

Abstract
For more than 40 years following its approval by the Food and Drug Administration (FDA) as an anesthetic, ketamine, a non-competitive N-methyl-D-aspartic acid (NMDA) receptor antagonist, has been used as a tool of psychiatric research. As a psychedelic drug, ketamine induces psychotic symptoms, cognitive impairment, and mood elevation, which resemble some symptoms of schizophrenia. Recreational use of ketamine has been increasing in recent years. However, little is known of the underlying molecular mechanisms responsible for ketamine-associated psychosis. Recent animal studies have shown that repeated ketamine administration significantly increases NMDA receptor subunit gene expression, in particular subunit 1 (NR1 or GluN1) levels. This results in neurodegeneration, supporting a potential mechanism where up-regulation of NMDA receptors could produce cognitive deficits in chronic ketamine abuse patients. In other studies, NMDA receptor gene variants are associated with addictive behavior. Here, we focus on the roles of NMDA receptor gene subunits in ketamine abuse and ketamine psychosis and propose that full sequencing of NMDA receptor genes may help explain individual vulnerability to ketamine abuse and ketamine-associated psychosis.
Keywords: N-Methyl-D-aspartic acid (NMDA) receptor, gene expression, ketamine administration, ketamine-associated psychosis
Introduction
Ketamine was first synthesized by Parke-Davis in 1963, and was soon introduced to medicine as an important anesthetic and analgesic medication. Ketamine is derived from phencyclidine (piperidine, PCP) but has weaker affinity for the NMDA receptor (NMDAR) and a shorter half-life. Because of its safety profile, ketamine remains in medical use today. It has been further applied in psychiatry research for understanding the pathophysiology of schizophrenia and depression.
The psychological effect of ketamine has been linked to sensory deprivation, mood elevation, and cognitive impairments.1,2 Because of the liability of abuse, ketamine has become a street drug in many countries in the world, particular in Southeast Asia. On the other hand, in experimental studies conducted in humans, ketamine induces hallucinations, delusions, and negative symptoms that resemble the symptomatology of schizophrenia. Ketamine administration has been used as a pharmacological model of schizophrenia in animal models and humans.1,3
The glutaminergic pathway has been implied to explain the pathophysiology of schizophrenia and addiction and. However, the roles of glutaminergic genes, in particular the NMDAR subunit genes, in ketamine abuse and ketamine psychosis are unknown and research into understanding ketamine abuse and associated psychosis is in its infancy. In this review, we (1) outline the problem of ketamine abuse and ketamine-associated psychosis, (2) relate gene expression profiles in response to repeated ketamine administration in rodents with a specific focus on transcriptional control of NMDAR gene expression, and (3) briefly, review genetic studies of NMDAR subunit genes with schizophrenia and addiction to better understand individual vulnerability for ketamine abuse and ketamine psychosis.
Ketamine abuse and related symptoms
While ketamine anesthesia was first given to American soldiers during the Vietnam War, it was later recognized as a problematic drug of abuse in North America.4 Although it is difficult to estimate exact prevalence of ketamine use worldwide, the incidence of ketamine seizures significantly increased from 2003 to 2006 according to World Drug Report 2010 by the United Nations Office on Drugs and Crime. Ketamine abuse has become the second most common drug abuse in an area of central China5 and is the most popular recreational drug in Hong Kong and Taiwan in youths.6,7 Outside of Asia, from 1999 to 2003 the prevalence of ketamine use among “club drug” users in the United Kingdom increased from 25% to 40%.8
Consistent with the profile of other substance abusers, most ketamine users are men and tend to be young. A survey of over 9000 adolescences in Taiwan showed an average age among ketamine users was 15 years.6 This trend is particularly concerning because young users are also more likely to inject ketamine intravenously and thus have a higher incidence of comorbid hepatitis C infection.9,10 Another feature of ketamine abuse is that more than 50% of ketamine users have less than 10 years of formal education. Together, these data suggest that adolescents are a vulnerable population. However, preventing ketamine abuse in this population is challenging.9
Dependence
Behaviorally, animal and human studies showed that acute ketamine administration produces ethanol-like effects. In animal studies, ketamine can be self-administered in monkeys or rats in a dose- and setting-dependent manner.11,12 In addition, ketamine injection in either healthy human subjects or recently detoxified alcohol-dependent individuals, caused subjective ethanol “high” effects, particularly in the subjects without a family history of alcohol dependence.13–16 Thus, ketamine demonstrates behavioral hallmarks for producing dependence.
It is likely that the dopamine reward pathway may play an important role in ketamine dependence. This idea is supported by biochemical findings showing that ketamine also has a high affinity for the dopamine D2 receptor and that a single sub-anesthetic dose of ketamine increased dopamine release in rat prefrontal cortex.17 Acute and chronic ketamine administration significantly increases dopamine release, with chronic ketamine injection increasing dopamine receptor 1 and 2 gene expression.18 Daily ketamine administration of 30 mg/kg ketamine for three months produced a 2.8-fold increase in dopamine level in mouse midbrain relative to chronic saline-treated mice.17 In addition, there was a significant 1.8-fold increase in tyrosine hydroxylase (TH) mRNA levels and increased TH immunoreactivity in midbrain of mice chronically treated with ketamine for three months.17 This finding was confirmed by another study that chronically exposed mice to ketamine for 10 days.18 Since TH is the rate-limiting enzyme in the synthesis of catecholamines such as dopamine, changes in its synthesis will likely affect overall flux through the dopamine biosynthetic pathway. In the same study, Tan et al. discussed that protein levels of a neuronal survival and differentiation factor, the neurotrophin brain-derived neurotrophic factor (BDNF), were significantly increased in cortical-subcortical regions following chronic ketamine administration.17 This observation suggested that a major signaling mechanism, mediated by the BDNF-TrkB signal transduction pathway, may play a role in long term up-regulation of TH.19 Taken together, these findings define the basis for changes in reward pathways and may lead to therapies for chronic ketamine abusers.
Withdrawal symptoms
Few studies of withdrawal symptoms in animal models of ketamine addiction have been conducted. The most recent study assessed physiological and behavioral withdrawal symptoms following repeated oral ketamine administration in Cynomolgus macaques.20 A study in mice reported that chronic ketamine administration significantly enhanced immobility during force swimming test, a measure of learned helplessness thought to mimic some clinical features of depression in humans.21 The immobility of mice persisted for 10 days after withdrawing ketamine. Pre-treating the mice with atypical antipsychotic medications, clozapine and risperidone, reduced the chronic ketamine-induced immobility.21 The effect was not observed in the acute ketamine injection paradigm. The persistent behavioral alteration after ketamine withdrawal indicates that long-term ketamine administration may also produce homeostatic changes to other aspects affecting the neuro-biology of behavior.
