

Abstract
Inactivation of the tumor suppressor PTEN is recognized as a major event in the pathogenesis of the brain tumor glioblastoma. However, the mechanisms by which PTEN loss specifically impacts the malignant behavior of glioblastoma cells including their proliferation and propensity for invasiveness remain poorly understood. Genetic studies suggest that the transcription factor STAT3 harbors a PTEN-regulated tumor suppressive function in mouse astrocytes. Here, we report that STAT3 plays a critical tumor suppressive role in PTEN-deficient human glioblastoma cells. Endogenous STAT3 signaling is specifically inhibited in PTEN-deficient glioblastoma cells. Strikingly, reactivation of STAT3 in PTEN-deficient glioblastoma cells inhibits their proliferation, invasiveness, and ability to spread on myelin. We also identify the chemokine IL8 as a novel target gene of STAT3 in human glioblastoma cells. Activated STAT3 occupies the endogenous IL8 promoter and directly represses IL8 transcription. Consistent with these results, IL8 is upregulated in PTEN-deficient human glioblastoma tumors. Importantly, IL8 repression mediates STAT3-inhibition of glioblastoma cell proliferation, invasiveness, and spreading on myelin. Collectively, our findings uncover a novel link between STAT3 and IL8 whose deregulation plays a key role in the malignant behavior of PTEN-deficient glioblastoma cells. These studies suggest that STAT3 activation or IL8 inhibition may have potential in patient-tailored treatment of PTEN-deficient brain tumors.
Keywords: STAT3, IL8, Astrocyte [Astroglia], gliogenesis, Glioma, Tumor
Introduction
Malignant gliomas are the most common primary tumors of the brain and among the most aggressive human tumors (Holland, 2001; Konopka and Bonni, 2003; Louis, 2006; Furnari et al., 2007). Despite intense efforts to find effective treatments, malignant gliomas are still incurable. Glioblastoma tumor cells display phenotypic characteristics of astrocytes and their precursors, the neural stem cells (Louis et al., 2007). Both neural stem cells and astrocytes have been proposed to represent the cells of origin of glioblastoma (Holland, 2001; Bachoo et al., 2002; Uhrbom et al., 2002; Bajenaru et al., 2003). Collectively, these studies suggest that deregulation of mechanisms that control the normal biology of neural stem cells and astrocytes, including the differentiation of neural stem cells into astrocytes, might contribute to the pathogenesis of glioblastoma.
The transcription factor STAT3 plays an instructive role in astrocyte differentiation during normal brain development (Bonni et al., 1997; Rajan and McKay, 1998). These observations raised the possibility that STAT3 signaling might be deregulated in glioblastoma. In genetic studies in mouse astrocytes, we recently identified STAT3 as an important regulator of glial cell transformation (de la Iglesia et al., 2008). STAT3 harbors a tumor suppressive or oncogenic function depending on the genetic background of the tumor. STAT3 is required for astrocyte transformation upon expression of the oncogenic form of the epidermal growth factor receptor, EGFRvIII. Conversely, STAT3 displays tumor suppressive properties in astrocytes deficient in the tumor suppressor PTEN (de la Iglesia et al., 2008). A tumor suppressive role of STAT3 is consistent with the function of STAT3 as a transcription factor dedicated to the differentiation of neural stem cells into astrocytes during normal brain development (Bonni, 2003).
Genetic studies in mouse astrocytes suggest that deregulation of the PTEN-Akt pathway triggers astrocyte transformation by inhibiting the LIFRβ-STAT3 pathway (de la Iglesia et al., 2008). Consistent with these results, PTEN deficiency in human glioblastoma specimens correlates with LIFRβ downregulation and inhibition of STAT3 phosphorylation at the key regulatory site Tyr705 (de la Iglesia et al., 2008). However, the effect of manipulating STAT3 activity on the malignant behavior of glioblastoma tumor cells and the mechanism by which STAT3 might mediate its tumor suppressive effects in glioblastoma cells remained unknown.
In this study, we establish a tumor suppressive function of STAT3 in human glioblastoma cells. Activation of endogenous STAT3 in wild type PTEN-expressing glioblastoma cells inhibits cell proliferation. Conversely, expression of a constitutive active form of STAT3 in PTEN-deficient glioblastoma cells significanlty inhibits cell proliferation, invasiveness, and spreading on myelin. We also report a mechanism by which STAT3 triggers tumor suppressive effects in human glioblastoma cells. The chemokine IL8 is identified as a direct repressed target gene of STAT3 in human glioblastoma cells. The repression of IL8 mediates the ability of STAT3 to inhibit glioblastoma cell proliferation, invasiveness, and spreading on myelin. Taken together, our results define the STAT3-IL8 signaling link as a novel PTEN-regulated mechanism that suppresses glioblastoma cell proliferation and invasiveness. These findings provide new targets for potential patient-tailored therapeutic intervention in glioblastoma. Activation of STAT3 and inhibition of IL8 might represent useful strategies for the treatment of PTEN-deficient malignant gliomas.
Materials and Methods
Cell Culture
U87 glioblastoma cells were infected with MSCV-IRES-GFP/S3C, MSCV-IRES-GFP/mS3C or empty vector retroviruses and a population of infected cells for each construct was collected by FACS-sorting cells for GFP expression. IL8 knockdown U87 cells were obtained by infection with pLL3.7-Puro/IL8i1 or pLL3.7-Puro/IL8i2 lentiviruses and selection with puromycin (4µg/ml).
Plasmids
The MSCV-IRES-GFP/mS3C construct was generated by site-directed mutagenesis of RcCMV/S3C (Bromberg et al., 1999) using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and subcloning into MSCV-IRES-GFP.
Virus Production and Infection
Recombinant retroviruses were made by transfecting 293T cells with pMD.MLV gag.pol, pHDM.G (VSVG pseudotype), and the transfer plasmid (e.g. MSCV-IRES-GFP). Cells were infected with equal amounts of retroviruses in the presence of 8µg/ml polybrene (Sigma).
