

Abstract
Purine biosynthesis requires ten enzymatic transformations to generate inosine monophosphate. PurF, PurD, PurL, PurM, PurC, and PurB are common to all pathways, while PurN or PurT, PurK/PurE-I or PurE-II, PurH or PurP, and PurJ or PurO catalyze the same steps in different organisms. X-ray crystal structures are available for all 15 purine biosynthetic enzymes, including seven ATP-dependent enzymes, two amidotransferases and two tetrahydrofolate-dependent enzymes. Here we summarize the structures of the purine biosynthetic enzymes, discuss similarities and differences, and present arguments for pathway evolution. Four of the ATP-dependent enzymes belong to the ATP-grasp superfamily and two to the PurM superfamily. The amidotransferases are unrelated with one utilizing an NTN-glutaminase and the other utilizing a triad glutaminase. Likewise the tetrahydrofolate-dependent enzymes are unrelated. Ancestral proteins may have included a broad specificity enzyme instead of PurD, PurT, PurK, PurC, and PurP, and a separate enzyme instead of PurM and PurL.
Keywords: purine biosynthesis, protein evolution, ATP-grasp superfamily, PurM superfamily, amidotransferases
Introduction
In most organisms, inosine 5′-monophosphate (IMP) is formed from small molecule precursors via the purine biosynthetic pathway (Figure 1). The details of the basic biosynthetic pathway were worked out by Buchanan and coworkers in the 1950’s using pigeon and chicken liver enzymes [1]. This pathway requires ten enzymatic steps while consuming four ATP molecules (Figure 2). IMP is converted to guanosine 5′-monophosphate (GMP) or adenosine 5′-monophosphate (AMP) by subsequent enzymes. IMP, AMP and GMP are also generated via the purine salvage pathway, which is the sole pathway to purine nucleotides in some parasitic organisms. Because of the critical role of purine nucleotides in the synthesis of RNA and DNA, the purine biosynthetic pathway has long been considered an important target for anticancer, antiviral and antimicrobial drug development.
Figure 1.

Precursors of inosine 5′-monophosphate. R5P is ribose 5′-phosphate.
Figure 2.

The purine biosynthetic pathway. Steps 1–10 define the original pathway worked out by Buchanan. Steps 3a, 4a, 6a, 6b, 9a and 10a represent variations of the pathway found in other organisms.
In recent years several variations to the basic purine biosynthetic pathway found in vertebrates have been reported (Figure 2). In Escherichia coli and related organisms, purine biosynthesis requires an additional enzyme, PurK, and thus utilizes 11 enzymatic steps while consuming five molecules of ATP [2]. In mammals, the third step of the pathway (PurN) utilizes the cofactor N10-formyltetrahydrofolate (N10-formylTHF) as the source of the C(8) carbon atom. E. coli can also utilize an alternate enzyme (PurT), which catalyzes the ATP-dependent ligation of formate and GAR [3], and some archeabacteria rely entirely on PurT. Most recently M. jannaschii, which lacks the gene for the folate dependent bifunctional AICAR transformylase/IMP cyclohydrolase (PurHJ), has been shown to express an ATP-dependent FAICAR synthetase (PurP) and a separate IMP cyclohydrolase (PurO) [4]. Currently, 15 unique enzymes are associated with purine biosynthesis; however, it is possible that other variations in the basic purine biosynthetic pathway will be discovered as the results of additional genome sequencing projects become available.
Variations are also found in gene organization. For example, in E. coli all of the enzymes except those for steps 9 and 10 are separate gene products, while in higher organisms the enzymes for steps 6 and 7 are fused, as are the enzymes for steps 2, 3 and 5. Other variations are distributed throughout the species. Within the purine biosynthetic pathway, some mechanistic strategies are used in more than one step, suggesting evolutionary links between the corresponding enzymes. For example, four enzymes (PurD, PurT, PurK and PurP) are structurally homologous and each utilizes an acylphosphate intermediate to catalyze the coupling of an amino group of one substrate to the carboxylate group of the other (Figure 3). This structural and mechanistic similarity extends beyond the purine biosynthetic pathway and includes biotin carboxylase [5], D-alanine-D-alanine ligase [6] and many others. This enzyme superfamily has been named ATP-grasp [7]. Despite the importance of the acylphosphate intermediate, it has not yet been trapped in an enzyme crystal structure. Class I and II PurEs represent another example of structurally and functionally related purine biosynthetic enzymes [8]. Class II enzymes catalyze the direct carboxylation of AIR to form CAIR, while class I enzymes require NCAIR, which is the product of PurK.
Figure 3.

The proposed acylphosphate intermediate in the reactions catalyzed by PurD, PurK, PurT and PurP. Nu: for PurD is the amino group of PRA, for PurK, the 5-amino group of AIR, for PurT, the amino group of GAR and for PurP, the amino group of AICAR. R5P is ribose 5′-phosphate. The amide group formed in each reaction is indicated by a dashed box.
The structural representatives for all known purine biosynthetic enzymes were worked out over the last two decades. The available structures provide insight into drug design, enzyme mechanism and protein evolution. In this review we will summarize the structures of the 15 known purine biosynthetic enzymes. We will also discuss the structural similarities and differences, and their implications for protein evolution. The naming of the purine biosynthetic enzymes and intermediates is sometimes confusing. Throughout this review enzymes are designated according to their gene names and pathway intermediates by their abbreviations. Table 1 (intermediate names) and Table 2 (enzyme names) are provided as a reference for the pathway shown in Figure 2.
Table 1.
Intermediates of purine biosynthesis
| intermediate abbreviation | product of step N | intermediate name |
|---|---|---|
| PRPP | - | 5-phosphoribosylpyrophosphate |
| PRA | 1 | 5-phospho-D-ribosylamine |
| GAR | 2 | glycinamide ribonucleotide |
| FGAR | 3, 3a | N-formylglycinamide ribonucleotide |
| FGAM | 4, 4a | N-formylglycinamidine ribonucleotide |
| AIR | 5 | aminoimidazole ribonucleotide |
| NCAIR | 6a | N5-carboxyaminoimidazole ribonucleotide |
| CAIR | 6, 6b | carboxyaminoimidazole ribonucleotide |
| SAICAR | 7 | N-succinocarboxamide-5-aminoimidazole ribonucleotide |
| AICAR | 8 | aminoimidazole-4-carboxamide ribonucleotide |
| FAICAR | 9, 9a | 5-formamido-4-imidazolecarboxamide ribonucleotide |
| IMP | 10, 10a | inosine monophosphate |
Table 2.
Enzymes of purine biosynthesis
| gene product | step N | enzyme name |
|---|---|---|
| PurF | 1 | amidophosphoribosyltransferase |
| PurD | 2 | GAR synthetase |
| PurN | 3 | GAR transformylase |
| PurT | 3a | FGAR synthetase |
| PurL | 4 | FGAM synthetase I |
| PurLQS | 4a | FGAM synthetase II |
| PurM | 5 | AIR synthetase |
| PurE II | 6 | AIR carboxylase |
| PurK | 6a | NCAIR synthetase |
| PurE I | 6b | NCAIR mutase |
| PurC | 7 | SAICAR synthase |
| PurB | 8 | adenylosuccinate lyase |
| PurH | 9 | AICAR transformylase |
| PurP | 9a | FAICAR synthetase |
| PurJ | 10 | IMP cyclohydrolase I |
| PurO | 10a | IMP cyclohydrolase II |
Step 1: Biosynthesis of phosphoribosylamine (PRA)
The first committed step of de novo purine biosynthesis is the conversion of PRPP to PRA catalyzed by PurF [9,10]. This transformation involves two half-reactions; glutamine is hydrolyzed to glutamate and ammonia, and ammonia is then transferred to PRPP to generate PRA while releasing pyrophosphate [11]. The two half-reactions occur in separate domains, which are connected by an ammonia channel, and the catalysis is tightly coupled [12,13]. Besides its catalytic function, PurF is also involved in regulation of purine biosynthesis via allosteric feedback inhibition by guanine and adenine nucleotides [14,15]. Two types of PurF have been characterized. The first type, present in higher vertebrates, plants, flies, cyanobacteria, and Gram-positive bacteria, contains a structural Fe4S4 cluster and a cleavable N-terminal propeptide, and is exemplified by Bacillus subtilis PurF in literature [10,16,17]. The second type, lacking both the metal center and the propeptide, is found in yeast, Neurospora, Haemophilus, and Gram-negative organisms, and the enzyme from E. coli has been characterized [11].
PurF has been studied extensively in both E. coli and B. subtilis, and seven crystal structures describing various complexes in both organisms are available [12–14,16,18]. The two enzymes share 40% sequence identity and are structurally homologous with the highest degree of conservation found in the catalytic regions and the ammonia channel [18]. While in the crystal structures PurF forms a tetramer (Figure 4A), in solution both the dimeric and tetrameric states have been observed [17,19]. A lesser degree of conservation between the two organisms is observed for the monomer interfaces as well as the regulatory sites. The binding site for the Fe4S4 cluster in B. subtilis is filled with water in the E. coli equivalent.
Figure 4.

