

Abstract
The compartmentalization and transport of proteins and solutes across the endothelium is a critical biologic function altered during inflammation and disease, leading to pathology in multiple disorders. The impact of tissue damage and subsequent extracellular matrix (ECM) fragmentation in regulating this process is unknown. We demonstrate that the collagen-derived matrikine acetylated proline-glycine-proline (N-α-PGP) serves as a critical regulator of endothelial permeability. N-α-PGP activates human endothelial cells via CXC-chemokine receptor 2 (CXCR2), triggering monolayer permeability through a discrete intracellular signaling pathway. In vivo, N-α-PGP induces local vascular leak after subcutaneous administration and pulmonary vascular permeability after systemic administration. Furthermore, neutralization of N-α-PGP attenuates lipopolysaccharide-induced lung leak. Finally, we demonstrate that plasma from patients with acute respiratory distress syndrome (ARDS) induces VE-cadherin phosphorylation in human endothelial cells, and this activation is attenuated by N-α-PGP blockade with a concomitant improvement in endothelial monolayer impedance. These results identify N-α-PGP as a novel ECM-derived matrikine regulating paracellular permeability during inflammatory disease and demonstrate the potential to target this ligand in various disorders characterized by excessive matrix turnover and vascular leak such as ARDS.
INTRODUCTION
The endothelium is a monolayer of cells lining the luminal surface of blood vessels, serving as a barrier between the circulation and underlying tissue. The ability of the endothelium to regulate paracellular transport of proteins and fluid across this monolayer is thought to serve as a critical developmental feature delineating the increasingly complex cellular functions of higher-order organisms. Although selective endothelial permeability is considered critical to the maintenance of health in humans, many diseases are characterized by excessive vascular leak. Often, these conditions feature considerable extracellular matrix (ECM) turnover and inflammation, suggesting a potential role for specific ECM fragments as regulators of vascular endothelial function.
Previous research has suggested that fragmentation of ECM can lead to the formation of small peptides (termed matrikines), which have the capacity to regulate inflammatory cell phenotypes both in vitro and in vivo (1). For example, we and others have demonstrated that alkali hydrolysis or proteolytic digestion of the ECM protein collagen results in production of the tripeptide matrikine proline-glycine-proline (PGP), which can be N-terminal acetylated (N-α-PGP) through direct chemical modification (2); both are bioactive, but because of different stabilities in vivo, N-α-PGP was used to model total bioactive PGP peptides (3–5). N-α-PGP can activate cell signaling via the CXC-chemokine receptor 2 (CXCR2) (6, 7). To date, the focus of studies on the cellular effects of N-α-PGP have centered on its role as a neutrophil chemoattractant (8, 9), whereas the potential effects on other CXCR2-expressing cell populations have not been evaluated.
N-α-PGP is the first matrikine reported in the airway and plasma of patients with chronic inflammatory lung disease and in the intestines of patients with inflammatory bowel disease (10–13). These conditions are characterized by increased protease activity and ECM turnover, and in a variety of animal models of disease, the blockade of N-α-PGP or its generating proteases has led to amelioration of disease (3, 14).
Our group recently described the increased presence of critical proteases required for N-α-PGP generation in the airway secretions from patients with the acute respiratory distress syndrome (ARDS) (15). ARDS is an acute inflammatory lung disorder initially characterized by a neutrophil-rich inflammatory response and dysregulated vascular permeability, leading to impaired gas exchange. Although various stimuli can trigger ARDS, common features of the condition are ongoing matrix remodeling and dysregulated vascular permeability, leading to disease progression and increased morbidity and mortality. Whether matrix remodeling and vascular leak are mechanistically integrated in ARDS or not remains unclear. We hypothesized that N-α-PGP is a critical link in the development of disease-mediated tissue damage and paracellular endothelial cell permeability. Here, we show that N-α-PGP can selectively activate primary human endothelial cells to induce monolayer permeability, in the absence of broad inflammatory signaling, in a CXCR2-dependent manner. Furthermore, we demonstrate that N-α-PGP induces both local and systemic vascular leak when directly administrated and functions as a critical component of lipopolysaccharide (LPS)–mediated vascular permeability. Finally, we highlight N-α-PGP as an important mediator of endothelial permeability in clinical specimens from patients with ARDS.
RESULTS
N-α-PGP activates endothelial permeability signaling via CXCR2
We first determined whether different tissue-specific endothelial cell lines displayed signaling through CXCR2. Human coronary artery, umbilical vein, pulmonary microvascular, and aortic endothelial cells were stimulated with the CXCR2 ligand interleukin-8 (IL-8), and the phosphorylation of extracellular signal–regulated kinase (ERK), a known downstream effector of IL-8–stimulated CXCR2 signaling (16), was determined. IL-8 treatment induced ERK phosphorylation in all cell lines tested, indicating that they could all support CXCR2-mediated cell signaling (fig. S1). To determine whether the novel CXCR2 agonist N-α-PGP could activate permeability signaling in endothelial cells, we stimulated human umbilical vein endothelial cells (HUVECs) with N-α-PGP for 0 to 60 min, and we assessed Rac1 activation and p21-activated kinase (PAK), ERK, and VE-cadherin phosphorylation because each has been implicated in IL-8–induced, CXCR2-mediated, endothelial permeability (17). Figure 1A demonstrates that N-α-PGP rapidly activated Rac1 as determined by glutathione S-transferase–p21 activated kinase binding domain (GST-PBD) pull-down assays. N-α-PGP increased phosphorylation of ERK and PAK (Fig. 1B), and subsequent phosphorylation of VE-cadhedrin (Fig. 1C). The latter is a major component of adherens junctions found specifically in endothelial cells, and phosphorylation of which is associated with paracellular permeability both in vivo and in vitro (18). Stimulation of HUVECs with a control peptide, proline-glycine-glycine (PGG), did not result in similar increases in Rac1 activation or ERK phosphorylation (fig. S2), thus demonstrating the specificity of N-α-PGP in activating endothelial cells.
Fig. 1. N-α-PGP activates endothelial cell signaling through CXCR2.

(A to C) HUVECs were serum-starved for 2 hours before stimulation with N-α-PGP (0.5 mg/ml) for indicated times, and activation of Rac1 (A) and phosphorylation of PAK (pPAK) and ERK (pERK) (B) and VE-cadherin (pVE-cad) (C) were determined by Western blot. Shown are representative Western blots together with quantification. Bar graphs show means ± SEM (n = 3). *P < 0.05 relative to time 0 by one-way analysis of variance (ANOVA) with Tukey post-test. (D) HUVECs were untreated or treated with N-α-PGP (0.5 mg/ml) (30 min) alone or after pretreatment with 200 nM SB225002, and Rac1 activity and phosphorylation of ERK, PAK, and VE-cadherin were determined by Western blot. Shown are representative Western blots together with quantification. Bar graphs show means ± SEM (n = 3). *P < 0.05 relative to time 0, #P < 0.05 relative to PGP by one-way ANOVA with Tukey post-test.