Cloning of recombinant lentiviruses coding for a short hairpin RNA directed against STAT3 or human IL8 was carried out using a modified pLL3.7 vector which encodes resistance to puromycin (pLL3.7 Puro). The following complementary oligonucleotides were inserted into pLL3.7 Puro: Stat3i fw, 5’-TGG GCA GTT TGA GTC GCT CAT TCA AGA GAT GAG CGA CTC AAA CTG CCC TTT TTG GAA AC-3’, Stat3i rev, 5’-TCG AGT TTC CAA AAA GGG CAG TTT GAG TCG CTC ATC TCT TGA ATG AGC GAC TCA AAC TGC CCA-3’; IL8i 1 fw, 5'-TGA ACT TAG ATG TCA GTG CAT TCA AGA GAT GCA CTG ACA TCT AAG TTC TTT TTG GAA AC-3', IL8i 1 rev, 5'-TCG AGT TTC CAA AAA GAA CTT AGA TGT CAG TGC ATC TCT TGA ATG CAC TGA CAT CTA AGT TCA-3'; IL8i 2 fw, 5'-TGA ACT GAG AGT GAT TGA GAG TTC AAG AGA CTC TCA ATC ACT CTC AGT TCT TTT TGG AAA C-3', IL8i 2 rev, 5'-TCG AGT TTC CAA AAA GAA CTG AGA GTG ATT GAG AGT CTC TTG AAC TCT CAA TCA CTC TCA GTT CA-3'. Hairpin structures containing the stem sequences (underlined) and the loops (bold italica) are indicated. Lentiviruses were generated by co-transfecting pLL3.7 and packaging vectors (VSVG, RSV-REV and pMDL g/p RRE) into 293T cells. Cells were infected with equal amounts of lentiviruses and selected with puromycin.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation analyses were done as described (Shi et al., 2003). Following immunoprecipitation, a PCR reaction was used to amplify the IL8 promoter with the following primers: IL8, 1st PCR reaction, human IL8 fw, 5'-TCT CAC TCC ATC CCT TTT GC-3', human IL8 rev, 5'-AGT GGC AGG TGT TAG AAC AAG A-3', nested PCR reaction, human IL8 nest fw, 5'- CTC CAT CCC TTT TGC TAG TGA-3', human IL8 nest rev, 5'-ACA GAT GCT ATC ATG ATG GTG AA-3'. Primers designed to amplify the E-cadherin promoter were used as negative controls for the PCR reaction: human E-cad fw, 5’-TAG CCT GGC GTG GTG GTG TGC ACC TG-3’, human E-cad rev, 5’-GTG CGT GGC TGC AGC CAG GTG AGC C-3’.
Matrigel Invasion assays
Matrigel pre-coated invasion chambers (BD) with a 8 µm pore size membrane were utilized according to the manufacturer’s instructions. 2.5×104 cells in 500µl of serum-free DMEM were added to each of the inserts and incubated at 37°C for 22 hours. Cells on the lower surface of the membrane which had migrated through the matrigel were fixed, stained with crystal violet, and counted. Non-invasive NIH3T3 cells were used as a negative control. Equivalent numbers of NIH3T3 failed to invade the matrigel.
Microarray Analysis
RNA was extracted from U87 stable cell lines using Trizol, followed by an additional purification step using an RNAeasy kit (Qiagen) according to the manufacturer’s instructions. Biotinylated cRNAs from each cell line were generated from 15µg of total RNA and hybridized to the Affymetrix U133A chips. Microarray procedures were conducted at the Dana-Farber Cancer Institute Microarray Core Facility, Boston, MA (http://chip.dfci.harvard.edu). Each cell line was used in three separate experiments. Gene expression data was analyzed using Vector Xpression software (InforMax Inc.). Raw expression values were normalized by linear scaling so that the mean array intensity was identical for all scans. Intensity thresholds were set a min=20 and max=16,000 units, resulting in 12,284 probe sets for subsequent analysis. These remaining 12,284 probe sets were then subjected to the t-test using Vector Xpression for the identification of differentially expressed transcripts. Fold change expression data was diagrammatically represented using GeneCluster software (http://www.broad.mit.edu/cancer/software/genecluster2/gc2.html).
Results
STAT3 suppresses human glioblastoma cell proliferation
The identification of STAT3’s tumor suppressive function in genetic studies of PTEN-deficient mouse astrocytes raises the major question of the role of STAT3 in human glioblastoma. To investigate STAT3 signaling in human glioblastoma cells, we first characterized the potential of the cytokine leukemia inhibitory factor (LIF) to induce the phosphorylation of endogenous STAT3 at the key regulatory site Tyr705 in PTEN-expressing and PTEN-deficient glioblastoma cells. We found that while LIF induced STAT3 Tyr705 phosphorylation in the wild type PTEN-expressing SF188 and LN229 glioblastoma cells, LIF failed to effectively induce the STAT3 phosphorylation in the PTEN-deficient U87 and A172 glioblastoma cells (Fig. 1A). These results suggest that PTEN deficiency suppresses the endogenous STAT3 signaling pathway in human glioblastoma cells.
Figure 1. Endogenous STAT3 inhibits proliferation of wild type PTEN-expressing glioblastoma cells.