Little is known about ketamine withdrawal symptoms in humans. This is probably because there is not a well-established and objective withdrawal scale to assess withdrawal symptoms. Clinical observation suggests that craving is the most common of withdrawal symptoms although anxiety is also commonly encountered after discontinuing ketamine use. Since ketamine produces ethanol-like effects and both ketamine and ethanol are NMDAR antagonists, compensatory changes underlying withdrawal symptoms of ketamine abuse may be similar to alcohol withdrawal mechanisms. Among these compensatory mechanisms are increases in NMDARs and a concomitant decrease in GABAA receptors. In the case of alcohol-dependent individuals, abstinence from alcohol leads to a rebound effect, producing increased excitability.
Ketamine induced transient and persistent psychosis
It is well known that transient psychosis can be induced by ketamine injection under laboratory controlled conditions and has been applied as a pharmacological model to test the hypoglutamatergic function hypothesis for schizophrenia.1,22,23 Compared to other psychotomimetic drugs, a low dose of ketamine infusion in healthy subjects induces psychotic symptoms more effectively and produces not only positive but also negative symptoms and cognitive impairment.1 However, psychotic symptoms during laboratory administration are transient and do not persistent. Although frequent ketamine users exhibit delusional thoughts that resemble prodromal symptoms, no persistent psychosis has been seen in low dose, short-term ketamine administration among more than 2000 healthy subjects.24
While an extensive study of ketamine-induced persistent psychosis among chronic ketamine users has not been reported, it is known that chronic ketamine use increases the severity of delusional ideation among individuals that were followed for up to one year and that delusions persisted even when ketamine was discontinued.25 The association between NMDAR antagonism and persistent psychosis is supported by the fact that another NMDAR antagonist, PCP, induces persistent psychosis.4,26 Like many recreational drugs, that are used consecutively or in combination, it seems that the most persistent psychotic symptoms among ketamine abusers are observed in the context of co-substance abuse. In a medical record review of 350 ketamine co-abusers of ecstasy (3, 4-methylenedioxy-N-methylamphetamine, MDMA), psychosis was the most common reason for individuals to seek inpatient treatment.27 The comorbidity of psychosis and ketamine/ MDMA use is as high as 77.1% among the ketamine users who were hospitalized in a psychiatric hospital.28 Thus, while ketamine-induced persistent psychosis as a model of schizophrenia is particularly interesting, it is not clear how ketamine-induced psychosis is manifested. Among the possibilities to be considered are: (1) chronic, high dose ketamine increases glutaminergic function, causing neurotoxicity, (2) chronic ketamine use manifests genetic vulnerability on those individuals carrying risk variants for psychosis, (3) co-abuse of ketamine with other drugs, such as psychotomimetic MDMA, changes dopaminergic reward pathway function.
Hence, individual genetic variation is expected to impact the above-mentioned pathways and others in developing psychotic symptoms. Variation in response to ketamine may relate to genetic variation impacting transcriptional control of NMDARs, pre-mRNA processing, and/or stability to NMDAR subunit specific mRNAs.
Transcriptional control of NMDAR subunit, gene expression and its potential role in ketamine abuse and ketamine psychosis
An understanding of how NMDARs are affected by ketamine, in particular on how expression of the receptors and their downstream signaling pathways are affected, may lead to cellular changes that underlie the symptoms seen among ketamine abusers. In addition, an understanding of the regulation of NMDAR subunits is important to identify genetic risk factors for ketamine abuse and ketamine-induced psychosis. This section focuses on NMDAR function, the transcriptional regulation of individual subunits, and the effects of NMDAR antagonists on expression of NMDAR subunits and on cell survival.
NMDA receptors
The NMDAR is a major mediator of excitatory neurotransmission. NMDARs are heteromeric complexes comprised of subunits that include GluN1 (encoded by the NR1 gene, also abbreviated as GRIN1), GluN2 (NR2, GRIN2 gene name) and GluN3 (NR3, GRIN3A, 3B gene names). Activation of NMDAR requires both glutamate and glycine binding. Recent evidence also suggests that D-serine is the coagonist for NMDA receptors.29 The binding sites for glutamate and glycine are found on different subunits – glycine binds to the GRIN1 subunit while glutamate binds to the GRIN2 subunit. Each binding site is located in the ligand binding domain of the extracellular portion of their respective receptor subunit.30 The carboxyl-terminal domain of NMDAR subunits contains multiple serine/threonine phosphorylation sites that act as sites of protein–protein interaction for intracellular substrates for cAMP-dependent protein kinase A (PKA), protein kinase C (PKC), protein kinase B (PKB), CaMKII, cyclin-dependent kinase-5 (Cdk5) and casein kinase II (CKII).31 For example, activation of PKA and PKC increases NMDAR-mediated currents and Ca2+ permeability,32,33 while phosphorylation of the carboxyl-terminal domain by Src family protein tyrosine kinases increases NMDAR function.34 Adding another layer of complexity to NMDAR function is the fact that alternative splicing of GRIN1 mRNA leads to a receptor with altered binding sites for intracellular proteins.
Intracellular signaling is also dictated by cellular location of NMDARs, as they are found in synaptic and extrasynaptic locations, where they assume different cellular functions. Extrasynaptic NMDAR activation leads to a “CREB-shut off” pathway, a repressive event that leads to inhibition of cyclic AMP binding protein (CREB) and nuclear import of class II histone deacetylases (HDACs) to reduced gene transcription.33,35
Transcriptional regulation of NMDAR subunit genes: GRIN1, GRIN2A, and GRIN2B
The level of expressed NMDARs is reflected by transcription, translation, mRNA stability, protein stability, and receptor assembly and presentation at the neuronal cell surface. The combination of subunits expressed by a particular neuron determines its synaptic phenotype. Promoter region sequence motifs (cis-acting elements) that control transcription factor binding and participate in chromatin remodeling have been identified over the past 20 years. These elements affect expression in neurons, either inducing or repressing transcription.
GRIN1
A number of consensus and non-consensus transcription factor binding sites have been identified in the NMDAR subunit genes, mostly in the proximal 5′ flanking region (the so called “core region”), relative to exon 1. Many are conserved across species, supporting their functional significance. The specific cis-acting elements that activate transcription include members of the AP family, CREB (cyclic AMP response element binding protein), NF-κB, and members of the Sp (specific protein) family.
An important regulatory region of the human GRIN1 gene is also located within the 5′flanking region, approximately 7 kb upstream of the transcription start site (TSS) for the gene. As shown in Figure 1a, this region is enriched in H3K27Ac marks, a histone modification that across the genome has been identified with transcriptional activation. This region also contains multiple conserved transcription factor binding sites, including CREB, AP-1, Sp1, and RE1 sites, and is enriched of in CpG islands. Recently, the ENOCDE project identified a long RNA binding site.36 Other CRE and RE1 sites are also located downstream of the TSS.