N-α-PGP induces signaling through CXCR2. We therefore tested to see whether a CXCR2 inhibitor, SB225002 (19), could abolish these signaling effects in endothelial cells. SB225002 inhibited N-α-PGP–induced Rac activation and similarly prevented PAK, ERK, and VE-cadherin phosphorylation (Fig. 1D). The ERK-related dependency of this pathway was further confirmed with U0126, a highly selective inhibitor of both mitogen-activated protein kinase (MAPK) kinase 1 and 2 (MEK1/2), to block ERK activation and downstream phosphorylation of VE-cadherin after N-α-PGP administration (fig. S3). N-α-PGP–dependent stimulation of ERK, VE-cadherin, and PAK phosphorylation was confirmed in pulmonary microvascular endothelial cells (PMVECs), also confirming a potential role of this signaling pathway in ARDS permeability mechanisms (fig. S4, A to E).
N-α-PGP induces paracellular permeability in endothelial cells
To test whether N-α-PGP could induce monolayer permeability, primary HUVECs were left untreated or treated with N-α-PGP, PGG, or vascular endothelial growth factor (VEGF) as a positive control. As shown in Fig. 2A, N-α-PGP induced a significant increase in horseradish peroxidase(HRP)leak across HUVEC monolayers, whereas the control peptide PGG had no effect compared to control. N-α-PGP–induced HRP leak was dependent on CXCR2 as demonstrated by the preservation of monolayer integrity with the addition of the CXCR2-specific inhibitor SB225002 (Fig. 2B). This pro-permeability effect of N-α-PGP was independently confirmed using the xPERT permeability assay (20). As seen in Fig. 2C, N-α-PGP induced a significant increase in the Alexa 488–positive area, demonstrating a loss of endothelial barrier integrity as compared to untreated cells; this permeability was prevented by SB225002 (Fig. 2D). Similarly, N-α-PGP increased permeability in PMVECs in a dose-dependent manner and was inhibited by SB225002 (fig. S4, F and G). These results mechanistically link N-α-PGP–mediated permeability signaling with a notable phenotype of paracellular permeability.
Fig. 2. N-α-PGP induces endothelial permeability through CXCR2.
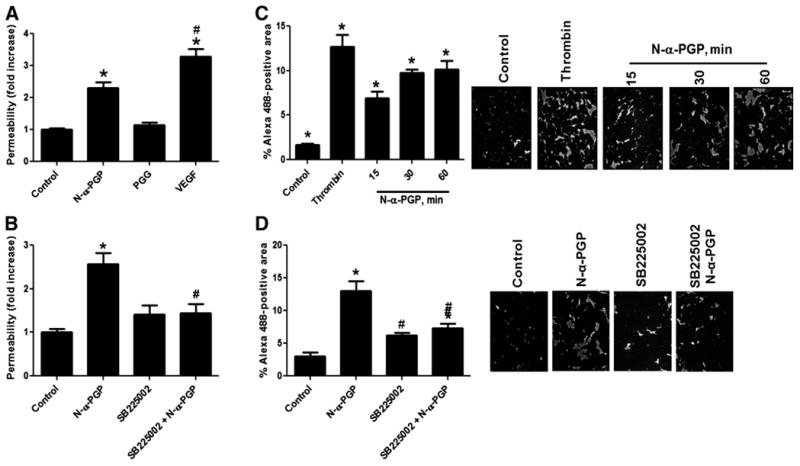
(A) HUVECs were grown on 3.0-μm Transwell membranes and left untreated or stimulated with N-α-PGP (0.5 mg/ml), PGG (0.5 mg/ml), or VEGF (50 ng/ ml) for 30 min. (B) HUVECs were cultured as before, and some cells were pretreated with 200 nM SB225002 for 30 min before stimulation with N-α-PGP (0.5 mg/ml). HRP leak from the upper chamber to the lower chamber was measured as described in Materials and Methods. (C) As a separate readout of permeability, HUVECs were grown on biotinylated collagen and stimulated with N-α-PGP (0.5 mg/ml) for 0 to 60 min or with thrombin (0.2 U/ml) for 10 min, and monolayer leak was determined by staining with streptavidin–Alexa 488. (D) HUVECs were treated with N-α-PGP (0.5 mg/ml) with or without 200 nM SB225002 for 30 min with streptavidin–Alexa 488 staining. *P < 0.05 versus control; #P < 0.05 versus N-α-PGP. Data are means ± SEM [n = 6 for (A) and (B), and n = 3 for (C) and (D)].
N-α-PGP treatment did not increase phosphorylation of the p65 subunit of nuclear factor κB (NFκB), intercellular adhesion molecule–1 (ICAM-1)/vascular cell adhesion molecule–1 (VCAM-1) expression, or monocyte adhesion, whereas tumor necrosis factor–α (TNF-α) induced potent increases in all of these readouts (fig. S5). Moreover, N-α-PGP had no effect on proinflammatory cytokine release from HUVECs or PMVECs (fig. S5). These data indicate that the effects of N-α-PGP on endothelial cells are selective for permeability-related pathways and are not of a broad inflammatory nature.
N-α-PGP induces permeability in vivo
To determine whether N-α-PGP induced local permeability in vivo, we used the Miles assay as a model (17). C57BL/6 mice were injected via the tail vein with Evans blue and followed by abdominal subcutaneous injection with either phosphate-buffered saline (PBS) alone or PBS containing N-α-PGP, PGG, or VEGF. As seen by the representative images in Fig. 3A, both N-α-PGP and VEGF significantly increased Evans blue leak to skin tissue, whereas PGG had no effect above PBS control. Total Evans blue per gram of tissue was quantified and confirmed that N-α-PGP treatment induced increased leak (Fig. 3B). Next, we wanted to determine whether in vivo administration of N-α-PGP could induce similar protein phosphorylation as seen in HUVECs. Mice were again administered with PBS alone or with N-α-PGP, PGG, or VEGF. Western blot analysis of skin tissue lysates for VE-cadherin phosphorylation (as an endothelial cell–specific readout) indicated that both N-α-PGP and VEGF increased phosphorylation above levels of PBS or control peptide (Fig. 3C). These data confirm that N-α-PGP can induce local vascular permeability and endothelial cell activation in vivo.
Fig. 3. N-α-PGP induces skin and pulmonary microvascular permeability.