Structures of PurF (step 1), PurD (step 2), PurN (step 3) and PurT (step 3a). (A) Ribbon diagram of the E. coli PurF (PDB: 1ECB) tetramer. Each monomer is in a different color, and the inhibitor, GMP, is in ball-and-stick representation, colored by atom type (with carbon black, oxygen red, nitrogen blue, sulfur yellow, and phosphorus orange, throughout). (B) Ribbon diagram of a monomer of E. coli PurF (PDB: 1ECC), color-coded by secondary structure. Ligands, Gln and cPRPP, bound in the active sites are in ball-and-stick representation. (C) Ribbon diagram of Geobacillus kaustophilus PurD (PDB: 2YS6), color-coded by secondary structure. Glycine and AMP, found in the active site, are in ball-and-stick representation. (D) Ribbon diagram of E. coli PurN (PDB: 1CDE), color-coded by secondary structure. The substrate glycinamide ribonucleotide and the inhibitor 5-deaza-5,6,7,8-tetrahydrafolate are in ball-and-stick representation. (E) Ribbon diagram of the E. coli PurT dimer (PDB: 1KJ8), color coded by monomer. The substrate, glycinamide ribonucleotide (GAR), and ATP are shown in ball-and-stick representation. (F) Ribbon diagram of the E. coli PurT monomer, color-coded by secondary structure. GAR and ATP are shown in ball-and-stick representation.
The PurF monomer consists of two domains (Figure 4B). The N-terminal glutaminase domain belongs to the N-terminal nucleophile (NTN) hydrolase family, the fold being a four-layer structure of two anti-parallel β-sheets sandwiched between two layers of α-helices [16]. The C-terminal domain has a fold characteristic of type I phosphoribosyltransferases (PRT); the main feature is a parallel β-sheet packed between helices [16].
The active site of the glutaminase domain has been mapped out in the presence of a covalent intermediate formed by a glutamine analog with the key N-terminal cysteine, the nucleophile responsible for ammonia liberation (Figure 5A) [12,13]. The reaction is thought to proceed through a tetrahedral intermediate, and two invariant residues, Asn101 and Gly102 (E. coli numbering), are thought to be critical for stabilizing the oxyanion. Active site Arg73 ligates the carboxylate moeity of the substrate, and is important for substrate specificity and coupling of catalysis.
Figure 5.

Active sites of PurF. (A) Schematic diagram of the active site residues and the covalently bound ligand, Gln, in the PurF glutaminase domain. Dashed lines represent hydrogen bonds. (B) Schematic representation of the active site residues and the bound ligand, cPRPP, in the PurF phosphoribosyl transferase domain.
The active site of the PRT domain forms completely only in the catalytically active state of the enzyme and has been described in the presence of a substrate analog bound with a divalent metal ion (Figure 5B) [13]. Six basic and two acidic residues create a highly-charged active site pocket, complementary with the highly-charged substrate. The divalent metal ion, important for catalysis, binds to the pyrophosphate tail and is coordinated by four oxygen atoms of the substrate and two water molecules. The role of the PRT active site is to provide residues for proper positioning of the substrate rather than those of specific catalytic function. When in the active state, the enzyme binds glutamine in a catalytically relevant conformation and ammonia is passed through a transiently formed channel. Allosteric inhibition of PurF by GMP and ADP is achieved via stabilization of the inactive state of the enzyme with an inaccessible glutaminase active site and only partially formed ammonia channel [9,13,14,18].
Step 2: Biosynthesis of glycinamide ribonucleotide (GAR)
Only one known enzyme, PurD in all organisms, catalyzes the conversion of PRA to GAR. In step two, glycine is ligated to the PRA amino group in an ATP-dependent manner and provides atoms C(4), C(5) and N(7) of the purine base. Only one structure has been published [20]; however, PurD structures for T. thermophilus, G. kaustophilus, Aquifex aeolicus and human forms have been deposited in Protein Data Bank (PDB). The human form consists of the N-terminal domain of the PurD-PurM-PurN trifunctional enzyme. The published structure from E. coli did not contain substrate or product; however, several of the deposited structures contain ADP or ATP, and one structure contains glycine.
The X-ray crystal structures reveal that PurD is a member of the ATP-grasp superfamily. The overall structure is monomeric and can be divided into three domains (A, B and C) (Figure 4C). The A and C-domains form a closely packed core. The B-domain extends away from the core and forms a flexible flap over the active site. The N-terminal A-domain consists of about 95 residues. The fold consists of a five-stranded β-sheet flanked by two α-helices on each side. The fourth helix of this domain is disrupted after the first turn before returning to an α-helical conformation. The A-domain is joined to the B-domain by a pair of α-helices that are separated by a single residue. The torsion angles for this residue are such that the two helices are roughly perpendicular to each other. The B-domain consists of about 70 residues with a four-stranded antiparallel β-sheet. One side of the sheet faces the active site while the other side is flanked by two α-helices. The C-domain contains about 235 residues. The fold consists of an eight-stranded antiparallel β-sheet flanked by six α-helices. The C-terminal domain can further be divided into two subdomains (C1 and C2). The first consists of the first five β-strands and the first four α-helices, while the second consists of the final three β-strands and the last two α-helices. This subdomain also contains some small areas of an additional β-sheet.
The active site of PurD can be divided into three subsites: the ATP binding site, the PRA binding site and the glycine binding site (Figure 6). The ATP binding site forms between the A and B-domains. The B-domain contains a flexible phosphate binding loop (P-loop) that is generally disordered in structures lacking ATP while ordered or partially ordered in those with ATP bound. The adenine location is consistent among the X-ray structures but the ribose and phosphate sites are variable, suggesting that the ATP is not fully ordered until all three substrates are bound and the B-domain flap has closed. The glycine site is located near the predicted ATP γ-phosphate site and is hydrogen bonded to a conserved arginine residue. None of the X-ray structures contain the substrate PRA or product GAR; however, the location of the PRA site can be inferred from the positions of ATP and glycine, and from the presence in some PurD structures of a fortuitously-bound sulfate or phosphate that is near the expected site of the 5′-phosphate group of the substrate. PurD is thought to function through a glycylphosphate intermediate that is generated by ATP-dependent phosphorylation of the glycine carboxylate group. Subsequent nucleophilic attack by the PRA amino group generates the final products, GAR and phosphate. The arrangement of ligand binding sites is consistent with this mechanism.
Figure 6.