A, Lysates of glioblastoma cells that express wild type PTEN (SF188 and LN229) or glioblastoma cells that harbor PTEN mutations (U87 and A172) were immunoblotted with STAT3 and Tyr 705-phosphorylated STAT3 (pYS3) antibodies. Cells were serum starved for 24 hours and then treated with 100ng/ml CNTF or 10ng/ml LIF for 15 minutes. LIF treatment robustly induced STAT3 tyrosine phosphorylation in control SKNMC neuroblastoma cells and in the wild type PTEN-expressing glioblastoma cells, but failed to effectively induce the STAT3 phosphorylation in the PTEN-deficient glioblastoma cells. B, Growth curves of PTEN-mutant (U87 and A172) and PTEN-expressing (SF188) glioblastoma cells that were treated with LIF (10ng/ml daily) or left untreated. LIF did not affect cell population growth of U87 (representative experiment shown) or A172 cells (two representative experiments shown), but significantly reduced SF188 cell population growth (n=3; paired t-test, *p<0.05). C, Cell population growth of SF188 cells infected with a retrovirus containing an IRES-GFP cassette alone or that also encodes wild type STAT3 or a dominant negative STAT3 (S3D). LIF (10ng/ml daily) led to a significant reduction in population growth of control-vector or wild type STAT3-expressing cells (n=3, ANOVA, *p<0.05, ¶p<0.01, §p<0.001), but not in S3D-expressing SF188 cells. D, Left panel: lysates of LN229 cells infected with either control lentivirus or lentivirus encoding a small interfering hairpin RNA directed at STAT3 and selected with puromycin were immunoblotted with a STAT3 antibody. Actin was used as loading control. The Stat3i hairpin RNA induced knockdown of endogenous human STAT3. Right panel: time-course of the growth of LN229 cells infected with control lentivirus or lentivirus encoding Stat3i and selected with puromycin. LIF (10ng/ml daily) led to a significant reduction in growth in control-infected (n=3; ANOVA, *p<0.05), but not Stat3i hairpin RNA-expressing LN229 cells.
PTEN deficiency suppresses STAT3 signaling in in mouse astrocytes via downregulation of the cytokine receptor LIFRβ (de la Iglesia et al., 2008). We therefore analyzed the expression of LIFRβ in human glioblastoma cells. Consistent with results in mouse astrocytes (de la Iglesia et al., 2008), we found very low levels of LIFRβ protein in the PTEN-deficient U87 glioblastoma cells (Supplemental Fig. 1). These results are consistent with the interpretation that LIFRβ downregulation may represent a mechanism by which PTEN deficiency suppresses STAT3 signaling in the U87 glioblastoma cells. Two lines of evidence suggested that inhibition of STAT3 signaling was specific to LIFRβ deficiency in U87 cells. First, treatment of U87 cells with the PI3K inhibitor LY294002 restored LIFRβ levels (Supplemental Fig. 2A). Second, reconstitution of LIFRβ signaling in U87 cells by expression of a chimeric protein consisting of the intracellular domain of LIFRβ fused to the ectodomain of epidermal growth factor receptor (EGFR) allowed the induction of STAT3 phosphorylation at Tyr705 in response to EGF (Supplemental Fig. 2B). These findings suggest that the absence of LIFRβ in the PTEN-deficient U87 glioblastoma cells may underlie the inactivation of STAT3 signaling in these cells. These results corroborate our findings in murine astrocytes (de la Iglesia et al., 2008). However, as a caveat that not all human PTEN-deficient glioblastoma cells behave in this fashion, we find that LIFRβ expression is intact in A172 cells (Supplemental Figure 1), suggesting that PTEN deficiency may inhibit STAT3 signaling independently of regulation of LIFRβ expression in these cells.
We also measured the levels of STAT3 phosphorylation at Ser727 in the human glioblastoma cells. These experiments revealed that STAT3 was phosphorylated at Ser727 in the human glioblastoma cells regardless of their PTEN status and treatment with LIF had little or no effect on the Serine 727 phosphorylation in these cells (Supplemental Fig. 3). These results suggest that the PTEN status of human glioblastoma cells predominantly affects the phosphorylation of STAT3 at Tyr705.
We next asked if LIF had distinct effects on proliferation in glioblastoma cells in parallel with the differential phosphorylation of endogenous STAT3 at Tyr705 depending on the PTEN status of these cells. Remarkably, while LIF inhibited the proliferation of the PTEN-expressing SF188 and LN229 glioblastoma cells (Fig. 1B–D), LIF had little or no effect on the proliferation of the PTEN-deficient A172 and U87 glioblastoma cells (Fig. 1B), suggesting that LIF-inhibition of cell proliferation correlates with LIF-induction of endogenous STAT3 in PTEN-expressing glioblastoma cells.
To determine the role of endogenous STAT3 activation in LIF-inhibition of cell proliferation in PTEN-expressing glioblastoma cells, we expressed in SF188 cells a dominant interfering form of STAT3 (S3D) that forms dimers with endogenous STAT3 that fail to bind to promoters of cytokine-responsive genes (Nakajima et al., 1996; Bonni et al., 1997). We found that the expression of S3D but not wild type STAT3 significantly reduced the ability of LIF to inhibit SF188 glioblastoma population growth (Fig. 1C). We also used a lentiviral DNA-based template method of RNA interference to knockdown STAT3 protein in the PTEN-expressing LN229 glioblastoma cells. STAT3 knockdown was found to significantly inhibit LIF-suppression of proliferation in LN229 glioblastoma cells (Fig. 1D). In other experiments, STAT3 knockdown in SF188 glioblastoma cells also blocked LIF-inhibition of SF188 cell proliferation (data not shown). Taken together, our results indicate that LIF-induction of endogenous STAT3 in wild type PTEN-expressing glioblastoma cells inhibits cell proliferation. These results are also consistent with the possibility that inhibition of endogenous STAT3 signaling upon PTEN loss may serve to promote glioblastoma cell proliferation.
To directly assess the significance of PTEN loss-suppression of STAT3 signaling in the malignant behavior of glioblastoma cells, we determined the effect of reactivation of STAT3 in PTEN-deficient glioblastoma cells. Using a retroviral approach, we expressed a constitutively active form of STAT3 (S3C) in these cells. S3C contains two point mutations whereby A661 and N663 are replaced with cysteine. S3C thus forms dimers through sulfhydryl bonds independently of Tyr705 phosphorylation, and translocates to the nucleus independently of cytokine stimulation (Bromberg et al., 1999). We generated a mutant form of activated STAT3 (S3C) in which the DNA binding region of S3C was mutated (mS3C) (Fig. 2A). When expressed in 293T cells, activated STAT3 (S3C) but not the mutant form mS3C robustly induced transcription of a reporter gene driven by STAT binding sites suggesting that mS3C serves as a transcriptionally-impaired control mutant of S3C (Supplemental Fig. 4).
