cocultures with mAbs to PD-L1 or PD-1 [25, 26, 31]. These results demonstrated that in vitro, CD80-Fc is more effective in sustaining T cell activation than mAbs to PD-L1 or PD-1.
CD80-Fc enhances T cell activation by simultaneously inhibiting PD-1 suppression and costimulating
CD80 was originally identified as a ligand that delivers a costimulatory signal to T cells via its receptor CD28 [32]. To determine whether CD80 was also functioning as a costimulatory molecule in our tumor systems, two in vitro approaches were used. (i) Using human tumor cells and PBMC from healthy donors, CD80–CD28 costimulation was prevented by inclusion of mAbs to CD28. (ii) In a murine system, costimulation was eliminated by using T cells from CD28-deficient mice. By blocking or eliminating CD28, costimulation was prevented and CD80 could only facilitate T cell activation by preventing PD-1 suppression. Therefore, IFNγ production in cultures blocked for CD28 represents activity exclusively due to inhibiting PD-1 suppression, while the differential in IFNγ levels between CD28 blocked and unblocked cultures represents activity due to costimulation. In both human and mouse systems, CD80-Fc maintained T cell production of IFNγ in the presence of PD-L1+ tumor cells; however, IFNγ levels were not as high as when costimulation was not inhibited [26, 31]. Therefore, blocking costimulation reduces, but does not eliminate the ability of CD80 to maintain T cell activation, indicating that CD80-Fc acts by both costimulating and blocking the PD-1 suppressive pathway.
CD80-Fc delays murine tumor progression and increases tumor-infiltrating T cells (TIL) more effectively than mAbs to PD-L1
In mouse models, mAbs to PD-L1 or PD-1 augment immunotherapy approaches such as adoptive T cell therapy and cancer vaccines [11, 15, 16, 33–37], and in some cases, antibody monotherapy reduces tumor progression and increases survival [9, 38–40]. However, some tumors are refractory or minimally responsive to PD-L1 or PD-1 monotherapy [34, 37, 41, 42]. To determine whether CD80-Fc is more effective in vivo than mAb monotherapy, we compared the efficacy of mAbs to PD-L1 vs. CD80-Fc for impacting tumor progression in mice with CT26 tumors. BALB/c mice were inoculated in the flank on day 0 with 5x105 PD-L1+ CT26 colon carcinoma cells and subsequently untreated or treated intraperitoneally with CD80-Fc, PD-L1 mAb 10F.9G2, or rat IgG2b isotype control (200 μg/mouse) on days 3, 6, 9, and 22. Tumor diameters were measured twice a week. Surviving mice were killed on day 42, and tumors were excised and weighed, and then analyzed by immunohistochemistry for TIL following staining with mAbs to CD3. All of the CD80-Fc-treated mice were alive at day 42, whereas only 67, 0, and 50 % of the PD-L1 mAb-treated mice, isotype control mice, and untreated mice, respectively, were alive. At day 42, the tumors in the CD80-Fc-treated group were approximately half the volume of the tumors in the surviving mice of the other groups. Immunohistochemistry revealed that the tumors from the CD80-Fc-treated group were highly infiltrated with TIL and contained many more TIL than tumors from the other groups (Fig. 3).
Fig. 3.

Tumors of mice treated with soluble CD80 (CD80-Fc) contain large areas of tumor-infiltrating CD3+ T cells. Tumors were excised on day 40 (Isotype control group) or 42 (all other groups) post-CT26 inoculation, embedded and frozen in O.C.T, and subsequently sectioned and stained for immunohistochemistry with mAbs to CD3. Magnification is ×200
Conclusions
Since many human tumors, as well as activated T cells and DC, express PD-L1, the PD-1 suppressive pathway is a significant obstacle to both natural antitumor immunity and immune therapies aimed at activating a patient’s immune system. Inhibitors of the p38 MAP kinase pathway reduce transcription of PD-L1 by DC and may thereby enhance DC-based cancer vaccines [43]. However, these drugs cannot be used in vivo, so other therapy strategies are needed. Binding of CD80 to PD-L1 in both murine and human systems has been known since 2007 and 2008, respectively [17, 18]. However, prior to the studies reviewed here, it was not appreciated that CD80 binding to PD-L1 prevents PD-L1/ PD-1 interactions and therefore neutralizes PD-1-mediated immune suppression. Because CD80 is also a potent costimulatory molecule that facilitates T cell activation when bound to its receptor CD28, CD80 serves the dual function of activating T cells while simultaneously inhibiting PD-1 pathway immune suppression (Fig. 1). This combination of effects by a single reagent may explain the heightened efficacy of soluble CD80 as compared to anti-PD-L1 or PD-1 monotherapy.
PD-L1 also tolerizes T cells by reverse signalling through T cell-expressed CD80, so soluble CD80 has the potential to also neutralize this additional suppressive mechanism. Whether or not soluble CD80 prevents suppression by reverse signalling remains to be tested.