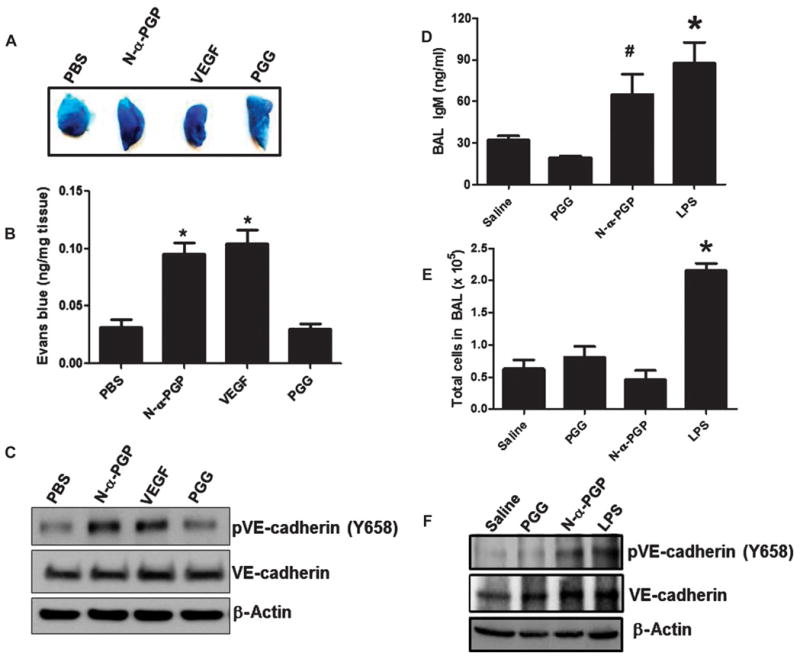
(A to C) Mice (n = 4 to 5 per group) were injected via the tail vein with Evans blue and then received abdominal subcutaneous injection of PBS alone, N-α-PGP (250 μg), PGG (250 μg), or VEGF (50 ng). Evans blue leak to the skin tissue was visually assessed (A) and quantified (B), and VE-cadherin phosphorylation after treatment was determined by Western blot (representative image) (C). *P < 0.05 versus PBS control by one-way ANOVA with Tukey post-test. (D to F) Mice (n = 4 to 5 per group) were intraperitoneally injected with saline alone or containing N-α-PGP (250 μg), PGG (250 μg), LPS (75 μg) once a day for 4 days, and then the total IgM in BAL fluid was measured by immunoassay (D) and total BAL cell (E), and VE-cadherin phosphorylation in lung lysate was measured by Western blot (F). *P < 0.05 versus saline or PGG, #P < 0.05 versus PGG by one-way ANOVA with Tukey’s multiple comparison post-test. All values represent means ± SEM.
Whereas local administration of N-α-PGP induces the endothelial changes outlined above, the effects of short-term systemic administration of N-α-PGP on lung vascular permeability are unknown. We addressed this question by administering saline, N-α-PGP, PGG, or LPS for 4 days intraperitoneally to mice and by analyzing bronchoalveolar lavage (BAL) fluid. With measurement of immunoglobulin M (IgM) levels, a sensitive marker of vascular leak in the lung (21), N-α-PGP significantly increased IgM levels in BAL relative to saline or PGG control and comparable to that observed after treatment with LPS, a known regulator of pulmonary vascular leak (22) (Fig. 3D). N-α-PGP had no effect on BAL cell count (Fig. 3E), suggesting no proinflammatory response, consistent with our observations described above (fig. S5). Moreover, the ratio of phosphorylated VE-cadherin to total VE-cadherin in lung homogenates was increased over control in mice treated with N-α-PGP, highlighting lung endothelial cell activation in vivo (Fig. 3F). Overall, these data demonstrate that N-α-PGP activates pro-permeability pathways and endothelial leak independent of its role as a neutrophil chemoattractant in vivo.
N-α-PGP acts as a critical regulator in LPS-mediated lung injury
Recent data have demonstrated that endothelial cell expression of CXCR2 is required for vascular leak in an LPS-induced model of acute lung injury (23). CXCR2 does not act as a ligand for LPS, suggesting that other mediators are involved in the mechanism of CXCR2-dependent vascular permeability in this model. To determine whether N-α-PGP is a critical regulator for LPS-mediated endothelial permeability, mice were administered LPS intraperitoneally every day for 4 days with or without RTR (arginine-threonine-arginine) peptide, which sequesters and inhibits PGP-containing peptides (14). Figure 4A shows that LPS treatment increased serum PGP peptides, which is ameliorated by RTR. Mice were injected with Evans blue, and their lungs were isolated to assess for vascular leak. Mice that were concomitantly treated with RTR and LPS demonstrated gross attenuation of Evans blue in the lungs as compared to LPS alone (Fig. 4B), which was found to be comparable to that observed in saline-treated mice (Fig. 4B). RTR treatment also inhibited IgM accumulation in the BAL and decreased VE-cadherin phosphorylation compared to lungs from mice treated with LPS alone (Fig. 4, C and D). Overall, these studies demonstrate the capability of N-α-PGP to act as a regulator of endothelial permeability in vivo and its relative importance in LPS-mediated lung injury.
Fig. 4. RTR attenuates LPS-induced pulmonary microvascular permeability.
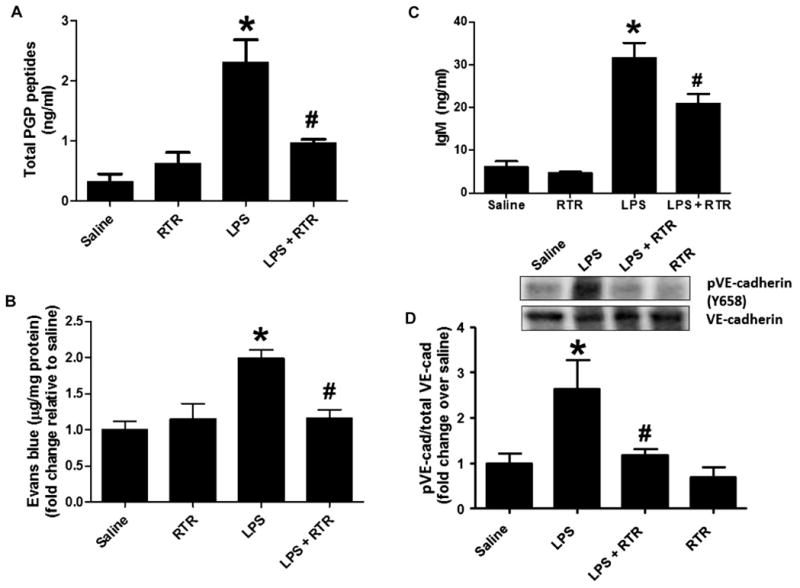
Mice were injected via the tail vein with 50 μl of PBS alone or containing 50 μg of RTR and then intraperitoneally administered with 75 μg of LPS (in 100 μl of PBS) once a day. (A) After 4 days of treatment, mice were sacrificed for serum measurements of PGP (n = 6) or injected via the tail vein with Evans blue. (B) Evans blue leak to the lung was quantified and normalized to protein; data show fold change relative to saline (n = 7 to 11). (C) IgM levels were measured in the BAL (n = 4 to 6). (D) VE-cadherin phosphorylation in lung homogenates was assessed by Western blot. Representative image. *P < 0.05 versus saline control, #P < 0.05 relative to LPS for (A) to (C) by one-way ANOVA with Tukey post-test; #P < 0.05 relative to LPS by t test for (D). All values represent means ± SEM.