Figure 2. Expression of activated STAT3 inhibits proliferation PTEN-deficient glioblastoma cells.

A, Schematic of activated STAT3 (S3C) or a DNA-binding mutant version of S3C (mS3C). B, Left panel: lysates of U87 glioblastoma cells infected with a control retrovirus containing an IRES-GFP cassette or retroviruses that also encode FLAG-tagged S3C or mS3C were immunoblotted with STAT3 and FLAG antibodies, and antibodies to STAT1 or Akt as loading controls. Right panel: cell population growth curves of U87 glioblastoma cells. S3C-expressing U87 cells proliferated significantly more slowly than mS3C-expressing U87 cells or those infected with control virus (n=3; ANOVA, *p<0.05). C, BrdU labeling of U87 glioblastoma stable cells measured as a percentage of the total number of cells. Incorporation of BrdU was significantly reduced in U87-S3C cells compared to both vector and mS3C (n=4; ANOVA, *p<0.05). D, Cell population growth curves of PTEN-deficient U87 cells or isogenic PTEN-expressing U87 cells infected with the S3C or control retrovirus. Expression of S3C significantly reduced the proliferation of U87 but not of isogenic PTEN-expressing U87 glioblastoma cells (representative experiment of 3 independent experiments performed in triplicate, ANOVA, *p<0.001, **p<0.0001).
We stably expressed activated STAT3 (S3C) or its mutant form mS3C in PTEN-deficient U87 glioblastoma cells. In cell growth assays, activated STAT3 (S3C)- but not mS3C-expressing U87 glioblastoma cells increased in number at a significantly lower rate than control vector-infected U87 cells (Fig. 2B). There was less than 1% cell death in control vector-infected U87 glioblastoma cells, and the expression of activated STAT3 (S3C) or mS3C had no effect on cell death in the U87 cells (data not shown). By contrast, the rate of BrdU incorporation in activated STAT3 (S3C)- but not mS3C-expressing glioblastoma cells was reduced when compared to control vector-infected cells (Fig. 2C). These results suggest that activated STAT3 inhibits the proliferation of PTEN-deficient glioblastoma cells. In other cell population growth experiments, we found that exogenous expression of PTEN in the PTEN-deficient U87 cells reduced the capacity of these cells to proliferate (Figure 2D). In addition, whereas expression of S3C reduced the proliferative capacity of the parental U87 cells, S3C failed to effectively reduce the proliferation of the isogenic PTEN-expressing U87 cells (Figure 2D). These results suggest that reactivation of STAT3 specifically inhibits the ability of PTEN deficiency to promote the proliferation of human glioblastoma cells.
STAT3 suppresses glioblastoma cell invasiveness and spreading on myelin
Having established STAT3 as a suppressor of glioblastoma cell proliferation, we considered the possibility that STAT3 might also control glioblastoma cell invasiveness. We determined if reactivation of STAT3 in the PTEN-deficient U87 glioblastoma cells alters their invasiveness. In matrigel invasion assays, U87 cells efficiently invaded the matrigel layer (Fig. 3A). However, expression of activated STAT3 (S3C) in U87 glioblastoma cells significantly reduced the ability of these cells to invade matrigel (Fig. 3A). STAT3-inhibition of U87 glioblastoma cell invasiveness was not secondary to the effect of activated STAT3 on cell proliferation as the invasive potential of these cells was measured at a time (22 hours after plating) prior to the inhibitory effects of activated STAT3 on U87 cell proliferation were manifest (see Fig. 2B). These results suggest that reactivation of STAT3 suppresses the invasive property of PTEN-deficient glioblastoma cells.
Figure 3 . STAT3 decreases glioblastoma cell invasiveness and spreading on myelin.

A, Matrigel invasion assay of U87 glioblastoma stable cell lines. Significantly more U87-vector cells invaded the matrigel substrate than U87-S3C cells (n=3; t-test, *p<0.05). STAT3-inhibition of U87 glioblastoma cell invasiveness was not secondary to the effect of activated STAT3 on U87 cell proliferation as the invasive potential of these cells was measured at a time (22 hours after plating) prior to the inhibitory effects of activated STAT3 on U87 cell proliferation (see Figure 2B). Equivalent numbers of NIH3T3 cells failed to invade the matrigel (data not shown). B, Phalloidin red staining of actin stress fibers of stable U87 glioblastoma cells plated onto coverslips coated with either polyornithine (PO) or polyornithine together with myelin (20µg/ml). U87-S3C glioblastoma cells failed to spread on myelin (middle bottom panel), compared to a spread appearance on a polyornithine control substrate (middle top panel). U87-vector and U87-mS3C glioblastoma cells spread and formed stress fibers on myelin (right and left bottom panels). C, Quantification of cell spreading of U87 stable cells on myelin. Significantly fewer U87-S3C glioblastoma cells spread on myelin compared to U87-vector glioblastoma cells (n=3; ANOVA, *p<0.0001). Cells were counted in a blinded manner in three independent experiments, and the percent spreading was determined by calculating the number of spread cells over the total number of cells.
To determine if STAT3 regulates glioblastoma cell behavior on a substrate that is relevant to the selective property of gliomas to invade brain tissue along white matter tracts, we tested the effect of activated STAT3 (S3C) on the ability of glioblastoma cells to adhere and spread on adult rat brain myelin (Amberger et al., 1998). Unlike NIH-3T3 cells which spread on polynornithine but not on myelin (data not shown), U87 glioblastoma cells spread equally well on polyornithine or myelin substrate forming actin stress fibers around the cell cortex (Fig. 3B). The selective ability of glioblastoma cells but not other tumor or immortalized cells to spread on myelin substrate provides a powerful assay that may reflect the propensity of malignant gliomas to migrate along brain white matter tracts (Amberger et al., 1998). Strikingly, by contrast to parental U87 glioblastoma cells or those infected with the control GFP virus or the mS3C virus, the majority of S3C-expressing U87 glioblastoma cells spread on polyornithine substrate but failed to spread or form stress fibers in their periphery on myelin (Fig. 3B,C). Thus, activated STAT3 specifically inhibits glioblastoma cell adhesion and spreading on myelin.