A potential limitation of soluble CD80 as a therapeutic agent is its binding to CTLA-4, another molecule capable of suppressing activated T cells [44] (Figs. 1, 2c). However, the enhancement of T cell activation that is routinely observed with soluble CD80, plus our preliminary data using antibodies to block CTLA-4 (Horn and Ostrand-Rosenberg, unpublished), indicates that CD80-Fc does not cause immune suppression by binding to CTLA-4. Given that the predicted binding sites for PD-L1 and CTLA-4 on CD80 are in the same region, and that the dissociation constant for CTLA-4–CD80 interactions is smaller than that for PD-L1–CD80 binding, it is surprising that CD80-Fc does not appear to suppress T cell activation through CTLA-4. The apparent lack of suppression through CTLA-4 could be due to the inability of CD80-Fc to cross-link CTLA-4, or because binding of CD80 to PD-L1 perturbs the CTLA-4 binding site. Additional studies are clearly needed to resolve this issue.
A phase I trial of superagonist mAb TGN1412 to CD28 created a cytokine storm (cytokine release syndrome) in all six volunteers [45]. Recent studies demonstrated that this effect involved TGN1412 binding to the Fc gamma receptor IIb (FcγRIIb) on monocytes, which is up-regulated by high-density culture [46]. Our in vitro studies have not produced elevated levels of cytokines characteristic of a cytokine storm. Likewise, tumor-bearing mice treated with CD80-Fc did not display symptoms of cytokine release syndrome. The absence of these symptoms is most likely because the binding constant of CD80-Fc for CD28 is approximately five orders of magnitude lower than mAb binding to CD28 [47]. However, use of an Fc domain that does not bind to the FcγRIIb could overcome the potential for cytokine release syndrome.
Clearly, more extensive studies in experimental animal models and clinical trials are needed to establish whether a soluble form of CD80 will be more effective than current mono or combination antibody therapies for patients with cancer. However, our initial studies indicate that soluble CD80 has efficacy and supports the continued testing of soluble CD80 as a novel cancer immunotherapy.
Acknowledgments
This work was supported by grants from the U.S. National Institutes of Health (RO1CA84232) and the State of Maryland Technology Development Corp. (TEDCO; 1000308). The authors thank Ms. Virginia Clements for her excellent technical support and Dr. Robert C. Rosenberg for the structural analyses of Fig. 2.
Abbreviations
- APC
Antigen-presenting cell
- CD80-Fc
Soluble CD80 (two extracellular domains of CD80 fused to the Fc domain of Ig)
- CTLA-4
Cytotoxic T lymphocyte antigen 4
- DC
Dendritic cells
- IgC
Immunoglobulin-like constant region
- IgV
Immunoglobulin-like variable region
- mAbs
Monoclonal antibodies
- PD-1
Programmed death 1
- PD-L1
Programmed death ligand 1
- PBMC
Peripheral blood mononuclear cells
- PHA
Phytohemagglutinin
- TcR
T cell receptor for antigen
- TIL
Tumor-infiltrating T cells
- TROY-Fc
Toxicity and JNK inducer or TNFRSF19 protein fused to the Fc domain of Ig
Footnotes
This paper is a Focussed Research Review based on a presentation given at the Fourteenth International Conference on Progress in Vaccination against Cancer (PIVAC 14), held in Rome, Italy, 24th–26th September, 2014. It is part of a Cancer Immunology, Immunotherapy series of Focussed Research Reviews and meeting report.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 2.Robert C, Soria JC, Eggermont AM. Drug of the year: programmed death-1 receptor/programmed death-1 ligand-1 receptor monoclonal antibodies. Eur J Cancer. 2013;49:2968–2971. doi: 10.1016/j.ejca.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 4.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, Chen L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008;111:3635–3643. doi: 10.1182/blood-2007-11-123141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, Yao S, Tsushima F, Narazaki H, Anand S, Liu Y, Strome SE, Chen L, Tamada K. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010;116:1291–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 8.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 9.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 10.Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, Majdic O, Gajewski TF, Theobald M, Andreesen R, Mackensen A. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006;119:317–327. doi: 10.1002/ijc.21775. [DOI] [PubMed] [Google Scholar]
- 11.Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, Salomao D, Cheville J, Hirano F, Lin W, Kasperbauer JL, Ballman KV, Chen L. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–6505. [PubMed] [Google Scholar]
- 12.Gibbons RM, Liu X, Harrington SM, Krco CJ, Kwon ED, Dong H. B7-H1 signaling is integrated during CD8(+) T cell priming and restrains effector differentiation. Cancer Immunol Immunother. 2014;63:859–867. doi: 10.1007/s00262-014-1563-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013;73:6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duraiswamy J, Kaluza KM, Freeman GJ, Coukos G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013;73:3591–3603. doi: 10.1158/0008-5472.CAN-12-4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butte MJ, Pena-Cruz V, Kim MJ, Freeman GJ, Sharpe AH. Interaction of human PD-L1 and B7-1. Mol Immunol. 2008;45:3567–3572. doi: 10.1016/j.molimm.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baskar S, Glimcher L, Nabavi N, Jones RT, Ostrand-Rosenberg S. Major histocompatibility complex class II + B7-1 + tumor cells are potent vaccines for stimulating tumor rejection in tumor-bearing mice. J Exp Med. 1995;181:619–629. doi: 10.1084/jem.181.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998;58:1486–1493. [PubMed] [Google Scholar]