Figure 1.


Gene structure and annotation for GRIN1 (a), GRIN2A (b), and GRIN2B (c). Data are from the University of California Santa Cruz (UCSC) Genome Browser. Panel (a): the human GRIN1 gene has seven transcripts. At the 5′ end, there is a high methylation region with high density of H3K27Ac (a histone modification mark), and a CpG island. Interestingly, this critical region contains six conserved transcription factor binding sites including CREB that may involve the mechanism of neuroplasticity and addiction. A long RNA is identified across this region. Within this region, 13 common SNPs are identified. The SNP rs11146020 maps close to this region and was found associated with schizophrenia in some studies; Panel (b): GRIN2A has four transcripts. A possible regulatory region is located in intron 7. The region contains a high density of H3K27Ac marks, multiple conserved transcription factor binding sites, long RNA sequence found by ENCODE. A (TC)17 polymorphism is identified within this region; (c) GRIN2B has one mRNA transcript. The second intron has a high density of histone modification marks with conserved transcription factor binding sites, suggesting a potential regulation region. Multiple microsatellite polymorphisms are identified in GRIN2B. (A color version of this figure is available in the online journal.)
GRIN2A
Similar to GRIN1, much of our understanding of the regulation of the GRIN2A gene comes from understanding its function in rodents. The core promoter of GRIN2A lies within exon 1. In addition, several potential regulatory regions are identified within the gene (Figure 1b). For example, a region lying in intron 7 has a high density of H3K27Ac marks, and is enriched with conserved transcription factor binding sites. Like GRIN1, expression of the GRIN2A gene is activity dependent. A CRE site was identified in rat and mouse, supporting a role for this element in activity-dependent transcription. NF-κB and Sp family proteins are also positive regulators. In humans, a polymorphic GT dinucleotide repeat (GT)n suppresses transcription where increasing repeat length represses GRIN2A transcription in luciferase reporter gene assays.37,38 This observation was consistent with reduced MK801 binding in postmortem brain samples based on increased GT repeat length.
GRIN2B
The core promoter for the GRIN2B gene has been defined to include a 5′flanking region of approximately 600 bp based on neuron-specific activity observed in transgenic mice. Functional Sp1, CRE, AP-1, and NF-κB sites have also been shown. The AP-1 and NF-κB sites are located more 5′ of the core promoter. Like the GRIN2A promoter, activation-dependent transcription of GRIN2B is NF-κB-dependent.
Genetic variants within or nearby regulatory regions of these genes may determine differential gene expression and alter gene functions. Thus, regulatory region of the NMDA subunits genes are excellent candidate regions to seek functional variants.
NMDAR subunit gene expression following NMDAR antagonism by ketamine or MK-801 Repeated ketamine administration in animals
Unlike fast NMDA channel blocker, e.g. memantine, ketamine exhibits slow NMDA open-channel block effect through full trapping of the channel.39 The persistent NMDA block effect of ketamine might alter NMDA function and contribute to psychosis. Although no NMDAR gene expression in behaviorally addicted ketamine models has been reported, studies have demonstrated that repeated ketamine administration alters NMDAR subunit gene expression in adult and developing rats. Repeated ketamine administration with high dose in 6 h dramatically increased NMDAR subunit mRNA expression in the frontal cortex (specifically GRIN1, GRIN2A, and GRIN2C, but not GRIN2B or GRIN2D subunit mRNA levels) (Table 1).40–42 The increase in GRIN1 mRNA levels is associated with increased neuronal cell death in the frontal cortex in the developing neonatal rat brain.40 The study suggested that increased NR1 expression and neurotoxicity is dependent upon dose and ketamine exposure. A ketamine dose less than 10 mg/kg or less than six injections did not alter GRIN1 expression and neuronal death.40,41 Interestingly, co-incubation of ketamine and GRIN1 antisense significantly reduces neurotoxicity in frontal cortical cells derived from neonatal monkey, suggesting ketamine-induced neurotoxicity is caused by GRIN1 up-regulation.43,44 It has been hypothesized that repeated ketamine treatments cause compensatory up-regulation of NMDAR expression that allows accumulation of toxic levels of intracellular Ca2+, which leads to neurodegeneration.41,45 Because the up-regulation of GRIN1 expression is predominant in the frontal cortex,46 it implies an underlying mechanism of ketamine-induced persistent psychosis in human.
Table 1.
NMDA receptor and dopamine-related gene expression changes resulting from repeated ketamine and chronic ketamine
| Gene | Model | Repeated | Chronic | References |
|---|---|---|---|---|
| grin1 | PND7 Rat-ketamine | + | nd | 42–44 |
| grin2a | PND7 rat-ketamine | + | nd | 42 |
| grin2b | PND 7 rat-ketamine | − | nd | 42 |
| grin2c | PND7 rat-ketamine | + | nd | 42 |
| TH | Adult mice-Ketamine | nd | + | 18 |
| DRD1 | Adult mice-ketamine | nd | + | 18 |
| DRD2 | Adult mice-ketamine | nd | + | 18 |
| DAT | Adult mice-ketamine | nd | + | 18 |
+: increased specific gene mRNA level; –: decreased specific gene mRNA level; nd: not determined; PND7: seven-day-old Sprague Dawley rat; Grin 1: glutamate receptor, ionotropic, NMDA1; Grin2a: glutamate receptor, ionotropic, NMDA2a; Grin2b: glutamate receptor, ionotropic, NMDA2b; Grin2c: glutamate receptor, ionotropic, NMDA2c; TH: TH tyrosine hydroxylase; DRD1: dopamine receptor 1; DRD2: dopamine receptor 2; DAT: dopamine transporter.
Ketamine-induced neurotoxicity in rats appears to be age-dependent. It was previously believed that ketamine-induced neurotoxicity only occurred in adult brain. A study showed MK-801 caused cell death only in the rats older than four months.47 Rats younger than one month old were not sensitive to MK-801.47 However, more recent studies have shown that the developing brain is also vulnerable to NMDAR antagonism. Injection of 20 mg/kg ketamine for six successive doses in postnatal day 7 rats significantly increased NMDAR mRNA expression and increased expression of genes involved in apoptosis, suggesting that developing neurons are sensitive to ketamine-induced NMDAR alteration and neurotoxicity.40–42,48 This evidence suggests that ketamine abuse in adolescence may put individuals at greater risk of developing psychosis through an up-regulation of NMDAR expression. Future studies to understand the relationship between early age onset of ketamine use and psychosis is warranted.