STAT3 suppresses glioblastoma cell proliferation and invasiveness via repression of the chemokine IL8
Having identified the functional effects of STAT3 on glioblastoma cell proliferation and invasiveness, we next set out to elucidate the mechanism by which STAT3 mediates these biological effects. Since STAT3’s ability to inhibit glioblastoma cell proliferation and invasiveness correlated with its function as a transcription factor, we reasoned that activated STAT3 triggers changes in the expression of specific genes that mediate STAT3’s glioblastoma suppressive properties. We therefore subjected the activated STAT3 (S3C)-expressing and control GFP-expressing U87 glioblastoma cells to microarray analyses using Affymetrix chips. In these analyses, the most profound alteration was a 5-fold repression of the gene encoding interleukin-8 (IL8) in S3C-expressing cells compared to control cells (Fig. 4A,B).
Figure 4. IL-8 is a direct STAT3-repressed target gene.

A, Diagrammatic representation of the top 50 genes in three independent microarray analyses of control or S3C-expressing U87 cells, ranked according to fold change, that were repressed or induced upon S3C expression. B, Top 10 repressed (left) or induced (right) genes upon S3C expression. The fold change is indicated, and GenBank numbers are in parenthesis. C, Upper panel: immunoblotting of lysates of human glioblastoma samples (GBM) with an IL8 antibody. GAPDH was used as loading control (GAPDH immunoblotting of some of the GBM samples is also shown in (de la Iglesia et al., 2008). Eight of the tumors (# 4, 8, 11, 13, 16, 19, 24, 25) displayed very high levels of IL8. The majority of these tumors (6/8) had also low PTEN levels (de la Iglesia et al., 2008). Lower panel: Fisher’s exact test of IL8-positive and IL8-negative tumors revealed a strong correlation between IL8 expression and low PTEN levels (OR(odds ratio)=14, p<0.01).D, Left panel: Northern analysis of control vector-infected U87 glioblastoma cells or those expressing S3C using an IL8 probe. 18S RNA levels indicate equal loading. Middle panel: immunoblotting of lysates of control U87 glioblastoma cells or those expressing S3C or mS3C with an IL8 antibody. Asterisk indicates a non-specific band. Right panel: sandwich ELISA chemiluminescent analysis of medium from the control U87 glioblastoma cells or those expressing S3C using two antibodies specific for human IL8 (R&D Systems). Shown are mean IL8 concentration ± SEM (pg IL8/mg cell protein; n=3; t-test, *p<0.01). E, STAT3 represses the IL8 promoter. Pten-/- astrocytes were transfected with increasing amounts of an expression plasmid encoding S3C or a control plasmid together with an IL8 promoter-controlled luciferase reporter gene and a renilla expression plasmid, and subjected to dual luciferase assay 48 hours after transfection. Activated STAT3 significantly reduced IL8 promoter activity (n=3; ANOVA, *p<0.0001). F, Chromatin immunoprecipitation analysis at the endogenous IL8 promoter in S3C-expressing U87 glioblastoma cells using two different STAT3 antibodies (m, mouse; r, rabbit). Mouse and rabbit HA antibodies were used as controls. Negative controls for the PCR reaction were performed with primers for the E-cadherin gene. STAT3 directly bound to the endogenous IL8 gene.
IL8 is a chemokine that regulates neutrophil activation and chemoattraction (Baggiolini et al., 1994). In addition, IL8 has been implicated in the promotion of angiogenesis in a variety of tumors including gliomas (Heidemann et al., 2003; Garkavtsev et al., 2004). Consistent with a role for IL8 in glioblastoma progression, although IL8 expression is very low in normal brain, IL8 is upregulated in gliomas and in particular in high grade astrocytoma and glioblastoma (Yamanaka et al., 1994). In a panel of human glioblastoma tumors we have characterized by immunoblotting, we found that eight out of twenty five (32%) tumors displayed high levels of IL8 (Fig. 4C). PTEN deficiency tightly correlates with inactivation of STAT3 signaling in these human glioblastoma samples (de la Iglesia et al., 2008). Importantly, the majority of the IL8-expressing tumors (6/8) had low PTEN levels (Fig. 4C; see Fig.6 in (de la Iglesia et al., 2008) for PTEN levels). Fisher’s exact test of IL8-positive and IL8-negative tumors revealed a strong correlation of IL8 expression with low PTEN levels (Fig. 4C). Together, these observations raised the question of whether the repression of IL8 mediates STAT3-inhibition of glioblastoma cell proliferation and invasiveness.
First, we confirmed the microarray data in Northern analyses in which the amount of IL8 mRNA was robustly suppressed in activated STAT3 (S3C)-expressing cells (Fig. 4D). Consistent with these results, immunoblotting of U87 glioblastoma cell extracts revealed that IL8 protein was significantly downregulated upon expression of activated STAT3 (S3C). By contrast, IL8 protein was reduced to a lesser extent in mS3C-expressing cells suggesting that the transcriptional function of STAT3 is required for regulation of IL8 expression (Fig. 4D). The secretion of mature soluble IL8 into the medium by these glioblastoma cells correlated tightly with the results of Northern and Western analyses (Fig. 4D). Together, these results established that STAT3 inhibits the expression of IL8 in glioblastoma cells.