PGP peptides participate in endothelial cell dysfunction observed in ARDS
ARDS is a complex disease characterized by increased lung vascular permeability and altered systemic inflammatory response (24). We sought to recapitulate our in vitro and in vivo animal findings in human disease. Plasma was collected from intubated subjects with ARDS secondary to gram-negative sepsis and intubated subjects without lung disease. Plasma samples were collected within 24 hours of intubation, and PGP peptide values were measured. Patient demographics for the two groups are shown in Table 1, with differences noted between groups for gender, arterial O2/inspired oxygen (PaO2/FIO2) ratio, Acute Physiology and Chronic Health Evaluation (APACHE) II score, and 30-day all-cause mortality. Figure 5A shows that total PGP peptide levels were higher in ARDS specimens compared to non-lung disease controls (0.53 ± 0.14 ng/ml and 0.14 ± 0.08 ng/ml, respectively), with N-α-PGP comprising between 5 and 10% of total PGP peptide levels. Figure 5B shows the N-α-PGP dose-dependent stimulation of PMVEC permeability, with 1 ng/ml causing a nearly twofold increase relative to control. Figure 5B (inset) shows that both N-α-PGP and PGP (both at 1 ng/ml) elicit similar increases in PMVEC permeability. ARDS versus non-lung disease plasma samples (n = 3 per group) were then pooled and added to HUVECs, and induction of phosphorylated VE-cadherin was assessed. As observed in Fig. 5C, ARDS plasma induces a notable increase in the ratio of phosphorylated VE-cadherin to total VE-cadherin compared to non-lung disease plasma as early as 15 min after exposure, consistent with previous in vitro stimulation experiments. Using this time point, we next compared the phosphorylated VE-cadherin/total VE-cadherin ratio between individual plasma samples incubated with RTR to the same plasma samples treated with RTR vehicle. Figure 5 (D and E) highlights a significant reduction in the phosphorylation of VE-cadherin with RTR coincubation with ARDS plasma, with levels comparable to nonstimulated endothelial cells. Notably, non-lung disease plasma demonstrated no notable increase in phosphorylated VE-cadherin/total VE-cadherin ratio compared to control (Fig. 5C).
Table 1.
Demographics for ARDS and non-lung disease ICU patients.
| Group | Age | Sex | Race | PaO2/FIO2 | APACHE II score | 30-Day mortality |
|---|---|---|---|---|---|---|
| ARDS (n = 6) | 59.5 (6.8) | 50% male, 50% female | 66% white, 34% black | 149.5 (60) | 25.5 (2.6) | 66% |
| Non-lung disease (n = 6) | 46.1 (9.7) | 66% male, 34% female | 66% white, 34% black | 469.2 (80) | 11.0 (4.9) | 0% |
| P (between groups) | <0.05 | ns | ns | <0.05 | <0.05 | <0.05 |
Values represent means ± SD. All ARDS subjects were secondary to documented gram-negative sepsis and intubated because of respiratory failure. Non-lung disease control subjects were intubated for various reasons unrelated to respiratory failure (one for airway protection secondary to gastrointestinal bleed, two for metabolic acidosis secondary to diabetic ketoacidosis, two for overdose, and one for airway protection status post–cerebrovascular accident). ns, no significant difference.
Fig. 5. ARDS plasma induces endothelial activation, which is attenuated by RTR.
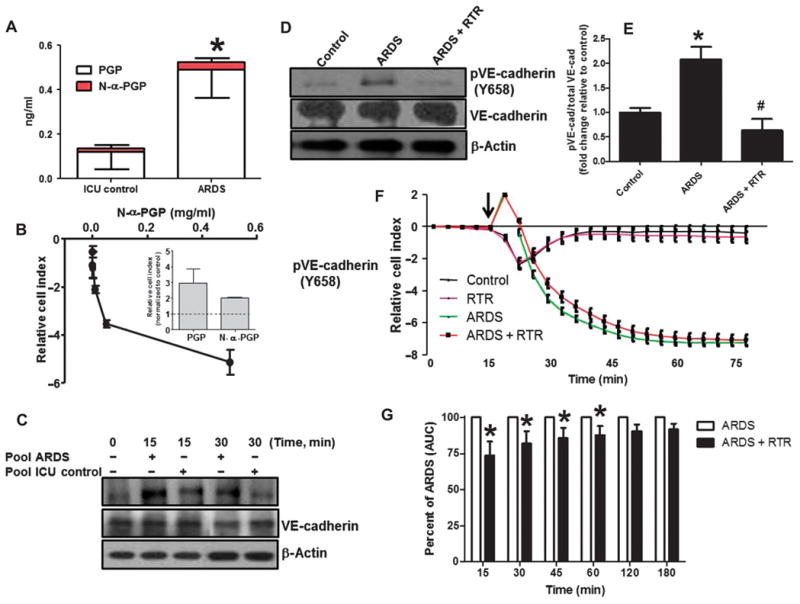
(A) Plasma was collected from patients with ARDS and normal control, and plasma PGP and N-α-PGP levels were measured via electrospray ionization–liquid chromatography–tandem mass spectrometry. *P < 0.05 by t test between ARDS and normal for all PGP peptides (n = 6). PMVECs were treated with N-α-PGP, and permeability was assessed (see fig. S4F). (B) Maximal changes in relative cell index as a function of N-α-PGP concentrations. Inset compares the effects of N-α-PGP and PGP (both at 1 ng/ml). (C) After 2 hours of serum starvation, HUVECs were treated with pooled ARDS (from three patients) or non-lung disease intensive care unit (ICU) patient (n = 3) plasma at two different time points, and VE-cadherin phosphorylation was measured by Western blot. Representative image. HUVECs were treated as above for 15 min with plasma collected from individual ARDS with or without RTR (30 μg/ml), and changes in VE-cadherin phosphorylation were assessed. (D) Representative Western blot. (E) Quantitation. *P < 0.05 relative to control, #P < 0.05 relative to ARDS by one-way ANOVA with Tukey post-test (n = 3). Measurements of cellular impedance were made in HUVECs over 75 min with ARDS plasma versus ARDS plasma + RTR. (F) Representative traces from one patient with four intraexperimental replicates. Arrow denotes addition of plasma. (G) Percent attenuation by RTR on ARDS plasma time-dependent permeability changes. *P < 0.05 by two-way repeated-measures ANOVA with Bonferroni post-test. All values represent means ± SEM (n = 3). AUC, area under the curve.
We next wanted to determine whether RTR could reduce permeability associated with inflammatory factors, principally PGP peptides, in plasma of ARDS patients. To better understand the dynamics of how this may occur, we used a technique to measure cellular impedance, a measurement of junctional integrity, for these assays. HUVECs were serum-starved before being treated with RTR, plasma from ARDS patients, or plasma that had been incubated for 1 hour with RTR before addition to the cells for 30 min. Plasma from ARDS subjects significantly reduced impedance (presented as relative cell index) relative to cells alone, indicating an increase in permeability (Fig. 5F). When the ARDS plasma had been preincubated with RTR to inactivate PGP peptides, there was a significant attenuation of impedance reduction (Fig. 5G), which was sustained for over an hour. Overall, these results highlight the differences in the capacity of ARDS plasma versus non-lung disease plasma in activating permeability-related signaling in endothelial cells and the ability of RTR to block this activation and modulate endothelial permeability observed in ARDS.
DISCUSSION
The present data demonstrate that the CXCR2 agonist matrikine N-α-PGP induces endothelial permeability. Multiple reports have now demonstrated that N-α-PGP is increased in a variety of inflammatory conditions (10, 11, 18, 25). These studies have focused solely on the role of N-α-PGP as a neutrophil chemoattractant and have not addressed other potential physiological functions. Each disease model studied is associated with not only neutrophilic injury but also increased endothelial permeability, suggesting the possibility that N-α-PGP also directly regulates vascular function. Our findings provide the first evidence for circulating matrix fragments as biologic effectors of vascular endothelial function and, more specifically, demonstrate that N-α-PGP selectively increases vascular permeability via CXCR2-dependent signaling. The downstream signaling induced by N-α-PGP is similar to other proangiogenic factors that are associated with disruption of monolayer permeability that also fail to induce a broad inflammatory response (26). We focused on the Rac1-PAK-ERK signaling axis here, which is well established in mediating permeability (17, 26); the potential for N-α-PGP to mediate effects via additional signaling mediators downstream of CXCR2 (for example, p38 MAPK) remains to be tested.