Long-term neurotoxicity of MK-801 in rats
While long-term effects of repeated ketamine administration on NMDAR expression have not been described in rodents, long-term effects with another non-competitive NMDAR antagonist, MK-801 have been reported in neonatal rats. Harris et al. reported significant neuronal cell reduction in the CA1 subfield of the hippocampus lasted for 14 days after injection of NMDAR blockade MK-801.49 Grin1 (grin1, rodent gene designation) mRNA levels in the hippocampus were also altered in adults following MK-801 treatment, producing a differential response. Dorsally, grin1 mRNA levels were decreased but increased ventrally.49 This difference in expression may reflect a difference in connectivity that underlies behavioral alterations in these rats seen through development and may be relevant to the developmental etiology of schizophrenia.
NMDAR subunit genetic variants and schizophrenia/substance dependence
As discussed above, the complex regulation of NMDAR gene family underscores the essential functions of these receptors in complex neuroplasticity and neurotoxicity. Genetic variation at NMDAR subunit genes may contribute to individual vulnerability to ketamine abuse and ketamine psychosis. Although genetic studies of NMDAR with ketamine abuse and ketamine psychosis have yet to be reported, NMDAR genes are excellent candidates for schizophrenia and addiction of other substances. Here, we will briefly summarize genetic association studies of GRIN1, GRIN2A, and GRIN2B genes with schizophrenia and addiction.
Association of GRIN1 with schizophrenia and addiction
Among the 747 known polymorphisms of the GRIN1 gene, the SNP rs11146020 has attracted the most attention for association studies with psychiatric disorders. This SNP is located within the 5′ flanking region of the gene that is close to the critical regulatory region of GRIN1 as discussed above (Figure 1). The C >G substitution changes a consensus binding site for the p50 subunit of the nuclear factor kappa B (NF-κB) transcription factor, presumably altering GRIN1 gene expression. Although the functionality of rs11146020 has not been examined, several studies have reported associations with schizophrenia with contradictory results. The minor C allele was found to increase risk for schizophrenia in Italian and Iranian populations,50,51 but was found to be protective for schizophrenia in a Chinese sample population.52 Two other SNPs of GRIN1 with reported associations with schizophrenia have also produced inconsistent findings.
Although no positive association of GRIN1 with substance dependence was reported, a recent study using array-based DNA methylation screen found a significant CpG island hypermethylation in alcohol-dependent subjects compared to control subjects, suggesting epigenetic mechanisms of GRIN1 may be involved in alcohol dependence.53 Interestingly, the hypermethylation of GRIN1 appears population specific seen in European Americans but not in African Americans.53 It is noteworthy that the 5′ critical region contains 13 SNPs, which may be associated with methylation of GRIN1 (Figure 1a). Future studies to examine the relationship of these 13 SNPs with methylation level and ketamine abuse may explain more variation of individual vulnerability to ketamine abuse and ketamine psychosis.
Association studies of GRIN2A with schizophrenia and addiction
In humans, a functional polymorphic GT dinucleotide repeat (GT)n was found to suppress GRIN2A transcription where increasing repeat length represses GRIN2A transcription. A case–control study of 375 schizophrenic patients and 378 control subjects showed weak association between increased repeat length and clinical severity of schizophrenia.37,54 Subsequent studies have shown a correlation between reduced hippocampal and amygdala volume with increasing repeat length.38 However, the number of repeat length differs in different studies.55 A family study in a Chinese sample revealed no association of D16S407 with schizophrenia.
The GRIN2A gene has also been associated with addictive behavior. Tag SNPs on the GRIN2A showed significant association with alcohol dependence. Furthermore, GRIN2A was associated with the severity of alcohol dependence, early age of alcohol drinking, and positive family history in independent sample populations.56 Interestingly, SNP rs9924016 that was found associated with alcohol dependence is located at the intron 7, a region compassing multiple regulatory transcription binding sites (Figure 1b). Recently, the functional promoter polymorphism D16S407 was examined for association with alcohol dependence in a small Caucasian population.57 The short repeat allele (GT)n <22 presented significantly higher frequency in control subjects compared to alcohol-dependent subjects. The result was replicated in a small independent sample. In a study of 130 candidate genes, four intronic SNPs (rs1650420, rs4587976, rs6497730, rs1070487) in GRIN2A were strongly associated with heroin addiction in an African American population.58 Furthermore, a haplotype in GRIN2A (rs4587976, rs1071502, rs1366076) was found with significantly higher frequency in heroin-dependent group.58 Taken together, this evidence suggests a role for GRIN2A in individual vulnerability to addiction and highlights the importance of regulatory regions of GRIN2A gene.
Association studies of GRIN2B with schizophrenia and addiction
A putative functional promoter SNP rs10193895 (200 T >G) was significantly associated with schizophrenia in Japanese and Caucasian populations,59,60 but was not predictive of clozapine treatment response.61 The rarer G allele was at higher frequency in patients with schizophrenia. In addition, a haplotype containing this SNP was present at significantly higher frequency in schizophrenia patients than controls.59 Of functional relevance, the SNP rs10193895 alters dinucleotide repeats in the 5′flanking region and predicts an altered Sp1 binding site (Figure 1c). In vitro, the G allele is associated with lower GRIN2B activity.60 Additionally, a silent coding SNP in GRIN2B rs1806201 (2664 C >T) was associated with schizophrenia and treatment response in Japanese and Chinese populations.62,63 Homozygous CC patients responded to higher doses of clozapine. However, the same SNP was not associated with tardive dyskinesia.64
The GRIN2B gene has been investigated for genetic associations with addiction. SNP rs1806201 was associated with alcohol dependence and early age onset of alcohol dependence.65 However, these findings were not replicated in an independent sample with alcohol dependence and early age onset.66 No association of rs1806201 with history of alcohol withdrawal induced seizure was reported. Another silent SNP on the same exonic region rs1806191 had no association with alcohol dependence.66 Among 60 3,4-methylenedioxymethamphetamine (MDMA, ecstasy) users, cognitive performance between genotypes for rs1806201 did not show significant group differences.67
Limitations of association studies with NMDAR
Current association studies of NMDAR genes with schizophrenia and addiction have focused on common variants with rare allele frequencies >5%. As discussed above, common variants of different NMDAR genes revealed inconclusive associations with phenotypes. Positive findings of association with loci at NMDAR genes have only shown small effect sizes with addiction (alcohol dependence) and schizophrenia (OR <2). There is a growing body of evidence supporting the concept that common gene variants play a mild to moderate role in complex diseases. A large portion of genetic heritability is hidden because a functional variant with a small effect size requires a larger sample population to achieve statistical significance for detecting an association with a phenotype. Thus, the strategy of using common SNPs is particularly challenging in psychiatric genetics which are predicted to be based on the presence of multiple genes having small individual effects on phenotype. In addition, a large sample size may contribute to increasing the heterogeneity within a phenotype and magnify the effect of hidden population admixture and inconsistent phenotype measurements. The limitation of focusing on common variants is further demonstrated by the fact that several large genome wide association studies resulted in no or marginally significant associations with schizophrenia and substance dependence.