Active site of PurD. Schematic diagram of the active site residues and ligands, AMP and glycine. Dashed lines represent hydrogen bonds.
Step 3: Biosynthesis of N-formylglycinamide ribonucleotide (FGAR)
Two different enzymes catalyze the third step of purine biosynthesis, which generates FGAR. PurN is found in most organisms and utilizes the cofactor N10-formylTHF as the source of the purine C(8) carbon atom. PurT is found together with PurN in E. coli and related microorganisms. PurT catalyzes the ATP-dependent ligation of GAR and formate to generate FGAR. The structure of E. coli PurN was determined almost simultaneously by two groups in 1992 [21,22] and was the first enzyme from the purine biosynthetic pathway to have its structure determined by X-ray crystallography (Figure 4D). In higher organisms PurN is the C-terminal domain of a trifunctional enzyme that also contains PurD and PurM activities, while separate genes encode these three enzymes in bacteria. Human PurN is a target for the design of anticancer agents and a number of crystal structures of human PurN-inhibitor complexes have been published [23–26].
The structure of PurN is formed by an αβα three-layer sandwich. The core β-sheet contains seven strands and is flanked by three α-helices on one side, two on the other and a sixth along the edge. The structure can be further divided into two domains. The N-terminal domain is compact and contains four parallel β-strands and the first four α-helices. The C-terminal domain is less compact and contains three antiparallel β-strands, two of which are parallel to the first five, and the remaining two α-helices. The active site is located at a cleft formed by these two domains (Figure 7) [21,27,28]. The N10-formylTHF binding site is formed primarily by three loops: one from the N-terminal domain and two from the C-terminal domain. The GAR binding site is slightly more exposed to the solvent and is positioned by the active site to accept the formyl group. A water molecule has been postulated to mediate the transfer of the formyl group [29].
Figure 7.

Active site of PurN. Schematic diagram of the active site residues, substrate GAR (A) and inhibitor, 5-deazatetrahydrfolate (B). Dashed lines represent hydrogen bonds.
The structure of PurT from E. coli belongs to the ATP-grasp superfamily [30,31]. PurT is a homodimer (Figure 4E) and like PurD each monomer contains A, B and C domains. The A and C-domains form the core of the structure, while the B-domain forms a flexible lid over the active site (Figure 4F). The N-terminal A-domain consists of approximately 120 residues that form a five-stranded parallel β-sheet flanked by two helices on each side. The A-domain is joined to the B-domain by two α-helices separated by only one residue. The B-domain is formed by approximately 65 residues. The domain contains a four-stranded antiparallel β-sheet. One side of the sheet faces the active site while the opposite side is flanked by two α-helices. The C-domain consists of approximately 195 residues. The central feature of the domain is an eight-stranded antiparallel β-sheet. The domain also contains two other regions of antiparallel β-sheet, one containing three strands and the other containing two, and four α-helices. The last 65 residues of the C-domain form a small subdomain.
The active site contains subsites for ATP, GAR and formate (Figure 8). The ATP binding site is formed primarily by the A and B-domains while the GAR binding site is formed primarily by the A and C-domains. Neither of the available crystal structures contains formate; however, the binding site is constrained to lie between the ATP γ-phosphate and the GAR amino group to which it will be transferred. The mechanistic strategy for PurT involves the formation of formylphosphate intermediate [3] followed by nucleophilic attack at the formyl carbon atom by the GAR amino group.
Figure 8.

Active site of PurT. Schematic diagram of the active site residues, GAR and ATP. Hydrogen bonds are indicated by dashed lines, except for those between the phosphate groups of ATP and Gln165, Arg114 and the P-loop, which are omitted for clarity.
Step 4: Biosynthesis of N-formylglycinamidine ribonucleotide (FGAM)
PurL catalyzes the fourth step of the purine biosynthetic pathway, ATP-dependent amidation of FGAR to FGAM. The amide oxygen of FGAR is activated by the γ-phosphate of an ATP molecule, resulting in formation of a proposed iminophosphate intermediate [32]. This intermediate is then attacked by nucleophilic ammonia generated from a glutamine molecule. FGAM, ADP and glutamate are the products of this transformation [33,34].
Two types of PurL have been characterized: large PurL and small PurL. Large PurL is found in eukaryotes and Gram-negative bacteria, and it is a multidomain protein consisting of a single polypeptide chain. The structure of a large PurL from Salmonella typhimurium described the domain organization of this enzyme [35] (Figure 9A). Large PurL has three main domains: the glutaminase, the FGAM synthetase, and the N-terminal domains. The glutaminase domain is responsible for the production of ammonia, which is then channeled to the FGAM synthetase domain. The N-terminal domain is thought important for the formation of the ammonia channel and coupling between the two catalytic domains. Small PurL is found in Gram-positive bacteria and archaea and requires two additional gene products, PurQ and PurS, for activity [36] (Figure 9B). Structures of all three components have been described [37–41] (Figure 9C). Small PurL possesses the FGAM synthetase activity and is structurally homologous to the FGAM synthetase domain of large PurL [39,41]. PurQ is the glutaminase, and a PurS dimer is structurally homologous to the N-terminal domain of large PurL [37,38]. The complex structure of PurL, PurQ, and PurS, in a ratio of 1:1:2, have been recently determined from Thermotoga maritima. Glutamine and a nucleotide, either ATP or ADP, are required for its formation (Figure 9B) [42].
Figure 9.

Structures of PurL (step 4), PurM (step 5) and PurK (step 6a). (A) Ribbon diagram of the large PurL structure from S. typhimurium (PDB: 1T3T) color-coded by secondary structure. Ligands, Gln and ADP, are in ball-and-stick representation. (B) Ribbon diagram of the PurLQS complex structure from T. maritima (PDB: 3D54), each monomer in the complex is in a different color. Ligands, Gln and ADP, are in ball-and-stick representation. (C) Ribbon diagrams of PurL, PurQ and PurS monomers from T. maritima, individually, color-coded by secondary structure. Ligands are in ball-and-stick representation. Ligands bound in the active of PurL are FGAR and AMPPCP (PDB: 2HS4), and the ligand in the auxiliary site is ATP (PDB: 2HS0). The ligand in PurQ is Gln. (D) Ribbon diagram of the PurM dimer from E. coli (PDB: 1CLI) color coded by monomer. (E) Ribbon diagram of the monomer of PurM, color-coded by secondary structure. (F) Ribbon diagram of the E. coli PurK dimer (PDB: 1B6S), color coded by monomer and with ADP in ball-and-stick representation. (G) Ribbon diagram of the PurK monomer, color-coded by secondary structure and with ADP in ball-and-stick representation.
Small PurL (the FGAM synthetase domain) is the largest of the three domains and consists of four subdomains all with an α/β structure (Figure 9C). The central two subdomains form an eight-stranded β-barrel surrounded by long α-helices. The two additional subdomains contain mixed β-sheets and small α-helices and flank the central β-barrel on each side. This organization results in a pseudo twofold symmetry with the twofold axis passing though the axis of the central β-barrel. The main feature of PurQ (the glutaminase domain) is a large mixed β-sheet, which is flanked by α-helices on both sides. The PurS monomer contains a three-stranded β-sheet and two long helices. In the PurS dimer (the N-terminal domain), the two β-sheets come together forming a six-stranded β-sheet with the four helices clustered on one side of it.
PurLQS contains two active sites: one in the FGAM synthetase domain and one in the glutaminase domain. The active site responsible for the FGAM production is located in one of the two clefts formed by the central β-barrel and the flanking subdomains of the FGAM synthetase domain. Five structures of the complexed small PurL from Thermotoga maritima describe the nature of binding of the substrates, FGAR and Mg2+-ATP, in this site (Figure 10A) [41]. The residues responsible for FGAR binding are polar and charged: a histidine and two serine residues ligate the 5′-phosphate, the hydroxyl groups of the ribose form hydrogen bonds to a glutamate residue, while the formyl glycine arm is hydrogen-bonded to two histidines, two glutamine residues, and an arginine residue. All of the residues involved in binding of FGAR are strictly conserved. The two histidine residues involved in the formyl glycine arm binding are involved in catalysis and are thought to function as general acid-bases in the formation of the proposed iminophosphate intermediate [41].
Figure 10.