In transient expression assays, expression of activated STAT3 (S3C) potently diminished IL8 promoter-mediated transcription (Fig. 4E). By chromatin immunoprecipitation analyses of activated STAT3 (S3C)-expressing U87 glioblastoma cells, activated STAT3 was specifically bound to the endogenous IL8 promoter in these cells (Fig. 4F). These findings suggest that IL8 is a direct gene target of STAT3 and that activated STAT3 represses IL8 transcription.
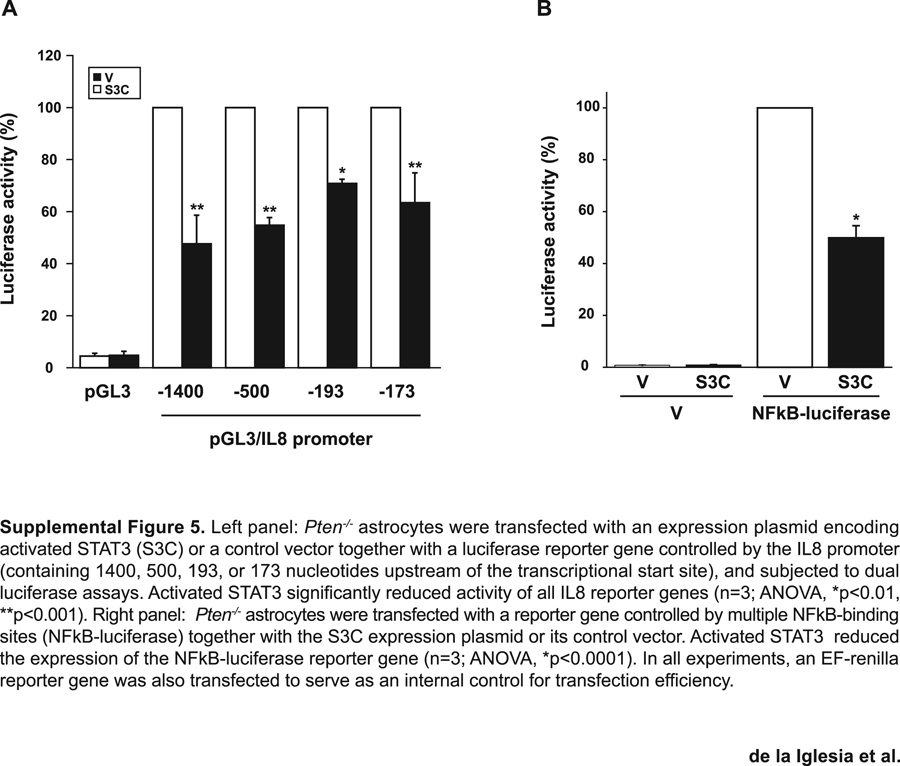
To investigate the mechanism by which STAT3 represses IL8 gene expression, we determined the region of the IL8 promoter necessary for activated STAT3 to inhibit IL8 transcription. Deletion analyses of the IL8 promoter in transient expression assays revealed that activated STAT3 (S3C) repressed a reporter gene controlled by a promoter fragment containing 173 nucleotides upstream of the IL8 transcriptional start site (Supplemental Fig. 5A). The −173 IL8 promoter fragment contains the NFkB response element that is critical for the expression of IL8 in diverse biological settings including malignant gliomas (Mukaida et al., 1990; Brat et al., 2005; Sarkar et al., 2008). We therefore asked if activated STAT3 might inhibit NFkB-dependent transcription in glial cells. Expression of activated STAT3 (S3C) inhibited a luciferase reporter gene controlled by multiple NFkB-binding sites (Supplemental Fig. 5B). Together, these results suggest that activated STAT3 may repress the IL8 gene via inhibtion of NFkB.
To determine if IL8 repression contributes to STAT3’s ability to inhibit glioblastoma cell proliferation, we used a lentiviral DNA template-based method of RNA interference to knockdown IL8 in U87 glioblastoma cells (Fig. 5A). Knockdown of endogenous IL8 in U87 glioblastoma cells by either of two hairpin RNAs targeting distinct regions of IL8 significantly reduced cell proliferation (Fig. 5B). IL8 knockdown also significantly reduced the ability of U87 glioblastoma cells to invade matrigel (Fig. 5C). In other experiments, we asked if IL8 restores the ability of activated STAT3 (S3C)-expressing U87 glioblastoma cells to spread on myelin substrate. Upon exposure to IL8, the S3C-expressing U87 glioblastoma cells spread as efficiently as control vector-infected U87 cells on myelin (Fig. 5D). Together, these results suggest that IL8 is a STAT3-repressed gene that promotes glioblastoma cell proliferation and invasiveness (Fig. 5E).
Figure 5. IL8 promotes glioblastoma cell proliferation, invasiveness, and spreading on myelin.

A, Lysates of U87 cells infected with IL8 RNAi-encoding lentiviruses (IL8i 1 or IL8i 2) or an empty vector and selected with puromycin were immunoblotted with IL8 and actin antibodies. Asterisk indicates a non-specific band. B, Cell population growth curves of IL8 knockdown or vector-control U87 glioblastoma cells. IL8 knockdown suppressed glioblastoma cell population growth (n=3; ANOVA, *p<0.001). C, Matrigel invasion assay of U87 glioblastoma cells infected with control or IL8 RNAi lentiviruses. Significantly more U87-vector glioblastoma cells invaded the matrigel substrate than U87-IL8i 1 or U87-IL8i 2 glioblastoma cells (ANOVA, *p<0.01). D, IL-8 addition rescues STAT3 suppression of glioma cell spreading on myelin. Quantification of cell spreading shows that in the presence of IL-8 significantly more U87-S3C cells spread on myelin compared to untreated U87-S3C cells plated on myelin (n=3; ANOVA, *p<0.0005). E, Model of the PTEN-regulated STAT3-IL8 signaling link in human glioblastoma cells. Left panel: in the presence of the tumor suppressor PTEN, STAT3 is activated by phosphorylation, binds to the IL8 promoter, and represses IL8 gene expression. Right panel: STAT3 is inhibited upon PTEN deficiency, thus relieving repression of the IL8 gene. Upregulation of IL8 promotes glioblastoma cell proliferation and invasiveness.