Future directions
Proposed model of ketamine abuse and psychosis
Although there is a lack of evidence to elucidate the neurobiological mechanism(s) underlying ketamine abuse and ketamine psychosis in humans, evidence from animal studies support a role for NMDARs in ketamine abuse and ketamine induced psychosis. NMDARs and DRD1 interaction play an important role in the mechanism of ketamine abuse. As described in Figure 2, NMDA receptors interact with DRD1 through a number of redundant and cooperative signaling transduction cascade. The most prominent involve protein kinase (PKA) and dopamine-and adenosine-3′,5′-monophosphate (cAMP)-regulated phosphoprotein of 32 kDa (DARPP-32), phosphorylation of NMDAR NR1 subunits, and activation of Ca2+ channels.68,69 Activation of NMDAR-dependent signaling pathways increase BDNF expression via activated CREB and/or NF-κB, enhancing neuroplasticity. We hypothesize that initial ketamine use enhances DRD1 function in order to augment neuroplastic changes in neurons. Enhanced DRD1 function then increases dopamine release in the striatum and other brain regions involving rewarding and reinforcement. On the other hand, chronic ketamine exposure leads to compensatory up-regulation of the GRIN1 subunit which increases Ca2+ influx to neurons. The overload of Ca2+ in mitochondria produces excessive levels of superoxide and activates an NF-κB-dependent pathway that causes cell death (Figure 2). Neurotoxicity in frontal cortex induced by chronic ketamine use may explain cognitive impairment and negative symptoms in ketamine abuse patients. We speculate that frontal cortex neurotoxicity may also involve the development of persistent delusion and hallucinations in chronic and heavy ketamine abusers.
Figure 2.
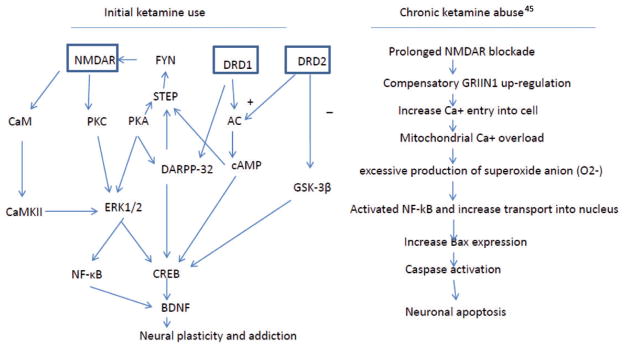
A proposed molecular mechanism describing initial ketamine use (left) and chronic abuse (right): initial ketamine use may involve altering two pathways: NMDA and dopamine receptors. Ketamine blocks NMDA receptor-dependent activation of NF-κB and CREB that promote synaptic plasticity via BDNF. However, ketamine also enhances dopamine function, via increased dopamine release and intracellular signaling, leading to activation of CREB. Increased CREB activation enhances BDNF transcription. In addition, ketamine enhances translation of BDNF mRNA, leading to increased neuroplasticity and addiction. Chronic ketamine exposure induces compensatory GRIN1 up-regulation that causes an influx of extracellular Ca2+ into the intracellular compartment. The overloaded Ca2+ infux produces superoxide anion that activates NF-κB, to promote Bax gene expression which is critical to active caspase, leading to apoptosis.45 (A color version of this figure is available in the online journal.)
Deep sequencing NMDAR gene family for extreme ketamine abuse and persistent psychosis patients
Additive effects from significant common variants explain only small proportion of variation for schizophrenia and addiction, suggesting that rare variants (frequency <1% in population) may underlie genetic vulnerability to complex phenotypes. Rare variants are only able to be detected by sequencing technologies. The cost of whole genome sequencing of large populations is still unaffordable. Sequencing candidate genes is an alternative strategy to detect rare variants that may have large effects. Deep target sequencing NMDAR gene family with focusing on regulatory regions in a larger sample population is a reasonable strategy to begin with the journey of uncovering genetic variants.
In the future, whole exome and whole genome sequencing will be routinely applied to complex diseases. Extreme individuals such as those with persistent psychosis and/or severe cognitive impairment induced by ketamine use may have heritable rare variants or de nova mutations. Sequencing those individuals may reveal candidate functional variants for molecular, cellular evaluation and for determining their impact in animal models of these disease phenotypes.
Other NMDAR-dependent gene effects mediated by ketamine
The focus of this article has been on uncovering the role of NMDAR gene variation with direct effects on NMDAR expression and determining their role in ketamine-mediated addiction and psychosis. NMDAR variation is also predicted to affect ketamine-induced effects on neurodevelopment. As examples of neurodevelopmental genes controlled via NMDA-mediated signaling, and represent targets of ketamine-induced changes in neurodevelopment, we draw on two examples, one a known schizophrenia risk gene (Disrupted in schizophrenia, DISC1) and the other, a NMDAR-regulated, CREB-responsive gene that encodes a micro RNA (miRNA).70 In the first case, DISC1, a gene conferring risk to schizophrenia and other neuropsychiatric disorders, was examined for changes in its expression when young adult mice were administered a single dose of the NMDAR antagonist memantine. Expression of DISC1 mRNA was significantly decreased in the dentate gyrus (DG). Decreased DISC1 mRNA was also seen with other NMDAR antagonists MK801 and CPP, although these inhibitors were not used in subsequent experiments. The effect of NMDAR antagonism was dose-dependent and ultimately revealed that NMDAR signaling governed migration of newly generated neurons in the DG by regulating DISC1 expression in that region of the hippocampus.71 These findings linked the role of excitatory neurotransmission in neurogenesis, acting through DISC1 protein.
In the second example, Miller et al. showed that a miRNA, miR-132, was significantly decreased in postmortem prefrontal cortical tissue from schizophrenic subjects versus controls.72 Analysis of miR-312 target gene levels in schizophrenia gene-expression arrays identified 26 genes that had increased expression in the schizophrenia samples, which correlated a dysregulation of genes normally suppressed by the miRNA. Consistent with the glutamatergic hypofunction hypothesis, repeated low-dose administration of the NMDA antagonist MK801 to adult mice resulted in decreased levels of miR-132 in the pre-frontal cortex. In addition, expression of miR-132 was reduced in adult mice by NMDAR antagonism during a brief postnatal period. A number of genes, including DNMT3A, which encodes a DNA methyltransferase, had increased expression. This correlative finding may be important as two genes associated with schizophrenia, GAD1 and REELIN (RELN), are hypermethylated. While direct evidence supporting a link between increased expression of the DNMT3A gene and hypermethylation of target genes, the finding suggests that miR-132 down-regulation in the prefrontal cortex is a common molecular feature of schizophrenia and overlaps with developmental and adult dysregulation brought about by NMDAR antagonism. Therefore, it is likely that ketamine administration will produce increased expression of DNMT3A, and is predicted to down regulate GAD1 and RELN. Further investigation will be required to determine if this mechanism plays a role in ketamine-induced addition and psychosis. However, a genome-wide approach is likely to identify gene effects down-stream of the NMDAR that are mediated by ketamine antagonism. Coupled with guided deep sequencing approaches to discover rare genetic variants in candidate genes, a more complete mechanistic picture of ketamine-induced abuse and psychosis is likely to emerge in the near future.