Published in vitro studies have suggested that CXCR2 signaling may play important roles in endothelial function including activation of small guanosine triphosphatase (GTPase) (17, 27), increased proliferation and migration (28, 29), promotion of angiogenesis (16, 30), and induction of monolayer permeability (17, 31, 32). However, to our knowledge, the current findings are the first to link a single CXCR2 ligand to endothelial permeability in cell culture and in animal models of disease. Moreover, data suggesting pro-permeability functions of PGP peptides in human plasma from ARDS patients highlight the potential clinical impact of this peptide in human disease as a therapeutic target.
Our in vivo studies demonstrate that local administration of N-α-PGP induces vascular leak and that systemic administration can lead to increased lung permeability similar to that observed with LPS administration. The importance of this signaling is highlighted by the prevention of LPS-mediated endothelial permeability through N-α-PGP inhibition and suggests a role for this peptide as a novel effector of endotoxin-induced injury. LPS has been previously shown to induce the extracellular activity and expression of numerous proteases implicated in PGP peptide generation (3, 6), strongly suggesting a feed-forward inflammatory system, which may serve as a critical target in sepsis and sepsis-related sequelae such as ARDS.
We also demonstrate increased PGP peptides in ARDS patient plasma. N-α-PGP was lower compared to PGP, which may reflect the relative activity of pathways, leading to peptide acetylation versus PGP breakdown. We note that the ratio between N-α-PGP and PGP varies in different acute and chronic disease states (3, 13, 33); understanding this balance in sepsis and ARDS is the subject of ongoing studies. We note the limited sample size for these measurements and that it is possible that additional confounding parameters because of the heterogeneity of ARDS pathogenesis could affect these data. However, these data do provide initial evidence as proof of concept that bioactive PGP peptides serve as potential targets in treating disease-related vascular permeability. The implication of N-α-PGP as a regulator of endothelial dysfunction in the plasma of patients with ARDS is of particular importance because recent evidence has coupled the amount of extravascular lung water as an independent predictor of mortality in the condition (34). Although previous interest in reducing alveolar lung water has focused on mobilizing fluid from the alveolar space after leak is established (35), there remains a need to target the dysfunctional endothelial paracellular leak from the vasculature. RTR is a tetramerized hydropathic peptide targeting both PGP and N-α-PGP. It has been tested in animal models in which PGP is operative, with inhibition of neutrophilic influx demonstrated (14). Our results present the first example of RTR targeting endothelial permeability in vivo, which suggests a role for endogenous bioactive PGP-containing peptides in mediating lung fluid accumulation in ARDS. Because RTR inhibits all PGP-containing peptides, future studies are needed to determine the relative contribution of PGP versus N-α-PGP in endothelial permeability and the potential effect of microenvironment in regulating PGP peptide bio-activity in vivo. However, we note that both PGP and N-α-PGP, at levels similar to those observed in vivo, stimulated endothelial permeability (Fig. 5B). Combined with RTR-dependent inhibition of permeability in vivo and of human disease samples ex vivo, these data underscore the potential that targeting PGP peptides may inhibit two key facets in the pathogenesis of ARDS: ongoing neutrophilic inflammation (6) and endothelial permeability.
Bioactive PGP peptides represent the first matrix-derived breakdown product found to be increased systemically in human inflammatory diseases that can modify vascular function. This finding is particularly interesting because these peptides represent a unique CXCR2 agonist that is regulated by direct tissue damage and not by transcriptional regulation (such as ELR+ CXC chemokines) and may therefore induce a feed-forward mechanism of ongoing vascular leak (36). It will be necessary to examine N-α-PGP levels as a potential regulator of progression in other diseases associated with matrix turnover and vascular dysfunction such as tissue fibrosis, ischemia-reperfusion injury, pulmonary vascular disease, kidney injury, and myocardial infarction. To conclude, we have identified a novel role for N-α-PGP as an activator of endothelial cell signaling, which actively couples vascular permeability and tissue injury. This peptide seems to be critically important in the endothelial activation associated with ARDS and may serve as a novel therapeutic target in this disease.
MATERIALS AND METHODS
Reagents
Primary HUVECs, human coronary arterial endothelial cells (HCoAECs), human aortic endothelial cells (HAECs), and human PMVECs (HPMVECs) were from Lonza, and THP-1 cells were from the American Type Culture Collection. MCDB-131, heat-inactivated fetal bovine serum (FBS), L-glutamine, and penicillin/streptomycin were from Invitrogen. N-α-PGP, PGG, and RTR were from Bachem. All other reagents were from Sigma-Aldrich unless otherwise noted.
Patient populations and plasma collection
ARDS patients secondary to gram-negative sepsis (n = 6) and non-lung disease patients (n = 6), who were also intubated and mechanically ventilated, were recruited from the University of Alabama at Birmingham (UAB) hospital. All subjects carried the diagnosis of ARDS based on accepted diagnostic criteria (37). Samples and health information were labeled using unique identifiers to protect subject confidentiality. Blood samples were collected in sodium citrate tube and centrifuged at 3500 rpm for 15 min, and plasma was stored at −80°C for later analysis. All human studies were approved by the UAB Institutional Review Board (protocol number F081016007).
In vitro experiments
Cell culture
Endothelial cells were maintained as previously described (38). For treatment, cells at 1 day after confluence were serum-starved for 2 hours before stimulation as described for each experiment. THP-1 cells were maintained in RPMI 1640 containing 10% FBS, penicillin (100 U/ml)/streptomycin (100 μg/ml), and 2 μM β-mercaptoethanol. For adhesion experiments, THP-1 monocytes were labeled with CellTracker Green (1 μM) for 15 min at 37°C.
Transwell permeability assay
Permeability assays were conducted as previously described (26). HUVECs were plated onto gelatin-coated 3.0-μm polycarbonate Transwell membranes (Corning Costar) and cultured for 4 days with medium changes every day. Cells were serum-starved for 2 hours in phenol red–free Dulbecco’s modified Eagle’s medium/F12 and treated as described for experiments. HRP was added to the upper chambers at a final concentration of 1.5 μg/ml, and after 30 min, the filters were removed and medium was harvested from the lower chamber. HRP leak was determined by absorbance at 450 nm after incubation with 0.5 mM guaiacol, 50 mM Na2HPO4, and 0.6 mM H2O2 for 15 to 30 min.
xPERT permeability assay
Experiments were performed with a modification of the previously reported protocol (20). Briefly, 12-mm glass coverslips were incubated with 2% 3-aminopropyltriehoxysilane isopropanol for 30 s, washed in isopropanol followed by washing with 100 mM sodium phosphate (pH 6.8), and then incubated with 5% glutaraldehyde in 100 mM sodium phosphate (pH 6.8) for 5 min. After washing in 100 mM sodium biocarbonate (pH 8.3), coverslips were coated with biotinylated collagen (0.25 mg/ml) in the aforementioned buffer for 2 hours at room temperature. Coverslips were then washed in PBS, and free aldehydes were quenched by incubation with complete growth medium. Cells were plated at 2 × 105 per well and, after 48 hours, were treated as described. To visualize permeability, 0.5 ml of 1:2000 dilution of streptavidin–Alexa 488 was added to wells for 3 min. Wells were then quickly washed with 3.7% paraformaldehyde and then incubated in 3.7% paraformaldehyde for 20 min at room temperature. After exhaustive washing with PBS, coverslips were mounted for viewing. Three random fields from three independent coverslips per condition were captured using a 20× objective, and the process was repeated at least three times. Data are represented as percent Alexa 488–positive area per field of view as assessed using the National Institutes of Health (NIH) ImageJ.