Active site of PurL. (A) Schematic diagram of the PurL active site showing the ligands, FGAR and AMPPCP, and the residues interacting with the ligands. (B) Schematic diagram of the PurQ active site showing the covalently bound ligand, Gln, and the residues interacting with the ligand. Dashed lines represent hydrogen bonds.
The ATP binding pocket contains hydrophobic residues accommodating the adenine base with N1 and N6 hydrogen-bonding to an asparagine residue; the ribose hydroxyl groups form no interactions, while the phosphate tail is bound primarily through interactions with two Mg2+ ions. The ligands to the Mg2+ ions include two highly conserved aspartates, a glutamate and an asparagine residue. The binding mode of the ATP molecule reveals a lack of positively charged residues generally involved in nucleotide binding. This characteristic leads to the proposal that phosphate transfer is dissociative with a lesser build up of the negative charge in the transition state [41].
In the active site of the glutaminase domain, a glutamyl-thioester intermediate is bound to a strictly conserved cysteine residue [35] (Figure 10B). This active site contains a classic Cys-His-Glu catalytic triad responsible for the catalysis of ammonia formation. The intermediate forms hydrogen bonds mostly with main chain atoms as well as a side chain of a glutamine residue.
The FGAM synthetase domain contains an auxiliary nucleotide binding site in addition to the active site. This site is located in the second cleft formed by the central β-barrel and the flanking subdomains, and it is related to the active site by the pseudo twofold axis. While in large PurL the auxiliary site is thought to be an evolutionary remnant, in small PurL this site is important for complex formation [36]. The PurLQS complex reveals a dramatic conformational change in the loop region of the FGAM synthetase associated with the auxiliary nucleotide binding, which then allows for docking of the glutaminase, PurQ. Additionally, the availability of structural information for both large PurL and the PurLQS complex allowed for the determination of the likely location for the ammonia channel formation and mechanistic proposals for catalytic coupling as well as the importance of the PurS domain in both processes.
Step 5: Biosynthesis of aminoimidazole ribonucleotide (AIR)
The fifth step in purine biosynthesis is the ATP-dependent conversion of FGAM to AIR catalyzed by PurM. In this reaction the five-membered ring of the purine base is formed; the formyl oxygen is activated by the γ-phosphate of the ATP for the nucleophilic attack by the N1 of FGAR resulting in ring closure [43]. This reaction is chemically very similar to that catalyzed by PurL (Figure 11), the enzyme involved in the preceding step, the major difference being that no exogenous ammonia source is necessary for this intramolecular conversion. In bacteria PurM is encoded separately, and in mammals it is part of a trifunctional enzyme, which includes PurD and PurN [44].
Figure 11.