In summary, we emphasize the role of NMDAR in ketamine abuse and associated psychosis based on current research on ketamine alteration of NMDAR expression and genetic associations of NMDAR genes with addiction and schizophrenia. We propose deep sequencing of NMDAR genes to identify individual risks of ketamine abuse and persistent psychosis. Like other psychiatric disorders, many genes and neuronal pathways are involved to describe such complex behavior. Future studies should expand to other NMDA-related genes and pathways.
Acknowledgments
This work is supported by the grant K12 DA000167 from National Institute on Drug Abuse, US, and APA/Merck Early Academic Career Award, US.
Footnotes
Author contribution: KX contributed to the literature review and the draft of the manuscript. RHL contributed to the literature review, manuscript preparation, and editing.
References
- 1.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 2.Jansen KLR. Ketamine: dreams and realities. In: Hanna Jon., editor. Multidisciplinary Association for Psychedelic Studies (MAPS) FL USA: Sarasota; 2004. [Google Scholar]
- 3.Moghaddam B, Krystal JH. Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull. 2012;38:942–9. doi: 10.1093/schbul/sbs075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen RC, Stillman RC. Phencyclidine: an overview. NIDA Res Monogr. 1978:1–17. [PubMed] [Google Scholar]
- 5.Deng Q, Tang Q, Schottenfeld RS, Hao W, Chawarski MC. Drug use in rural China: a preliminary investigation in Hunan Province. Addiction. 2012;107:610–13. doi: 10.1111/j.1360-0443.2011.03648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee KH, Yeh YC, Yang PC, Lin HC, Wang PW, Liu TL, Yen CF. Individual and peer factors associated with ketamine use among adolescents in Taiwan. Eur Child Adolesc Psychiatry. 2012;21:553–8. doi: 10.1007/s00787-012-0292-7. [DOI] [PubMed] [Google Scholar]
- 7.Li JH, Vicknasingam B, Cheung YW, Zhou W, Nurhidayat AW, Jarlais DC, Schottenfeld R. To use or not to use: an update on licit and illicit ketamine use. Subst Abuse Rehabil. 2011;2:11–20. doi: 10.2147/SAR.S15458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCambridge J, Winstock A, Hunt N, Mitcheson L. 5-Year trends in use of hallucinogens and other adjunct drugs among UK dance drug users. Eur Addict Res. 2007;13:57–64. doi: 10.1159/000095816. [DOI] [PubMed] [Google Scholar]
- 9.Lankenau SE, Bloom JJ, Shin C. Longitudinal trajectories of ketamine use among young injection drug users. Int J Drug Policy. 2010;21:306–14. doi: 10.1016/j.drugpo.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang WK, Liang HJ, Lau CG, Tang A, Ungvari GS. Relationship between cognitive impairment and depressive symptoms in current ketamine users. J Stud Alcohol Drugs. 2013;74:460–68. doi: 10.15288/jsad.2013.74.460. [DOI] [PubMed] [Google Scholar]
- 11.De Luca MT, Badiani A. Ketamine self-administration in the rat: evidence for a critical role of setting. Psychopharmacology. 2011;214:549–56. doi: 10.1007/s00213-010-2062-x. [DOI] [PubMed] [Google Scholar]
- 12.Carroll ME, Stotz DC. Oral d-amphetamine and ketamine self-administration by rhesus monkeys: effects of food deprivation. J Pharmacol Exp Ther. 1983;227:28–34. [PubMed] [Google Scholar]
- 13.Krystal JH, Petrakis IL, Webb E, Cooney NL, Karper LP, Namanworth S, Stetson P, Trevisan LA, Charney DS. Dose-related ethanol-like effects of the NMDA antagonist, ketamine, in recently detoxified alcoholics. Arch Gen Psychiatry. 1998;55:354–60. doi: 10.1001/archpsyc.55.4.354. [DOI] [PubMed] [Google Scholar]
- 14.Krystal JH, Petrakis IL, Limoncelli D, Webb E, Gueorgueva R, D’Souza DC, Boutros NN, Trevisan L, Charney DS. Altered NMDA glutamate receptor antagonist response in recovering ethanol-dependent patients. Neuropsychopharmacology. 2003;28:2020–28. doi: 10.1038/sj.npp.1300252. [DOI] [PubMed] [Google Scholar]
- 15.Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, Gelernter J, Krystal JH. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–82. doi: 10.1176/ajp.161.10.1776. [DOI] [PubMed] [Google Scholar]
- 16.Krystal JH, Petrakis IL, Krupitsky E, Schutz C, Trevisan L, D’Souza DC. NMDA receptor antagonism and the ethanol intoxication signal: from alcoholism risk to pharmacotherapy. Ann N Y Acad Sci. 2003;1003:176–84. doi: 10.1196/annals.1300.010. [DOI] [PubMed] [Google Scholar]
- 17.Tan S, Lam WP, Wai MS, Yu WH, Yew DT. Chronic ketamine administration modulates midbrain dopamine system in mice. PloS one. 2012;7:e43947. doi: 10.1371/journal.pone.0043947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatterjee M, Verma R, Ganguly S, Palit G. Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology. 2012;63:1161–71. doi: 10.1016/j.neuropharm.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 19.Fraga DB, Reus GZ, Abelaira HM, De Luca RD, Canever L, Pfaffenseller B, Colpo GD, Kapczinski F, Quevedo J, Zugno AI. Ketamine alters behavior and decreases BDNF levels in the rat brain as a function of time after drug administration. Braz J Med Biol Res. 2013;35:262–6. doi: 10.1590/1516-4446-2012-0858. [DOI] [PubMed] [Google Scholar]
- 20.Walgren JL, Carfagna MA, Koger D, Sgro M, Kallman MJ. Withdrawal assessment following subchronic oral ketamine administration in Cynomolgus macaques. Drug Dev Res. 2014;75:162–71. doi: 10.1002/ddr.21168. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee M, Ganguly S, Srivastava M, Palit G. Effect of ‘chronic’ versus ‘acute’ ketamine administration and its ‘withdrawal’ effect on behavioural alterations in mice: implications for experimental psychosis. Behav Brain Res. 2011;216:247–54. doi: 10.1016/j.bbr.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Newcomer JW, Farber NB, Jevtovic-Todorovic V, Selke G, Melson AK, Hershey T, Craft S, Olney JW. Ketamine-induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–18. doi: 10.1016/S0893-133X(98)00067-0. [DOI] [PubMed] [Google Scholar]