Real-time cell analysis experiments
Barrier integrity of HUVECs was analyzed using the xCELLigence Real-Time Cell Analyzer (ACEA Biosciences/Roche Applied Science). This technology measures electrical impedance as a readout for the barrier integrity in cells grown on microelectrodes to provide a constant assessment of barrier function. Decreases in cellular impedance (presented as area under the curve) reflect disruption in barrier integrity and increased paracellular permeability. Cells (5 × 104) were plated in each well of an E-Plate 16 (Roche Applied Science) and allowed to grow for 48 hours. Impedance readings were recorded at 10-min intervals for 1 hour to confirm that stable junctions were present. Cells were then changed to serum-free medium and monitored until junctions had again stabilized (about 2.5 hours) and treated as described. Area under the curve was calculated as the net difference in relative cell index for each treatment group as compared to control over selected time points using Microsoft Excel and plotted using GraphPad.
GTPase activity assay
Rac1 activity was determined by pull-downs using GST-PBD and GST-RBD (rhotekin-binding domain) (gift of A. W. Orr, Louisiana State University Health Sciences Center Shreveport). Cells were serum-starved for 2 hours before being stimulated as described, and washed twice with ice-cold PBS and lysed [50 mM tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, 10% glycerol, 1 mM NaF, 1 mM Na3VaO4, 1 mM phenylmethylsulfonyl fluoride (PMSF)] for 10 min on ice before clarification by centrifugation at 14,000g for 5 min. Lysates were incubated with 25 μg of GST-PBD or GST-RBD and 25 μl of packed GST-Sepharose (GE Life Sciences) for 45 min at 4°C with gentle shaking (10% of the lysates were reserved as input control). Beads were washed extensively with lysis buffer, and bound proteins were released by boiling in SDS–polyacrylamide gel electrophoresis sample buffer. Reserved whole-cell lysates and bound proteins were resolved by Western blot analysis as described below.
Western blot
Samples were resolved on 4 to 15% TGX gels (Bio-Rad) and transferred to polyvinylidene difluoride membranes. Blots were blocked with 5% milk in TBS + 0.1% Tween 20 (TBST) [except for phosphorylated VE-cadherin, which was blocked in 5% bovine serum albumin (BSA)–TBST] and incubated overnight at 4°C with antibodies against phosphorylated ERK (T202, Y204) (4370), ERK (4695), and ICAM-1 (4915) (Cell Signaling Technology); phosphorylated VE-cadherin (Y658) (441144G) and phosphorylated PAK (S141) (44940G) (Invitrogen); Rac1 (610650) (BD Transduction Labs); and VE-cadherin (sc-28644), γPAK (sc-1872), and VCAM-1 (sc-8304) (Santa Cruz Biotechnology) in 2% heat-denatured BSA-TBST. Blots were washed in TBST, incubated with species-appropriate HRP-conjugated secondary antibody, and washed again in TBST, and signals were detected using enhanced chemiluminescence and x-ray film.
THP-1 adhesion assay
Assays were performed as previously described (39). Briefly, HUVECs were grown in 48-well plates and treated as described for each experiment. Cells were washed with warm PBS and incubated with 6 × 104 CellTracker Green–labeled THP-1 monocytes for 30 min at 37°C. Plates were gently washed with PBS, and fluorescence was measured on a Victor2 Perkin-Elmer Fluorescent plate reader (excitation,485 nm; emission,535 nm).
In vivo experiments
Mice
C57BL/6 female mice (The Jackson Laboratory) were used in these studies. All animal studies were approved by the UAB Institutional Animal Care and Use Committee (animal protocol number 130709133).
Miles assay
In vivo skin permeability was determined as previously described (17). Six- to 8-week-old C57BL/6 mice had their abdominal fur removed by depilatory agents, were injected via the tail vein with 200 μl of 1% Evans blue, and were injected subcutaneously on the abdomen with 200 μl of PBS or 250 μg of N-α-PGP, 250 μg of PGG, or 50 ng of VEGF. One hour after injections, mice were sacrificed and abdominal skin was removed and weighed. Evans blue was extracted from skin tissue by overnight incubation in formamide at 56°C. Absorbance was measured at 620 nm, and Evans blue concentration was determined from a standard curve and presented compared to tissue mass.
Murine model of LPS-induced pulmonary microvascular permeability
Six- to 8-week-old C57BL/6 mice were injected with either saline vehicle or 50 μg of RTR daily for 4 days via tail vein. Thirty minutes after each dose, mice were administered 75 μg of LPS from Escherichia coli (Sigma-Aldrich) intraperitoneally to induce pulmonary permeability changes as previously described (40, 41). Control animals received saline instead of LPS in the same manner. Twenty-four hours after the last treatment, pulmonary microvascular permeability was assessed using the Evans blue dye extravasation technique as described previously (23). Briefly, Evans blue (20 mg/kg; Sigma-Aldrich) was injected intravenously 1 hour before isoflurane euthanasia. Lungs were perfused and removed, and Evans blue was extracted. Absorbance was measured at 620 nm, and Evan’s blue concentration was determined from a standard curve.
Murine model of N-α-PGP–induced lung injury
Saline (50 μl) containing 50 μg of N-α-PGP was intraperitoneally administered daily for 4 days to 6- to 8-week-old C57BL/6 mice. Saline and saline containing 50 μg of PGG and 75 μg of LPS served as controls. Twenty-four hours after the last dose, mice were anesthetized by intraperitoneal injection of ketamine (120 mg/kg) and xylazine (8 mg/kg). Through a 1-cm abdominal midline incision, blood was collected by intracardiac puncture before euthanasia. Blood samples were centrifuged at 3500 rpm for 15 min, and plasma was aspirated and stored at −80°C. BAL was collected using three 1.0-ml injections of saline through a tracheal cannula. To estimate pulmonary vascular permeability, BAL supernatant was analyzed for IgM using an enzyme immunoassay (Immunology Consultant Laboratory).