The proposed iminophosphate intermediate in reactions catalyzed by PurL and PurM. R5P is ribose 5′-phosphate. The amidine formed in each reaction is highlighted by a dashed circle.
The structure of PurM from E. coli has been published [45] and a structure of PurM from Bacillus anthracis has been deposited in the PDB. Both of these organisms encode an unfused PurM and are both homodimers (Figure 9D). The PurM monomer consists of two domains, and both domains have an α/β structure (Figure 9E). The N-terminal domain consists of a four-stranded mixed β-sheet flanked by four α-helices; the C-terminal domain contains a six-stranded mixed β-sheet, with seven α-helices located along one edge of the sheet. The dimer interface is primarily formed by the N-terminal domains of both monomers, resulting in the formation of an eight-stranded β-barrel as the central feature of the enzyme, flanked by the C-terminal domains, and with the twofold symmetry axis of the dimer running through the axis of the β-barrel.
The active site of PurM is located in the cleft between the N- and C-terminal domains, and there are two active sites per PurM homodimer. Although a detailed description of the active site of PurM is unavailable due to the lack of ligand-complexed structures, its location is known with fair certainty based on the primary sequence conservation, the homology with the PurL structure, and the presence of a sulfate ion binding pocket thought to be the location of the 5′-phosphate of FGAM.
The structure of PurM revealed two histidines, His190 and His247 (E. coli numbering), in the active site cleft that could be involved in catalysis analogous to PurL, as well as residues, Asp65 and Asp94, involved in ATP and Mg2+ ion binding; all of these residues are conserved in both PurM and PurL [45]. Importantly, structural characterization of PurM resulted in the discovery of a new family of ATP-binding enzymes, referred to as the PurM superfamily, which is discussed below.
Step 6: Biosynthesis of carboxyaminoimidazole ribonucleotide (CAIR)
The generation of CAIR occurs in different ways for bacteria and higher organisms. In bacteria, two enzymes are required. CAIR is first converted to NCAIR in an ATP-dependent manner by ligation of bicarbonate and the N(5) amino group by PurK.
NCAIR is subsequently converted to CAIR by class I PurE in an unusual mutase reaction. The carboxylate carbon becomes atom C(6) of the purine base and one of the carboxylate oxygen atoms becomes atom O(6). In vertebrates, AIR is directly carboxylated by CO2 at the C(4) position using class II PurE. Class I and class II PurEs are structurally homologous and share considerable sequence identity, with the main differences occurring in only two active site regions. In higher organisms PurC and PurE are fused to form a bifunctional enzyme. In bacteria PurE is usually a separate gene product but is sometimes fused with PurK. Finally, in methanogenic bacteria PurE is fused with PurE′, where PurE and PurE′ share about 35% sequence identity.
The structure of E. coli PurK showed that it is a member of the ATP-grasp superfamily [46]. PurK is a homodimer (Figure 9F) and, like other ATP-grasp superfamily members, the monomer can be divided into A, B and C domains (Figure 9G). The A and C-domains form a compact core structure and the B-domain extends away from the core and forms a flexible active site flap. The PurK A-domain consists of about 75 amino acids and is smaller than the corresponding A-domains of PurD and PurT. The PurK A-domain fold is a three-stranded parallel β-sheet flanked by two α-helices. The connector to the B-domain consists of two α-helices that are approximately perpendicular to each other. The B-domain contains about 65 amino acid residues and, like the PurD and PurK B-domains, consists of a four-stranded β-sheet facing the active site on one side and flanked by two α-helices on the other. A flexible P-loop is located between the second and third strands of the four-stranded β-sheet. The C-domain consists of a central eight-stranded antiparallel β-sheet flanked by six α-helices. The C-domain can be further divided into one subdomain containing the first five β-strands and the first three α-helices (C1) and another subdomain containing the last three β-strands and the last three α-helices (C2). The C1-subdomain is more similar to that of PurD and PurT than the C2-subdomain. The active site contains ATP, AIR and bicarbonate subsites. The ATP site as defined by the PurK-ADP complex is consistent with that of PurD, PurT and other ATP-grasp enzymes (Figure 12). The AIR binding site was not directly observed but modeling studies suggest that it is in the same location as the PRA site of PurD and the GAR site of PurT. The PurK reaction is thought to occur via a carboxyphosphate intermediate. Subsequent nucleophilic attack by the AIR amino group generates the products NCAIR and phosphate.
Figure 12.
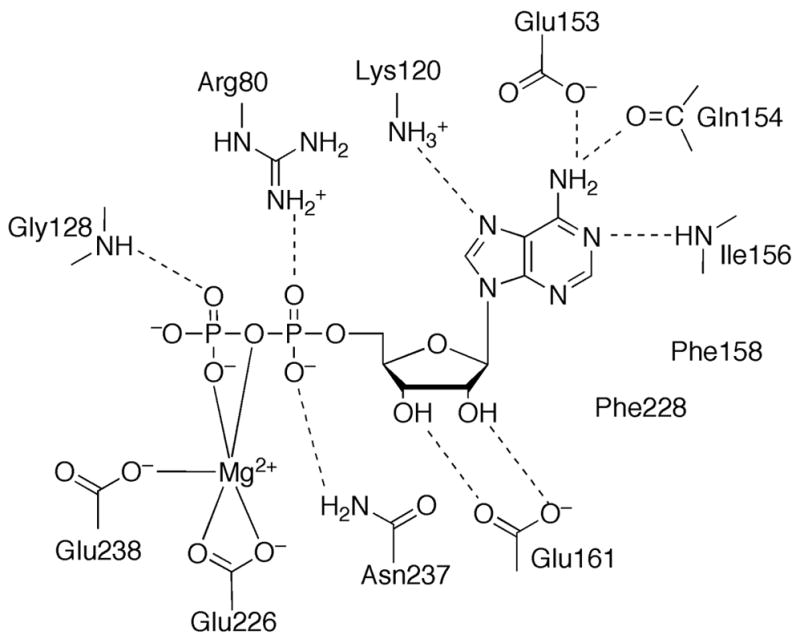
Active site of PurK. Schematic diagram of the active site residues and ADP. Dashed lines represent hydrogen bonds.
Structural studies reveal that both class I (Figure 13A) [8] and class II (Figure 13C) [47] PurEs are homooctamers with 422 symmetry. The class I and class II PurE monomers are very similar and have flavodoxin-like topologies. The core structure consists of a central five-stranded parallel β-sheet that is flanked by three α-helices on one side and two on the other (Figure 13B). A final C-terminal α-helix extends away from the core towards an adjacent monomer. Details of the active site come from the structures of E. coli PurE complexes [8,48] (Figure 14). Each of the eight active sites is located in a cleft formed by three monomers. The phosphate binding site is the most exposed and utilizes hydrogen bonds to two serine residues and an arginine residue. The ribose hydroxyl groups form hydrogen bonds with an aspartate residue and a glycine amide group. The imidazole forms hydrogen bonds with two backbone amide groups, two backbone carbonyl groups and an absolutely conserved serine residue. A conserved histidine is believed to serve as the acid for the decarboxylation at N(5) and as the base for the carboxylation at C(5). Structural studies suggest that after decarboxylation a small rotation of the imidazole properly positions C(5) for the carboxylation step [48]. The structure of bifunctional human PurCE was determined without bound ligands [47]. Biochemical studies demonstrated that class II PurE does not accept NCAIR as a substrate but only catalyzes the carboxylation of AIR, which is equivalent to the second half of the class I PurE reaction. Superposition of the class I and class II PurE active sites suggests that the two enzymes use different residues in the phosphate binding site which may play a role in determining substrate specificity. Several structures of Acetobacter aceti PurE have also been reported [49,50].
Figure 13.

Structures of class I PurE (Step 6b), PurCE (Steps 6 and 7), PurC (Step 7) and PurB (Step 8). (A) Ribbon diagram of the E. coli PurE octamer (PDB: 2ATE) with the eight monomers shown in different colors. 4-Nitroaminoimidazole ribonucleotide (nitroAIR), an analog of the product carboxyaminoimidazole ribonucleotide (CAIR), is shown in ball-and-stick representation. (B) Ribbon diagram of the PurE monomer, color-coded by secondary structure and with nitroAIR shown in ball-and-stick representation. (C) The ribbon diagram of bifunctional human PurCE (PDB: 2H31). The octameric PurE core is in yellow, decorated with four PurC dimers in red. (D) Ribbon diagram of the E. coli PurC dimer (PDB: 2GQS) color coded by monomer. The substrate carboxyaminoimidazole ribonucleotide (CAIR) and associated ADP are shown in ball-and-stick representation. (E) Ribbon diagram of the PurC monomer color coded by secondary structure and with CAIR and ADP shown in ball-and-stick representation. (F) Ribbon diagram of the E. coli PurB tetramer (PDB: 2PTR) color-coded by monomer. PurB also catalyzes the second and final step in the conversion of IMP to AMP. The ligand adenylosuccinate bound at each active site is shown in ball-and-stick representation. (G) A monomer of PurB color-coded by secondary structure and with the ligand adenylosuccinate shown in ball-and-stick representation.
Figure 14.

Active site of Class I PurE. Schematic diagram of the active site residues associated with nitroAIR. Dashed lines represent hydrogen bonds.
Step 7: Biosynthesis of N-succinocarboxamide-5-aminoimidazole ribonucleotide (SAICAR)
In the seventh step of purine biosynthesis, PurC catalyzes the ligation of the carboxylate group of CAIR to the amino group of aspartate. The amino group provides the N(1) nitrogen atom of the final purine base and the remainder of the aspartate is eliminated as succinate in the subsequent step. In higher organisms PurC is fused to PurE to form a bifunctional enzyme referred to as PurCE (Figure 13C). In lower organisms PurC and PurE are not fused. The structure of the PurC from S. cerevesiae was the first structure to be determined for the seventh enzyme in the pathway [51]. Subsequently, the structures of PurCs from T. maritima [52] and E. coli [53] were determined and most recently the structure of the human PurCE fused enzyme was reported [47]. The yeast PurC was reported to be monomeric; however, all other PurCs appear to be dimeric (Figure 13D).
The structure of the PurC monomer (Figure 13E) resembles that of the ATP-grasp superfamily. The main core domain has a topology similar to that of the C-domain in PurD, PurT and PurK. PurC contains an ATP-binding flap domain reminiscent of the B-domain of the ATP-grasp superfamily but has a completely different topology and contains a five-stranded antiparallel β-sheet with topology 1↑2↓3↑5↓4↑. PurC lacks a domain corresponding to the A-domain of the ATP-grasp family. A number of PurC complexes have been reported [53,54] and map out the key active site residues (Figure 15). The ATP binding site is associated primarily with the flap domain. The CAIR binding site utilizes residues both from the flap domain and the core domain.
Figure 15.