- 23.Lahti AC, Koffel B, LaPorte D, Tamminga CA. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 24.Perry EB, Jr, Cramer JA, Cho HS, Petrakis IL, Karper LP, Genovese A, O’Donnell E, Krystal JH, D’Souza DC Yale Ketamine Study G. Psychiatric safety of ketamine in psychopharmacology research. Psychopharmacology. 2007;192:253–60. doi: 10.1007/s00213-007-0706-2. [DOI] [PubMed] [Google Scholar]
- 25.Morgan CJ, Muetzelfeldt L, Curran HV. Consequences of chronic ketamine self-administration upon neurocognitive function and psychological wellbeing: a 1-year longitudinal study. Addiction. 2010;105:121–33. doi: 10.1111/j.1360-0443.2009.02761.x. [DOI] [PubMed] [Google Scholar]
- 26.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–8. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 27.Tang A, Liang HJ, Ungvari GS, Tang WK. Referral patterns and clinical characteristics of subjects referred to substance abuse clinic of a regional hospital in Hong Kong. East Asian Arch Psychiatry. 2011;21:22–7. [PubMed] [Google Scholar]
- 28.Zhang Y, Lu C, Zhang J, Hu L, Song H, Li J, Kang L. Gender differences in abusers of amphetamine-type stimulants and ketamine in southwestern China. Addict Behav. 2013;38:1424–30. doi: 10.1016/j.addbeh.2012.06.024. [DOI] [PubMed] [Google Scholar]
- 29.Papouin T, Ladepeche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet JP, Oliet SH. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150:633–46. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 30.Geranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–73. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–8. doi: 10.1016/j.neuropharm.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–3. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- 33.Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–21. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 34.Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–28. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 35.Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–14. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 36.Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itokawa M, Yamada K, Yoshitsugu K, Toyota T, Suga T, Ohba H, Watanabe A, Hattori E, Shimizu H, Kumakura T, Ebihara M, Meerabux JM, Toru M, Yoshikawa T. A microsatellite repeat in the promoter of the N-methyl-D-aspartate receptor 2A subunit (GRIN2A) gene suppresses transcriptional activity and correlates with chronic outcome in schizophrenia. Pharmacogenetics. 2003;13:271–8. doi: 10.1097/00008571-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Inoue H, Yamasue H, Tochigi M, Suga M, Iwayama Y, Abe O, Yamada H, Rogers MA, Aoki S, Kato T, Sasaki T, Yoshikawa T, Kasai K. Functional (GT)n polymorphisms in promoter region of N-methyl-d-aspartate receptor 2A subunit (GRIN2A) gene affect hippocampal and amygdala volumes. Genes Brain Behav. 2010;9:269–75. doi: 10.1111/j.1601-183X.2009.00557.x. [DOI] [PubMed] [Google Scholar]
- 39.Kotermanski SE, Wood JT, Johnson JW. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J Physiol. 2009;587:4589–604. doi: 10.1113/jphysiol.2009.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Q, Guo L, Patterson TA, Dial S, Li Q, Sadovova N, Zhang X, Hanig JP, Paule MG, Slikker W, Jr, Wang C. Gene expression profiling in the developing rat brain exposed to ketamine. Neuroscience. 2010;166:852–63. doi: 10.1016/j.neuroscience.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu F, Paule MG, Ali S, Wang C. Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr Neuropharmacol. 2011;9:256–61. doi: 10.2174/157015911795017155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zou X, Patterson TA, Sadovova N, Twaddle NC, Doerge DR, Zhang X, Fu X, Hanig JP, Paule MG, Slikker W, Wang C. Potential neurotoxicity of ketamine in the developing rat brain. Toxicol Sci. 2009;108:149–58. doi: 10.1093/toxsci/kfn270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C, Sadovova N, Fu X, Schmued L, Scallet A, Hanig J, Slikker W. The role of the N-methyl-D-aspartate receptor in ketamine-induced apoptosis in rat forebrain culture. Neuroscience. 2005;132:967–77. doi: 10.1016/j.neuroscience.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 44.Wang C, Sadovova N, Hotchkiss C, Fu X, Scallet AC, Patterson TA, Hanig J, Paule MG, Slikker W., Jr Blockade of N-methyl-D-aspartate receptors by ketamine produces loss of postnatal day 3 monkey frontal cortical neurons in culture. Toxicol Sci. 2006;91:192–201. doi: 10.1093/toxsci/kfj144. [DOI] [PubMed] [Google Scholar]
- 45.Slikker W, Jr, Paule MG, Wright LK, Patterson TA, Wang C. Systems biology approaches for toxicology. J Appl Toxicol. 2007;27:201–17. doi: 10.1002/jat.1207. [DOI] [PubMed] [Google Scholar]
- 46.Zou X, Patterson TA, Divine RL, Sadovova N, Zhang X, Hanig JP, Paule MG, Slikker W, Jr, Wang C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int J Dev Neurosci. 2009;27:727–31. doi: 10.1016/j.ijdevneu.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 47.Farber NB, Wozniak DF, Price MT, Labruyere J, Huss J, St Peter H, Olney JW. Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: potential relevance to schizophrenia? Biol Psychiatry. 1995;38:788–96. doi: 10.1016/0006-3223(95)00046-1. [DOI] [PubMed] [Google Scholar]
- 48.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 49.Harris LW, Sharp T, Gartlon J, Jones DN, Harrison PJ. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur J Neurosci. 2003;18:1706–10. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- 50.Galehdari H, Pooryasin A, Foroughmand A, Daneshmand S, Saadat M. Association between the G1001C polymorphism in the GRIN1 gene promoter and schizophrenia in the Iranian population. J Mol Neurosci. 2009;38:178–81. doi: 10.1007/s12031-008-9148-5. [DOI] [PubMed] [Google Scholar]
- 51.Begni S, Moraschi S, Bignotti S, Fumagalli F, Rillosi L, Perez J, Gennarelli M. Association between the G1001C polymorphism in the GRIN1 gene promoter region and schizophrenia. Biol Psychiatry. 2003;53:617–9. doi: 10.1016/s0006-3223(02)01783-3. [DOI] [PubMed] [Google Scholar]