Discussion
In this study, we have uncovered a novel STAT3-dependent tumor suppressive mechanism in human glioblastoma cells (see model in Fig. 5E). We found that cytokine-induced endogenous STAT3 signaling was specifically impaired in PTEN-deficient but not PTEN-expressing glioblastoma cells. Inhibition of endogenous STAT3 blocked cytokine-suppression of proliferation in PTEN-expressing glioblastoma cells. Conversely, reactivation of STAT3 in PTEN-deficient glioblastoma cells inhibited their proliferation, invasiveness, and spreading on myelin. We also identified the chemokine IL8 as a direct repressed target gene of STAT3 in glioblastoma cells. IL8 repression mediated the ability of STAT3 to inhibit glioblastoma cell proliferation and invasiveness. Thus, deregulation of the STAT3-IL8 signaling link may confer PTEN loss with the ability to stimulate glioblastoma cell proliferation and invasiveness.
A major conclusion of our study is that STAT3, a protein with oncogenic behavior outside the brain (Bromberg et al., 1999; Bowman et al., 2000), exerts tumor suppressor effects in human glioblastoma cells. This finding provides pathophysiological significance to our recent studies indicating that STAT3 suppresses malignant transformation of mouse astrocytes (de la Iglesia et al., 2008). In some reports, it has been suggested that STATs may promote human glioblastoma tumor cell proliferation and survival (Rahaman et al., 2002; Konnikova et al., 2003), and STAT3 activation has been reported in human glial tumors (Weissenberger et al., 2004). On the other hand, other studies have provided evidence of the absence of STAT3 activation in human gliomas (Wang et al., 2004). We recently used a genetic approach in mouse astrocytes to directly assess STAT3 function in glial cell transformation. We found that the predominant role of STAT3 is to suppress malignant glial transformation including in the context of PTEN deficiency (de la Iglesia et al., 2008). In agreement with these mouse genetic studies, our present findings reveal that STAT3 suppresses the malignant properties of PTEN-deficient glioblastoma cells including their proliferation and invasiveness. Interestingly, expression of the glioblastoma-associated protein EGFRvIII in mouse astrocytes triggers an oncogenic switch in STAT3 function (de la Iglesia et al., 2008). In human glioblastomas, EGFRvIII expression correlates with STAT3 activation in both immunoblotting and immunohistochemical analyses (Mizoguchi et al., 2006; de la Iglesia et al., 2008). Therefore, it will be interesting to determine in future studies if and how STAT3 might promote the malignant behavior of EGFRvIII-expressing human glioblastoma cells.
Inhibition of the gliogenesis-promoting STAT3 signaling pathway in glioblastoma cells may serve to couple loss of a general tumor suppressor such as PTEN to the glioblastoma cell-specific behavior of invasiveness and adhesion and spreading on myelin. The PTEN loss-triggered suppression of STAT3 signaling in glioblastoma cells illustrates how interactions between ubiquitously-acting tumor suppressors and developmental signaling pathways implicated in cell differentiation and fate specification may produce tumor-specific pathological behaviors.
The identification of IL8 as a novel STAT3-repressed target gene in glioma cells, whose repression mediates STAT3-inhibition of glioma cell proliferation, invasiveness, and spreading on myelin defines a novel cytokine-chemokine connection (Fig. 5E). Although STAT3 is commonly known to induce transcription (Darnell, 1997; Levy, 2003), our findings suggest that STAT3 may also act as a transcriptional repressor in glial cells. STAT3 appears to repress the IL8 promoter via inhibition of the NFkB reponse element. Interestingly, STAT3 inhibits expression of the inducible nitric oxide synthase (iNOS) gene in endothelial cells by directly inhibiting NFkB (Yu et al., 2002; Yu and Kone, 2004). The DNA binding domain of STAT3 directly interacts with NFkB and thereby inhibits NFkB-dependent iNOS transcription (Yu and Kone, 2004). These results are consistent with the finding that the DNA binding domain of STAT3 is required for STAT3’s ability to repress IL8 expression in glioblastoma cells (Fig. 4D). The candidate glial tumor suppressor ING4 reportedly inhibits glioblastoma-induced angiogenesis via repression of IL8, whereby ING4 inhibits NFkB-dependent IL8 transcription (Garkavtsev et al., 2004). Collectively, these observations raise the possibility that STAT3 and ING4 may cooperate at the IL8 promoter to inhibit NFkB and thereby repress IL8 gene expression and thus control glioblastoma cell proliferation, invasiveness, and glioblastoma-induced angiogenesis.
The elucidation of STAT3 and IL8 in this study as critical regulators of glioblastoma cell proliferation and invasiveness provides new targets for therapeutic intervention. The chemokine IL8 and its cell surface receptors, CXCR1 and CXCR2, are particularly attractive as potential targets in the treatment of PTEN-deficient glioblastoma. These chemokine receptors have been studied in the context of acute inflammation (Rollins, 1997; Schraufstatter et al., 2001), and small molecule inhibitors of CXCR1 and CXCR2 have been characterized (White et al., 1998; Bertini et al., 2004). It will be important to determine the potential of CXCR1 and CXCR2 antagonists to inhibit the malignant features of PTEN-deficient glioblastoma cells. These studies may provide the foundation for patient-tailored therapy in the management of brain tumors.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Judith Stegmüller for providing adult rat brain myelin, Jeng-Shin Lee, Andrew Keates and Joan Brugge for providing plasmids. Supported by awards from the Stewart Trust of Washington, D.C. (A.B.), the Armenise-Harvard Foundation (A.B.), the Carolyn and Peter Lynch Research Fund (A.B.), NIH grants to D.N.L. and NIH grants to A.B. (NS051255, NS41021, and NS047188), and by a postdoctoral fellowship from the Fundación Ramón Areces, Spain, and a Taplin Postdoctoral fellowship (to N.d.I.). A.B. is the recipient of a fellowship from the Alfred P. Sloan Foundation, a Robert H. Ebert Clinical Scholar Award from the Esther A. and Joseph Klingenstein Fund, an EJLB Foundation award, and a Sidney Kimmel Foundation Award.