Active site of PurC. Schematic diagram of the active site residues associated with ADP and CAIR. Dashed lines represent hydrogen bonds.
The structure of human PurCE is an octamer with 422 point symmetry (Figure 13C) [47]. PurE (C-terminal half) forms the core and is structurally homologous to the unfused bacterial PurE octamer. PurC dimers span the twofold axes and pack tightly against the PurE core; however, the PurC dimers do not contact each other. The PurE active sites as viewed in Figure 13C are above or below the plane while the PurC active sites are on the four sides. The structure does not suggest any obvious communication between the PurE and PurC active sites.
Step 8: Biosynthesis of aminoimidazole-4-carboxamide ribonucleotide (AICAR)
The eighth step of the de novo purine biosynthesis pathway is catalyzed by the gene product of purB. Unique in the pathway, PurB has dual substrate specificity, and consequently is responsible for two non-consecutive reactions utilizing the same active site: (1) conversion of SAICAR to AICAR in step 8 and (2) catalysis of the second step in the conversion of IMP to AMP. Both reactions release fumarate as a result of β-elimination, and presumably share the same catalytic residues and mechanism.
The structures of PurB have been determined from T. maritima [55], P. aerophilum [56], P. viva [57], E. coli [58], C. elegans, B. anthracis, and H. sapiens, the last three being PDB depositions. The structures belong to the aspartase/fumarase superfamily, also called the β-elimination superfamily. Members of the family, including argininosucccinate lyase, 3-carboxy-cis, cis-muconate lactonizing enzyme, L-aspartase, class II fumarase, and PurB, are all biologically active as a homotetramer (Figure 13F), with each monomer displaying a three-domain, mostly helical architecture (Figure 13G). Domains 1 and 3 are relatively more compact, and comprise approximately 95 residues from the N-terminal and 80 residues from the C-terminal parts of the polypeptide chain. Domain 2, which is the core of the structure, consists of approximately 250 residues forming an elongated up-down five helical bundle. The active site is contributed by all three domains, each coming from a different monomer.
The β-elimination reaction catalyzed by PurB is proposed to occur via a general acid-base mechanism, with the base deprotonating the Cβ to form a carbanion intermediate, and the acid protonating the leaving nitrogen and resulting in scission of the C-N bond. Although two histidine residues, His91 and His171 for the E. coli enzyme, have been considered as the catalytic acid and base, respectively, recent structures of mutant-substrate (adenylosuccinate) and product (AMP and fumarate) complexes provided new insights for the identity of the catalytic residues [58]. Although uncommon as a base catalyst, a conserved serine residue from a flexible loop region that covers the active site, Ser295, is proposed to abstract the proton from Cβ of the substrate (Figure 16) [58]. The structures also revealed that His91 is unlikely to be the acid catalyst since it is more than 4 Å away; however, either His171 or a water molecule can be the proton donor to the N(6) or N(1) atom to facilitate the bond breakage [58].
Figure 16.

Active Site of PurB. The chain id for each residue is given in parentheses. Hydrogen bonds are indicated by dashed lines. Proposed catalytic residues are highlighted by rectangular frames.
Step 9: Biosynthesis of 5-formamido-4-imidazolecarboxamide ribonucleotide (FAICAR)
In the penultimate step of the de novo purine biosynthesis pathway, a formyl group is transferred to AICAR to produce FAICAR. The reaction is catalyzed in bacteria and eukaryotes by the gene product of purHJ, a bi-functional enzyme (AICAR transformylase/IMP cyclohydrolase), which also catalyzes the following step to form final product IMP. In archaea, PurHJ is missing and PurP and PurO are responsible for steps 9 and 10, respectively.
While formed by one single polypeptide chain, the AICAR transformylase (PurH) and IMP cyclohydrolase (PurJ) activities of PurHJ reside in two functionally independent domains. Each domain can be individually expressed as an enzymatically active fragment [59]; however, the coupling of the two domains appears to be essential as the AICAR transformylase reaction favors the reverse direction by itself and the irreversible cyclization of FAICAR to IMP drives the formyl transfer in the forward direction [60]. PurHJ is also a target for the development of anticancer therapeutics, and effort has been devoted to searching for specific anti-folate reagents [61,62] as well as non-folate inhibitors [60].
PurHJ is folate-dependent, like PurN in step 3, transferring a formyl group from the cofactor N10-formylTHF to AICAR to generate FAICAR. Several structures of PurHJ have been determined in complex with substrate, analogs or inhibitors from G. gallus [60,62–65] and H. sapiens [61,66] and in the unliganded form from T. maritima. PurHJ is a homodimer (Figure 17A), in which the two monomers intertwine significantly forming an extensive dimer interface.
Figure 17.

Structures of PurHJ (Steps 9 and 10), PurP (Steps 9a) and PurO (Step 10a). (A) Ribbon diagram of human PurHJ (PDB: 1P4R) color-coded by monomers. Ligands AICAR, folate-based inhibitor, and XMP are shown in ball-and-stick representation. (B) The PurH domain colored by secondary structure elements. Ligands AICAR and folate-based inhibitor are shown in ball-and-stick representation. The N-terminus connecting to the PurJ domain is indicated by the asterisk. (C) The PurJ domain colored by secondary structural elements. The asterisk indicates the C-terminus connecting to the PurJ domain. The substrate analog XMP is shown in ball-and-stick representation. (D) The hexamer of M. jannaschii PurP (PDB: 2R7L) colored by monomer and viewed down the 3-fold symmetry. (E) Ribbon diagram of a monomer of PurP colored by secondary structural elements. ATP and AICAR are shown in ball-and-stick representation. (F) Ribbon diagram of the M. thermoautotrophicus PurO tetramer (PDB: 2NTK). The diagram is colored by monomer, with IMP shown in ball-and-stick representation at each active site. (G) A PurO monomer colored by secondary structural elements, with IMP shown in ball-and-stick representation.
The PurH domain of PurHJ consists of the C-terminal approximately 400 residues of the polypeptide chain (Figure 17B), and can be further divided into three sub-domains. The two large domains presumably resulted from gene duplication and have a conserved topology characterized as five-stranded mixed β-sheet 2↑1↓3↑4↑5↑ flanked by four α-helices that is also found in cytidine deaminase. A linker domain composed of four α-helices is inserted between the two bigger domains. The two duplicated domains together form an extended eleven-stranded β-sheet.
The active site for PurH is located in a cleft at the dimer interface (Figure 18A). While AICAR is mostly interacting with residues from one monomer, the folate binding site is primarily formed by residues from the opposite monomer. Based upon the structures of PurHJ complexed with substrate and substrate analogs, a direct formyl transfer mechanism was proposed [64]. Key residue His268 (human numbering, +1 for avian) acts as a catalytic base to deprotonate the 5-amino group of AICAR, which attacks N10-formylTHF. Residue Lys266 is proposed to stabilize the oxyanion transition state, and protonate the N10-THF leaving group of THF. Residue Asn431 is also in the vicinity, and, especially in a structure of human PurHJ, is found directly interacting with both AICAR and a THF analog [61] to provide stabilizing side chain hydrogen bonds.
Figure 18.