- 21.Armstrong TD, Clements VK, Martin BK, Ting JP, Ostrand-Rosenberg S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc Natl Acad Sci USA. 1997;94:6886–6891. doi: 10.1073/pnas.94.13.6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chornoguz O, Gapeev A, O’Neill MC, Ostrand-Rosenberg S. Major histocompatibility complex class II + invariant chain negative breast cancer cells present unique peptides that activate tumor-specific T cells from breast cancer patients. Mol Cell Proteomics. 2012;11:1457–1467. doi: 10.1074/mcp.M112.019232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dolan BP, Gibbs KD, Jr, Ostrand-Rosenberg S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8 + T cells. J Immunol. 2006;177:6018–6024. doi: 10.4049/jimmunol.177.9.6018. [DOI] [PubMed] [Google Scholar]
- 24.Dolan BP, Gibbs KD, Jr, Ostrand-Rosenberg S. Tumor-specific CD4 + T cells are activated by “cross-dressed” dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J Immunol. 2006;176:1447–1455. doi: 10.4049/jimmunol.176.3.1447. [DOI] [PubMed] [Google Scholar]
- 25.Haile S, Bosch JJ, Agu N, Zeender A, Somasundaram P, Srivastava MK, Rodel S, Wolf J, Ksander BR, Ostrand-Rosenberg S. Tumor cell programmed death ligand-1-mediated T cell suppression is overcome by co-expression of CD80. J Immunol. 2011;186:6822–6829. doi: 10.4049/jimmunol.1003682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haile ST, Dalal SP, Clements V, Tamada K, Ostrand-Rosenberg S. Soluble CD80 restores T cell activation and overcomes tumor cell programmed death ligand 1-mediated immune suppression. J Immunol. 2013;191:2829–2836. doi: 10.4049/jimmunol.1202777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, Kroeger J, Eysmans C, Sarnaik AA, Chen YA. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31:4311–4318. doi: 10.1200/JCO.2013.51.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haile S, Horn LH, Ostrand-Rosenberg S. A soluble form of CD80 enhances anti-tumor immunity by neutralizing programmed death ligand-1and simultaneously providing costimulation. Cancer Immunol Res. 2014;2:610–615. doi: 10.1158/2326-6066.CIR-13-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 33.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, Dhurandhar N, Myers L, Berggren R, Hemminki A, Alvarez RD, Emilie D, Curiel DT, Chen L, Zou W. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 34.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U, Limmer A, Jonsson G, Holzel M, Tuting T. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4:674–687. doi: 10.1158/2159-8290.CD-13-0458. [DOI] [PubMed] [Google Scholar]
- 36.Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74:4042–4052. doi: 10.1158/0008-5472.CAN-13-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pilon-Thomas S, Mackay A, Vohra N, Mule JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol. 2010;184:3442–3449. doi: 10.4049/jimmunol.0904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Investig. 2014;124:687–695. doi: 10.1172/JCI67313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, Sharpe AH, Vallera DA, Azuma M, Levine BL, June CH, Murphy WJ, Munn DH, Blazar BR. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116:2484–2493. doi: 10.1182/blood-2010-03-275446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–2194. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cannon MJ, Goyne HE, Stone PJ, Macdonald LJ, James LE, Cobos E, Chiriva-Internati M. Modulation of p38 MAPK signaling enhances dendritic cell activation of human CD4 + Th17 responses to ovarian tumor antigen. Cancer Immunol Immunother. 2013;62:839–849. doi: 10.1007/s00262-013-1391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber J. Immune checkpoint proteins: a new therapeutic paradigm for cancer–preclinical background: CTLA-4 and PD-1 blockade. Semin Oncol. 2010;37:430–439. doi: 10.1053/j.seminoncol.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 46.Hussain K, Hargreaves CE, Roghanian A, Oldham RJ, Chan HT, Mockridge CI, Chowdhury F, Frendeus B, Harper KS, Strefford JC, Cragg MS, Glennie MJ, Williams AP, French RR. Upregulation of FcgammaRIIb on monocytes is necessary to promote the superagonist activity of TGN1412. Blood. 2015;125:102–110. doi: 10.1182/blood-2014-08-593061. [DOI] [PubMed] [Google Scholar]
- 47.Peach RJ, Bajorath J, Naemura J, Leytze G, Greene J, Aruffo A, Linsley PS. Both extracellular immunoglobin-like domains of CD80 contain residues critical for binding T cell surface receptors CTLA-4 and CD28. J Biol Chem. 1995;270:21181–21187. doi: 10.1074/jbc.270.36.21181. [DOI] [PubMed] [Google Scholar]
- 48.Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, Okazaki T, Honjo T, Minato N, Garboczi DN. The PD-1/ PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc Natl Acad Sci USA. 2008;105:3011–3016. doi: 10.1073/pnas.0712278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, Stahl ML, Seehra J, Somers WS, Mosyak L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 2001;410:608–611. doi: 10.1038/35069118. [DOI] [PubMed] [Google Scholar]