Electrospray ionization–liquid chromatography–tandem mass spectrometry
Plasma samples were processed by filtering through a Millipore 10,000 molecular weight cutoff centrifugal filter and then by washing with 40 μl of 1 mM HCl. BAL samples were not filtered. PGP peptides in plasma and BAL were quantified using two different devices: For BAL, a MDS Sciex (Applied Biosystems) API-4000 triple quadrupole mass spectrometer equipped with a Shimadzu high-performance liquid chromatography (HPLC) and a 2.0 × 150–mm Jupiter 4μ Proteo column (Phenomenex) was used. For plasma, a Finnigan TSQ Quantum Discovery Max triple quadrupole mass spectrometer was used with electrospray ionization (Thermo Fisher Scientific) on an Atlantis dC18 column [100 mm × 2.1 mm, particle diameter (dp) = 3 μm; Waters Chromatography] with an Atlantis dC18 precolumn (10 × 2.1 mm, dp = 3 μm; Waters), also coupled to a Shimadzu LC system. The mobile phase consisted in both sample types out of (A) 0.1% (v/v) formic acid in water and (B) 0.1% formic acid in acetonitrile using gradient elution: 0 to 0.5 min 5% B/95% A, then increased linearly over 0.5 to 2.5 min to 100% B/0% A. During the run, samples were kept at 4°C, and the column at 30°C. Positive electrospray mass transitions were at 312 to 140, 312 to 112, and 312 to 70 m/z (mass/charge ratio) for Ac-PGP and 270 to 70, 270 to 116, and 270 to 173 m/z for PGP. Peak area was measured, and PGP peptide concentrations were calculated using a relative standard curve method as previously described (6, 25).
Murine tissue preparation for Western blot analysis
All murine tissues assessed for Western blot were removed immediately after sacrifice and snap-frozen in liquid nitrogen. The tissue was lysed [50 mM tris (pH 7.4), 150 mM NaCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM NaF, 1 mM Na3VaO4, and 1 mM PMSF] and homogenized. Lysates were clarified by centrifugation at 14,000g for 10 min, and protein concentration was determined by BCA assay (Bio-Rad). Equal amounts of protein were analyzed by Western blot analysis as described above.
Statistics
For multiple comparisons, ANOVA and post hoc analysis (Tukey for one-way and Bonferroni for two-way) for multiple comparison tests were applied. The Student’s t test was used for comparisons of the mean values of two different samples. P ≤ 0.05 was considered significant.
Supplementary Material
Acknowledgments
We thank D. Sullivan for his technical assistance.
Funding: This work was supported by an American Heart Association predoctoral fellowship (D.W.S.), a Howard Hughes Medical Institute Med-to-Grad fellowship (D.W.S.), and NIH training grant 3T32CA009156 (D.W.S.). M.A.R. was supported by a Mosaic grant from the Netherlands Organization for Scientific Research (017.008.029). Additional funding from the National Heart, Lung, and Blood Institute (HL07783, HL090999, and HL087824 to J.E.B.; HL92624 to R.P.P.; and HL102371 to A.G.), the Veterans Administration (1 I01 BX001756 to A.G.), and the Ismail Moustapha Scholar Fund (to A.G.). Research reported in this publication was supported by the NIH and the Family Smoking Prevention and Tobacco Control Act. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or the Food and Drug Administration.
Footnotes
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/ full/1/3/e1500175/DC1
Fig. S1. Multiple endothelial cells demonstrate ERK phosphorylation with IL-8 stimulation.
Fig. S2. PGG stimulation does not activate HUVECs.
Fig. S3. Blockade of ERK mitigates N-α-PGP–mediated VE-cadherin activation in endothelial cells.
Fig. S4. Effects of N-α-PGP on PMVEC permeability.
Fig. S5. N-α-PGP does not induce proinflammatory signaling in endothelial cells.
Author contributions: C.S.H., D.W.S., X.X., M.A.R., J.M.W., L.V., G.A.P., C.J.W., and P.B. carried out experiments. C.S.H., D.W.S., X.X., M.A.R., P.L.J., R.W.S., F.A.R., G.F., J.E.B., R.P.P., and A.G. designed experiments and analyzed data. C.S.H., D.W.S., X.X., J.E.B., R.P.P., and A.G. wrote the manuscript.
Competing interests: The authors declare that they have no competing financial interests.
REFERENCES AND NOTES
- 1.Postlethwaite AE, Kang AH. Collagen-and collagen peptide-induced chemotaxis of human blood monocytes. J Exp Med. 1976;143:1299–1307. doi: 10.1084/jem.143.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardison MT, Brown MD, Snelgrove RJ, Blalock JE, Jackson P. Cigarette smoke enhances chemotaxis via acetylation of proline-glycine-proline. Front Biosci. 2012;4:2402–2409. doi: 10.2741/e552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, Clancy JP, Blalock JE. A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol. 2008;180:5662–5669. doi: 10.4049/jimmunol.180.8.5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfister RR, Haddox JL, Sommers CI, Lam KW. Identification and synthesis of chemotactic tripeptides from alkali-degraded whole cornea. A study of N-acetyl-proline-glycine-proline and N-methyl-proline-glycine-proline. Invest Ophthalmol Vis Sci. 1995;36:1306–1316. [PubMed] [Google Scholar]
- 5.O’Reilly PJ, Hardison MT, Jackson PL, Xu X, Snelgrove RJ, Gaggar A, Galin FS, Blalock JE. Neutrophils contain prolyl endopeptidase and generate the chemotactic peptide, PGP, from collagen. J Neuroimmunol. 2009;217:51–54. doi: 10.1016/j.jneuroim.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 7.Braber S, Overbeek SA, Koelink PJ, Henricks PA, Zaman GJ, Garssen J, Kraneveld AD, Folkerts G. CXCR2 antagonists block the N-Ac-PGP-induced neutrophil influx in the airways of mice, but not the production of the chemokine CXCL1. Eur J Pharmacol. 2011;668:443–449. doi: 10.1016/j.ejphar.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Lin M, Jackson P, Tester AM, Diaconu E, Overall CM, Blalock JE, Pearlman E. Matrix metalloproteinase-8 facilitates neutrophil migration through the corneal stromal matrix by collagen degradation and production of the chemotactic peptide Pro-Gly-Pro. Am J Pathol. 2008;173:144–153. doi: 10.2353/ajpath.2008.080081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overbeek SA, Henricks PA, Srienc AI, Koelink PJ, de Kruijf P, Lim HD, Smit MJ, Zaman GJ, Garssen J, Nijkamp FP, Kraneveld AD, Folkerts G. N-acetylated proline–glycine–proline induced G-protein dependent chemotaxis of neutrophils is independent of CXCL8 release. Eur J Pharmacol. 2011;668:428–434. doi: 10.1016/j.ejphar.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Reilly P, Jackson PL, Noerager B, Parker S, Dransfield M, Gaggar A, Blalock JE. N-α-PGP and PGP, potential biomarkers and therapeutic targets for COPD. Respir Res. 2009;10:38. doi: 10.1186/1465-9921-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardison MT, Galin FS, Calderon CE, Djekic UV, Parker SB, Wille KM, Jackson PL, Oster RA, Young KR, Blalock JE, Gaggar A. The presence of a matrix-derived neutrophil chemoattractant in bronchiolitis obliterans syndrome after lung transplantation. J Immunol. 