Active sites of PurH and PurJ. (A) Schematic diagram of the PurH active site formed at the dimer interface. Letters in parenthesis refer to the chain id for each residue. Hydrogen bonds are indicated by dashed lines. (B) Schematic diagram of the PurJ active site with XMP bound.
In archaea step 9 is carried out without the involvement of either folate or modified folates. Instead, as revealed by recent studies on M. jannaschii, a FAICAR synthetase, the product of the purP gene, catalyzes the formyl transfer reaction using ATP and formate [67]. M. jannaschii PurP has a hexameric arrangement of 32 symmetry as the biological unit (Figure 17D) and is a member of the ATP-grasp superfamily, with the signature A, B, and C three domain architecture for the monomeric fold [68] (Figure 17E). Domain A consists of 137 residues with a central four-stranded parallel β-sheet flanked by four α-helices plus an additional linker region of four α-helices connecting to domain B. Domain B, containing 67 residues, is the flap domain capping the substrate binding site and is formed by a four-stranded antiparallel β-sheet with one side facing the active site and the other side flanked by two α-helices and a 310 helix. Domain C is smaller than that of other ATP-grasp family members and is equivalent to the C1-domain of PurD, PurT and PurK. It contains 157 residues and is an αβα three layered sandwich, consisting of a central five-stranded antiparallel β-sheet flanked by a total of five α-helices and two 310 helices.
Both substrate and product-complexed structures of M. jannaschii PurP have been determined to locate the active site and elucidate structural features of the mechanism (Figure 19). The active site of PurP is at a cleft formed by all three domains of one monomer and the C-domains of two adjacent monomers related by 3-fold symmetry. The ATP binding of PurP is similar to that of other ATP-grasp superfamily members, including a mixture of stacking interactions and hydrogen bonds to the base, a pair of hydrogen bonds to the ribose hydroxyl groups by a glutamate residue, and hydrogen bond interactions to the phosphate groups by a glycine rich P-loop. Substrate AICAR and product FAICAR primarily bind through hydrogen bonds with the aminoimidazole-carboxamide moiety and the 5′-monophosphate, while the ribose hydroxyl groups are solvent exposed. PurP is thought to use a formylphosphate intermediate formed by an ATP-dependent phosphorylation, and a conserved residue, His27, as the catalytic base to deprotonate the 5-amino group of AICAR for nucleophilic attack. Two conserved arginine residues, Arg228 and Arg314, are also possibly involved in the catalysis, by orienting the formyl group and providing positive charge to stabilize the transition state.
Figure 19.

Active site of PurP. The active site of PurP is shown in schematic diagram, with hydrogen bonds indicated by dashed lines. The active site is comprised of residues from three adjacent monomers. The chain id for each residue is indicated in parenthesis.
Step 10: Biosynthesis of inosine monophosphate (IMP)
The final step of the de novo purine biosynthesis, which is a relatively facile cyclization reaction of FAICAR to generate IMP with elimination of water, is catalyzed by IMP cyclohydrolase. Two different IMP cyclohydrolases have been classified: (1) the bi-functional enzyme PurHJ for bacteria and eukaryotes, and (2) the archaeal enzyme PurO. The purO gene is found in only 11 archaeal organisms: five of which have a single purP gene.
The IMP cyclohydrolase domain (PurJ) of PurHJ consists of the N-terminal 199 residues of the polypeptide chain and is structurally independent from the PurH domain (Figure 17C). The two active sites are separated by approximately 50 Å, and there is no evidence of channeling or tunneling of FAICAR between the two functional domains. However, strong electropositive patches along the surface connecting the two active sites might facilitate the passage of FAICAR. The structure of PurJ resembles a Rossmann fold, with a central five-stranded parallel β-sheet (3↑2↑1↑4↑5↑) flanked by three α-helices on one side and seven α-helices on the other.
Structures of avian and human PurHJ have been determined with feedback inhibitor XMP [61,64,66] and various sulfonyl containing analog inhibitors that mimic the tetrahedral intermediate during the cyclization reaction [60] bound at the PurJ binding site (Figure 18B). Unlike the PurH domain, a complete active site for the PurJ domain is contained within each monomer. The unliganded active site shows a large open cavity, while upon ligand binding, significant changes at loops 65 – 67 and 103 – 108 are triggered to form a more tightly packed binding pocket. The bound nucleotide is almost completely buried and sequestered from the solvent, and interacts with the active site residues primarily via sidechain and backbone hydrogen bonds. In the structure complexed with the sulfonyl substituted nucleotide, the partially negatively charged sulfonyl group is stabilized by a helical dipole. Based upon the structural evidence, the active site rearrangement induced by substrate binding appears to be most important for the IMP cyclohydrolase activity, by correctly orienting the substrate to facilitate the intramolecular nucleophilic attack and subsequent cyclization. The helical dipole, which provides an oxyanion hole, is possibly involved in the polarization of the formyl group of FAICAR and stabilizing the tetrahedral intermediate. Active site residues Lys66, Tyr104, Asp125, and Lys137 (human numbering, +1 for avian) were also proposed to aid catalysis; however, they may not be essential for this relatively facile cyclization reaction [60,63,64].
PurO shows no similarity with PurHJ in sequence, structure, or the active site architecture, and utilizes a different IMP cyclohydrolase mechanism [69]. The structures of PurO have been determined from M. thermoautotrophicus with product IMP and substrate analog AICAR bound, as well as in the unliganded form [69,70]. The mutagenesis and kinetic studies have been mostly done for PurO from M. jannaschii, which shares 42% sequence identity with M. thermoautotrophicus PurO.
The biological unit of PurO is a tetramer (Figure 17F) and the monomeric structure adopts the N-terminal nucleophile (NTN) hydrolase fold [69,70] (Figure 17G). NTN hydrolase family members in general are translated as pre-enzymes and require an autocatalytic self-processing step to free up the nucleophilic residue, usually a cysteine, serine, or threonine, at the new N-terminus. However, PurO does not undergo the self-processing step and also lacks the nucleophilic residue at the corresponding position. The structure of PurO is characterized as an αββα four layer sandwich with a six-stranded mixed β-sheet and a seven-stranded mixed β-sheet, each of which can be divided into two antiparallel β-sheets that pack against each other to form the core structure, and two α-helices that flank one side of the double β-sheets, and three α-helices on the other side.
The active site of PurO resides in a deep pocket between the two β-sheets in each monomer (Figure 20). The nucleotides are bound at the active site mainly through hydrogen bond interactions to the aminoimidazole-carboxamide moiety, the ribose hydroxyl groups, and the 5′-monophosphate. Key residue Tyr59 is proposed to be responsible for ionization and tautomerization of the carboxamide group of FAICAR. Arg30 is thought to stabilize the partially negatively charged oxygen of the carboxamide group during the catalysis and Glu104 further assists the tautomerization after the ring closure. Two possible mechanisms that differ at the intermediates of tautomerization have been proposed and cannot be differentiated with current knowledge [69].
Figure 20.