References
- Amberger VR, Hensel T, Ogata N, Schwab ME. Spreading and migration of human glioma and rat C6 cells on central nervous system myelin in vitro is correlated with tumor malignancy and involves a metalloproteolytic activity. Cancer Res. 1998;58:149–158. [PubMed] [Google Scholar]
- Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, Tang Y, DeFrances J, Stover E, Weissleder R, Rowitch DH, Louis DN, DePinho RA. Epidermal growth factor receptor and Ink4a/Arf. Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1:269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines--CXC and CC chemokines. Adv Immunol. 1994;55:97–179. [PubMed] [Google Scholar]
- Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, Gutmann DH. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, Cervellera MN, Di Cioccio V, Cesta MC, Galliera E, Martinez FO, Di Bitondo R, Troiani G, Sabbatini V, D'Anniballe G, Anacardio R, Cutrin JC, Cavalieri B, Mainiero F, Strippoli R, Villa P, Di Girolamo M, Martin F, Gentile M, Santoni A, Corda D, Poli G, Mantovani A, Ghezzi P, Colotta F. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci U S A. 2004;101:11791–11796. doi: 10.1073/pnas.0402090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A. STATs in the central nervous system. In: Signal Transducers and Activators of Transcription (STATs) In: Sehgal PB, Levy DE, Hirano T, editors. Activation and Biology. Kluwer Academic Publishers; 2003. pp. 663–685. [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–2488. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol. 2005;7:122–133. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- de la Iglesia N, Konopka G, Puram SV, Chan JA, Bachoo RM, You MJ, Levy DE, Depinho RA, Bonni A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–462. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, Barnett GH, Jain RK. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004;428:328–332. doi: 10.1038/nature02329. [DOI] [PubMed] [Google Scholar]
- Giese A, Westphal M. Glioma invasion in the central nervous system. Neurosurgery. 1996;39:235–250. doi: 10.1097/00006123-199608000-00001. [DOI] [PubMed] [Google Scholar]
- Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, Otterson MF, Ota DM, Lugering N, Domschke W, Binion DG. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–8515. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- Holland EC. Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet. 2001;2:120–129. doi: 10.1038/35052535. [DOI] [PubMed] [Google Scholar]
- Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka G, Bonni A. Signaling pathways regulating gliomagenesis. Curr Mol Med. 2003;3:73–84. doi: 10.2174/1566524033361609. [DOI] [PubMed] [Google Scholar]
- Levy DE. STAT transcriptional activation mechanisms. In: Signal Transducers and Activators of Transcription (STATs) In: Sehgal PB, Levy DE, Hirano T, editors. Activation and Biology. Kluwer Academic Publishers; 2003. pp. 327–341. [Google Scholar]
- Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WKe. Lyon: IARC Press; World Health Organization Histological Classification of Tumours of the Central Nervous System. 2007 International Agency for Research on Cancer (IARC).
- Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006;65:1181–1188. doi: 10.1097/01.jnen.0000248549.14962.b2. [DOI] [PubMed] [Google Scholar]
- Mukaida N, Mahe Y, Matsushima K. Cooperative interaction of nuclear factor-kappa B-and cis-regulatory enhancer binding protein-like factor binding elements in activating the interleukin-8 gene by pro-inflammatory cytokines. J Biol Chem. 1990;265:21128–21133. [PubMed] [Google Scholar]
- Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, Kitaoka T, Fukada T, Hibi M, Hirano T. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. Embo J. 1996;15:3651–3658. [PMC free article] [PubMed] [Google Scholar]
- Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–8413. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- Rajan P, McKay RD. Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 1998;18:3620–3629. doi: 10.1523/JNEUROSCI.18-10-03620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Park ES, Emdad L, Lee SG, Su ZZ, Fisher PB. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008;68:1478–1484. doi: 10.1158/0008-5472.CAN-07-6164. [DOI] [PubMed] [Google Scholar]
- Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1094–L1103. doi: 10.1152/ajplung.2001.280.6.L1094. [DOI] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Thorsen F, Tysnes BB. Brain tumor cell invasion, anatomical and biological considerations. Anticancer Research. 1997;17:4121–4126. [PubMed] [Google Scholar]
- Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002;62:5551–5558. [PubMed] [Google Scholar]
- Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004 doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- Weissenberger J, Loeffler S, Kappeler A, Kopf M, Lukes A, Afanasieva TA, Aguzzi A, Weis J. IL-6 is required for glioma development in a mouse model. Oncogene. 2004;23:3308–3316. doi: 10.1038/sj.onc.1207455. [DOI] [PubMed] [Google Scholar]
- White JR, Lee JM, Young PR, Hertzberg RP, Jurewicz AJ, Chaikin MA, Widdowson K, Foley JJ, Martin LD, Griswold DE, Sarau HM. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J Biol Chem. 1998;273:10095–10098. doi: 10.1074/jbc.273.17.10095. [DOI] [PubMed] [Google Scholar]
- Yamanaka R, Tanaka R, Saitoh T, Okoshi S. Cytokine gene expression on glioma cell lines and specimens. J Neurooncol. 1994;21:243–247. doi: 10.1007/BF01063773. [DOI] [PubMed] [Google Scholar]
- Yu Z, Kone BC. The STAT3 DNA-binding domain mediates interaction with NF-kappaB p65 and inducible nitric oxide synthase transrepression in mesangial cells. J Am Soc Nephrol. 2004;15:585–591. doi: 10.1097/01.asn.0000114556.19556.f9. [DOI] [PubMed] [Google Scholar]
- Yu Z, Zhang W, Kone BC. Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J. 2002;367:97–105. doi: 10.1042/BJ20020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.