- 52.Zhao X, Li H, Shi Y, Tang R, Chen W, Liu J, Feng G, Shi J, Yan L, Liu H, He L. Significant association between the genetic variations in the 5′ end of the N-methyl-D-aspartate receptor subunit gene GRIN1 and schizophrenia. Biol Psychiatry. 2006;59:747–53. doi: 10.1016/j.biopsych.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 53.Zhang H, Herman AI, Kranzler HR, Anton RF, Zhao H, Zheng W, Gelernter J. Array-based profiling of DNA methylation changes associated with alcohol dependence. Alcohol Clin Exp Res. 2013;37:E108–15. doi: 10.1111/j.1530-0277.2012.01928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwayama-Shigeno Y, Yamada K, Itokawa M, Toyota T, Meerabux JM, Minabe Y, Mori N, Inada T, Yoshikawa T. Extended analyses support the association of a functional (GT)n polymorphism in the GRIN2A promoter with Japanese schizophrenia. Neurosci Lett. 2005;378:102–5. doi: 10.1016/j.neulet.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 55.Tang J, Chen X, Xu X, Wu R, Zhao J, Hu Z, Xia K. Significant linkage and association between a functional (GT)n polymorphism in promoter of the N-methyl-D-aspartate receptor subunit gene (GRIN2A) and schizophrenia. Neurosci Lett. 2006;409:80–82. doi: 10.1016/j.neulet.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 56.Schumann G, Johann M, Frank J, Preuss U, Dahmen N, Laucht M, Rietschel M, Rujescu D, Lourdusamy A, Clarke TK, Krause K, Dyer A, Depner M, Wellek S, Treutlein J, Szegedi A, Giegling I, Cichon S, Blomeyer D, Heinz A, Heath S, Lathrop M, Wodarz N, Soyka M, Spanagel R, Mann K. Systematic analysis of glutamatergic neurotransmission genes in alcohol dependence and adolescent risky drinking behavior. Arch Gen Psychiatry. 2008;65:826–38. doi: 10.1001/archpsyc.65.7.826. [DOI] [PubMed] [Google Scholar]
- 57.Domart MC, Benyamina A, Lemoine A, Bourgain C, Blecha L, Debuire B, Reynaud M, Saffroy R. Association between a polymorphism in the promoter of a glutamate receptor subunit gene (GRIN2A) and alcoholism. Addict Biol. 2012;17:783–5. doi: 10.1111/j.1369-1600.2011.00321.x. [DOI] [PubMed] [Google Scholar]
- 58.Levran O, Londono D, O’Hara K, Randesi M, Rotrosen J, Casadonte P, Linzy S, Ott J, Adelson M, Kreek MJ. Heroin addiction in African Americans: a hypothesis-driven association study. Genes Brain Behav. 2009;8:531–40. doi: 10.1111/j.1601-183X.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyatake R, Furukawa A, Suwaki H. Identification of a novel variant of the human NR2B gene promoter region and its possible association with schizophrenia. Mol Psychiatry. 2002;7:1101–6. doi: 10.1038/sj.mp.4001152. [DOI] [PubMed] [Google Scholar]
- 60.Hwang R, Souza RP, Tiwari AK, Zai CC, Muller DJ, Potkin SG, Lieberman JA, Meltze HY, Kennedy JL. Gene-gene interaction analyses between NMDA receptor subunit and dopamine receptor gene variants and clozapine response. Pharmacogenomics. 2011;12:277–91. doi: 10.2217/pgs.10.182. [DOI] [PubMed] [Google Scholar]
- 61.Chiu HJ, Wang YC, Liou YJ, Lai IC, Chen JY. Association analysis of the genetic variants of the N-methyl D-aspartate receptor subunit 2b (NR2b) and treatment-refractory schizophrenia in the Chinese. Neuropsychobiology. 2003;47:178–81. doi: 10.1159/000071211. [DOI] [PubMed] [Google Scholar]
- 62.Hong CJ, Yu YW, Lin CH, Cheng CY, Tsai SJ. Association analysis for NMDA receptor subunit 2B (GRIN2B) genetic variants and psychopathology and clozapine response in schizophrenia. Psychiatr Genet. 2001;11:219–22. doi: 10.1097/00041444-200112000-00007. [DOI] [PubMed] [Google Scholar]
- 63.Nishiguchi N, Shirakawa O, Ono H, Hashimoto T, Maeda K. Novel polymorphism in the gene region encoding the carboxyl-terminal intracellular domain of the NMDA receptor 2B subunit: analysis of association with schizophrenia. Am J Psychiatry. 2000;157:1329–31. doi: 10.1176/appi.ajp.157.8.1329. [DOI] [PubMed] [Google Scholar]
- 64.Liou YJ, Wang YC, Chen JY, Bai YM, Lin CC, Liao DL, Chen TT, Chen ML, Mo GH, Lai IC. Association analysis of polymorphisms in the N-methyl-D-aspartate (NMDA) receptor subunit 2B (GRIN2B) gene and tardive dyskinesia in schizophrenia. Psychiatry Res. 2007;153:271–5. doi: 10.1016/j.psychres.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 65.Wernicke C, Samochowiec J, Schmidt LG, Winterer G, Smolka M, Kucharska-Mazur J, Horodnicki J, Gallinat J, Rommelspacher H. Polymorphisms in the N-methyl-D-aspartate receptor 1 and 2B subunits are associated with alcoholism-related traits. Biol Psychiatry. 2003;54:922–8. doi: 10.1016/s0006-3223(03)00072-6. [DOI] [PubMed] [Google Scholar]
- 66.Tadic A, Dahmen N, Szegedi A, Rujescu D, Giegling I, Koller G, Anghelescu I, Fehr C, Klawe C, Preuss UW, Sander T, Toliat MR, Singer P, Bondy B, Soyka M. Polymorphisms in the NMDA subunit 2B are not associated with alcohol dependence and alcohol withdrawal-induced seizures and delirium tremens. Eur Arch Psychiatry Clin Neurosci. 2005;255:129–35. doi: 10.1007/s00406-004-0545-7. [DOI] [PubMed] [Google Scholar]
- 67.Cuyas E, Verdejo-Garcia A, Fagundo AB, Khymenets O, Rodriguez J, Cuenca A, de Sola Llopis S, Langohr K, Pena-Casanova J, Torrens M, Martin-Santos R, Farre M, de la Torre R. The influence of genetic and environmental factors among MDMA users in cognitive performance. PloS one. 2011;6:e27206. doi: 10.1371/journal.pone.0027206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tseng KY, O’Donnell P. Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci. 2004;24:5131–39. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, Malhotra AK. Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet. 2004;75:862–72. doi: 10.1086/425586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Namba T, Ming GL, Song H, Waga C, Enomoto A, Kaibuchi K, Kohsaka S, Uchino S. NMDA receptor regulates migration of newly generated neurons in the adult hippocampus via Disrupted-In-Schizophrenia 1 (DISC1) J Neurochem. 2011;118:34–44. doi: 10.1111/j.1471-4159.2011.07282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J, Willoughby D, Kenny PJ, Elsworth JD, Lawrence MS, Roth RH, Edbauer D, Kleiman RJ, Wahlestedt C. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci U S A. 2012;109:3125–30. doi: 10.1073/pnas.1113793109. [DOI] [PMC free article] [PubMed] [Google Scholar]