2009;182:4423–4431. doi: 10.4049/jimmunol.0802457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koelink PJ, Overbeek SA, Braber S, Morgan ME, Henricks PA, Roda MA, Verspaget HW, Wolfkamp SC, Te Velde AA, Jones CW, Jackson PL, Blalock JE, Sparidans RW, Kruijtzer JA, Garssen J, Folkerts G, Kraneveld AD. Collagen degradation and neutrophilic infiltration: A vicious circle in inflammatory bowel disease. Gut. 2013;63:578–587. doi: 10.1136/gutjnl-2012-303252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wells JM, O’Reilly PJ, Szul T, Sullivan DI, Handley G, Garrett C, McNicholas CM, Roda MA, Miller BE, Tal-Singer R, Gaggar A, Rennard SI, Jackson PL, Blalock JE. An aberrant leukotriene A4 hydrolase–proline-glycine-proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190:51–61. doi: 10.1164/rccm.201401-0145OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Houwelingen AH, Weathington NM, Verweij V, Blalock JE, Nijkamp FP, Folkerts G. Induction of lung emphysema is prevented by L-arginine-threonine-arginine. FASEB J. 2008;22:3403–3408. doi: 10.1096/fj.07-096230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kong MY, Li Y, Oster R, Gaggar A, Clancy JP. Early elevation of matrix metalloproteinase-8 and -9 in pediatric ARDS is associated with an increased risk of prolonged mechanical ventilation. PLOS One. 2011;6:e22596. doi: 10.1371/journal.pone.0022596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, Otterson MF, Ota DM, Lugering N, Domschke W, Binion DG. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–8515. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- 17.Gavard J, Hou X, Qu Y, Masedunskas A, Martin D, Weigert R, Li X, Gutkind JS. A role for a CXCR2/phosphatidylinositol 3-kinase γ signaling axis in acute and chronic vascular permeability. Mol Cell Biol. 2009;29:2469–2480. doi: 10.1128/MCB.01304-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monaghan-Benson E, Burridge K. VE-cadherin status as an indicator of microvascular permeability. Methods Mol Biol. 2013;1046:335–342. doi: 10.1007/978-1-62703-538-5_20. [DOI] [PubMed] [Google Scholar]
- 19.White JR, Lee JM, Young PR, Hertzberg RP, Jurewicz AJ, Chaikin MA, Widdowson K, Foley JJ, Martin LD, Griswold DE, Sarau HM. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J Biol Chem. 1998;273:10095–10098. doi: 10.1074/jbc.273.17.10095. [DOI] [PubMed] [Google Scholar]
- 20.Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest. 2013;93:254–263. doi: 10.1038/labinvest.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kantrow SP, Shen Z, Jagneaux T, Zhang P, Nelson S. Neutrophil-mediated lung permeability and host defense proteins. Am J Physiol Lung Cell Mol Physiol. 2009;297:L738–L745. doi: 10.1152/ajplung.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, Koch WJ, Wang H, Soboloff J, Gill DL, Madesh M. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest. 2013;123:887–902. doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reutershan J, Morris MA, Burcin TL, Smith DF, Chang D, Saprito MS, Ley K. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaggar A, Olman MA. Biologic markers of mortality in acute lung injury. Clin Chim Acta. 2006;372:24–32. doi: 10.1016/j.cca.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 25.Koelink PJ, Overbeek SA, Braber S, Morgan ME, Henricks PA, Roda MA, Verspaget HW, Wolfkamp SC, Te Velde AA, Jones CW, Jackson PL, Blalock JE, Sparidans RW, Kruijtzer JA, Garssen J, Folkerts G, Kraneveld AD. Collagen degradation and neutrophilic infiltration: A vicious circle in inflammatory bowel disease. Gut. 2014;63:578–587. doi: 10.1136/gutjnl-2012-303252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stockton R, Reutershan J, Scott D, Sanders J, Ley K, Schwartz MA. Induction of vascular permeability: βPIX and GIT1 scaffold the activation of extracellular signal-regulated kinase by PAK. Mol Biol Cell. 2007;18:2346–2355. doi: 10.1091/mbc.E06-07-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1094–L1103. doi: 10.1152/ajplung.2001.280.6.L1094. [DOI] [PubMed] [Google Scholar]
- 28.Singh S, Wu S, Varney M, Singh AP, Singh RK. CXCR1 and CXCR2 silencing modulates CXCL8-dependent endothelial cell proliferation, migration and capillary-like structure formation. Microvasc Res. 2011;82:318–325. doi: 10.1016/j.mvr.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369–3376. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 30.Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165:5269–5277. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 31.Dwyer J, Hebda JK, Le Guelte A, Galan-Moya EM, Smith SS, Azzi S, Bidere N, Gavard J. Glioblastoma cell-secreted interleukin-8 induces brain endothelial cell permeability via CXCR2. PLOS One. 2012;7:e45562. doi: 10.1371/journal.pone.0045562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petreaca ML, Yao M, Liu Y, Defea K, Martins-Green M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol Biol Cell. 2007;18:5014–5023. doi: 10.1091/mbc.E07-01-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A, Gaggar A, Shastry S, Rowe SM, Shim YM, Hussell T, Blalock JE. A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science. 2010;330:90–94. doi: 10.1126/science.1190594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jozwiak M, Silva S, Persichini R, Anguel N, Osman D, Richard C, Teboul JL, Monnet X. Extravascular lung water is an independent prognostic factor in patients with acute respiratory distress syndrome. Crit Care Med. 2013;41:472–480. doi: 10.1097/CCM.0b013e31826ab377. [DOI] [PubMed] [Google Scholar]
- 35.Perkins GD, McAuley DF, Thickett DR, Gao F. The β-agonist lung injury trial (BALTI): A randomized placebo-controlled clinical trial. Am J Respir Crit Care Med. 2006;173:281–287. doi: 10.1164/rccm.200508-1302OC. [DOI] [PubMed] [Google Scholar]
- 36.Xu X, Jackson PL, Tanner S, Hardison MT, Abdul Roda M, Blalock JE, Gaggar A. A self-propagating matrix metalloprotease-9 (MMP-9) dependent cycle of chronic neutrophilic inflammation. PLOS One. 2011;6:e15781. doi: 10.1371/journal.pone.0015781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Force ADT, Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS. Acute respiratory distress syndrome: The Berlin definition. JAMA. 2012;307:2526–2533. doi: 10.1001/jama.2012.5669. [DOI] [PubMed] [Google Scholar]
- 38.Scott DW, Patel RP. Endothelial heterogeneity and adhesion molecules N-glycosylation: Implications in leukocyte trafficking in inflammation. Glycobiology. 2013;23:622–633. doi: 10.1093/glycob/cwt014. [DOI] [PubMed] [Google Scholar]
- 39.Scott DW, Chen J, Chacko BK, Traylor JG, Jr, Orr AW, Patel RP. Role of endothelial N-glycan mannose residues in monocyte recruitment during atherogenesis. Arterioscler Thromb Vasc Biol. 2012;32:e51–e59. doi: 10.1161/ATVBAHA.112.253203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reddy AT, Lakshmi SP, Kleinhenz JM, Sutliff RL, Hart CM, Reddy RC. Endothelial cell peroxisome proliferator–activated receptor γ reduces endotoxemic pulmonary inflammation and injury. J Immunol. 2012;189:5411–5420. doi: 10.4049/jimmunol.1201487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim KH, Kwun MJ, Choi JY, Ahn KS, Oh SR, Lee YG, Christman JW, Sadikot RT, Han CW, Joo M. Therapeutic effect of the tuber of Alisma orientale on lipopolysaccharide-induced acute lung injury. Evid Based Complement Alternat Med. 2013;2013:863892. doi: 10.1155/2013/863892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.