Active site of PurO. The schematic diagram shows bound IMP. Hydrogen bonds to IMP are indicated by dashed lines.
ATP-grasp superfamily members in purine biosynthesis
The preceding sections described the structures and function of the 15 unique purine biosynthetic enzymes. The following sections highlight similarities and differences among these enzymes. Seven purine biosynthetic enzymes utilize ATP. Four of these (PurD, PurL, PurM and PurC) are found in all purine biosynthetic pathways and three additional enzymes (PurT, PurK and PurP) are found in prokaryotic pathways. Four of the seven enzymes (PurD, PurT, PurK and PurP) are members of the ATP-grasp superfamily while PurC has several structural elements in common with the ATP-grasp superfamily. PurM and PurL belong to a different structural superfamily (see below). In general, the ATP-grasp superfamily catalyzes the ligation of amino and carboxylate groups of small molecule metabolites, and examples of this function are widespread in primary metabolism. An acylphosphate intermediate is thought to be a common feature of the ATP-grasp superfamily.
Structurally, the ATP grasp superfamily is characterized by a three domain architecture (A, B and C); the C-domain can be further divided into two subdomains (C1 and C2) (Figure 21A). The A and C-domains form a core structure and provide the substrate binding sites. The B-domain provides the ATP binding site and forms a flap that covers the active site. The B-domain also contains a conserved phosphate binding P-loop and undergoes a disordered to ordered transition upon nucleotide binding. PurD, PurT and PurK contain all three domains and are structurally most similar to each other. PurP lacks the C2-domain but retains all other structural features. In the hexameric assembly for PurP, a domain from a threefold related monomer replaces C2. When PurD, PurT and PurK are superimposed using the secondary structure alignment algorithms found in the molecular graphics computer program Coot [71], the adenine nucleotides also superimpose, although some variation is observed in the position of the γ-phosphate. The substrate binding sites, where known, are located in the same general area compared to each other and are near the phosphate tail of the ATP. PurC is distantly related to the ATP-grasp superfamily. PurC contains the ATP-grasp C-domain, but lacks the A-domain. PurC contains an ATP-binding flap domain but both its location within the chain and its topology differ from the flap domain (B-domain) of the ATP-grasp superfamily. Nevertheless, when PurC is superimposed upon the ATP-grasp superfamily members, the ATP-binding sites and the flap domains also superimpose.
Figure 21.

Structural similarities and differences among purine biosynthetic enzymes. (A) Structure of the ATP-grasp superfamily members. The representative structure shown is PurD from E. coli (PDB: IGSO). (B) Ribbon diagram for the structural superposition of the R5P binding sites from PurL (blue) and PurM (red). FGAR, the ligand bound in the PurL structure and the sulfate ion present in the PurM structure are shown as a ball-and-stick representation. (C) The R5P binding motifs of PurT (1KJ8), PurK (1B6S), PurD (2QK4), PurP (2R7L), PurN (1CDE), and PurC (2GQS) illustrated in ribbon diagrams. The A, C1, C2-domains (or their equivalent) are colored in red, cyan, and light green, respectively. Active sites are indicated by arrows and substrates are shown as schematic diagrams. Ligands, where observed, are shown in ball-and-stick representation. Three structurally conserved residues Glu, Lys, and Arg for PurT and PurK are also shown in ball-and-stick representation. (D) A hypothetical common ancestor for PurM and PurL. In the absence of ADP, the ancestral enzyme binds FGAM and ATP, and functions as AIR synthetase (PurM). In the presence of ADP, the AIR synthetase activity is inhibited and the recruitment protein and glutaminase subunit bind causing the enzyme to function as FGAM synthetase (PurL).
The ATP-dependent PurM superfamily
PurLQS (and fused large PurL) and PurM catalyze the conversion of FGAR to FGAM, and FGAM to AIR, in the fourth and fifth steps of the purine biosynthetic pathway, respectively (Figure 2) [72]. For PurLQS, the actual FGAM synthesis takes place in PurL, while PurQ is a glutaminase, and PurS is an essential noncatalytic component. The chemical reactions catalyzed by PurL itself and PurM are similar; both reactions involve a transformation of a carbonyl moiety to an imine, and both enzymes utilize the γ-phosphate of an ATP molecule to activate the carbonyl oxygen as a leaving group. This activation results in a proposed iminophosphate intermediate formation and in subsequent attack of the intermediate by a nitrogenous nucleophile (Figure 11). The difference between these reactions is the source of the nucleophile. In PurL ammonia, formed upon glutamine hydrolysis in PurQ, acts as the nucleophile, while in PurM the N(1) nitrogen, part of the FGAM arm, serves this function resulting in an intramolecular transformation.
Besides the similarities in catalysis, PurL and PurM also share a similar overall fold and a conserved ATP-binding motif [35,45]. This ATP-binding motif defines the PurM superfamily and three additional members of the PurM superfamily are known: Ni-Fe hydrogenase maturation protein (HypE), selenophosphate synthetase (SelD), and thiamin phosphate kinase (ThiL) [73,74] (Figure 22). The PurM-like ATP-binding motif is characterized by a DXcGXXP signature sequence (Figure 23), with the aspartate of this sequence serving as a ligand for an Mg2+ ion, which in turn is responsible for stabilizing the phosphate tail of ATP [41,45]. Additional common features of the ATP-binding motif include lack of positively charged residues and utilization of solely Mg2+ ions in stabilization of the phosphate tail of ATP. Aside from the common ATP dependence, the reactions catalyzed by the superfamily members are diverse, and the γ-phosphate acceptor substrates specific to these enzymes vary substantially, as do the binding sites for the acceptors. Therefore, the transfer of the γ-phosphate of the ATP, thought to be dissociative, is hypothesized as the unifying feature for the PurM superfamily [41].
Figure 22.

Structural conservation within the PurM superfamily. The ribbon diagrams show members of the PurM-like ATP binding superfamily: T. maritima PurL (PDB: 2HS4), E. coli PurM (PDB: 1CLI), Desulfovibrio vulgaris HypE (PDB: 2Z1U), A. aeolicus SelD (PDB: 2YYE), and A. aeolicus ThiL (PDB: 3C9T). Monomers are in different shades. ATP molecules bound in the active sites of the superfamily members are in ball-and-stick representation.
Figure 23.

Signature sequence for the PurM superfamily. The sequences shown are EcPurM (E. coli), TmPurL (T. maritima), AaThiL (A. aeolicus), AaSelD (A. aeolicus) and EcHypE (E. coli). The boxed regions are residues absolutely conserved throughout the superfamily. The conserved aspartic acid serves as a ligand for a magnesium ion. The lengths of the spacers vary among species and the glycine residues outside the box show some species variation but are always small residues.
Comparison of R5P binding sites
All substrates and products for all enzymes in the purine biosynthetic pathway share a common moiety: ribose 5′-phosphate (R5P). The enzymes make use of two structural elements of R5P, the 5′-phosphate and the 2′- and 3′-hydroxyls of the ribose, when forming interactions. In general, because the folds of the enzymes within the pathway vary, so do the locations of the residues involved in binding R5P. The conformation of the ribose pucker as well as the position of 5′-phosphate relative to the ribose also change depending on the enzyme. However, direct interactions utilized in binding of R5P show some similarities.
For the 5′-phosphate moiety, PurF, PurN, and PurL, utilize helix polarity by binding to its N-terminal end and forming hydrogen bonds mostly with the mainchain amides and occasional hydroxyl groups of Ser and/or Thr residues. PurT, PurE, and PurB utilize sidechains of residues located in two or three loop regions, and the most commonly used residues are Arg, Ser, Lys, and Tyr. In PurP, these loops come from adjacent monomers. Finally, PurO and PurJ combine the two strategies and have elements of both binding modes.
In most purine biosynthetic enzymes, the 2′- and 3′-hydroxyl groups of the ribose form interactions with two moieties, an acidic residue and a mainchain amide or carbonyl. The two residues binding the hydroxyls are generally far removed from each other in the primary structure. PurF is different in its use of two consecutive Asp residues for binding of the ribose hydroxyls, while in PurP no protein interactions with the ribose are observed.
A conserved R5P binding motif formed by the A, C1 and C2-domains (Figure 21C) for three ATP-grasp enzymes, PurD, PurT, and PurK, was proposed by Kappock et al. [75]. At the time a structure of PurT was available in complex with the substrate GAR, while none of the PurK or PurD structures has substrate or analog bound. However, a structure comparison of the proposed R5P binding site of PurK and the GAR binding site of PurT revealed remarkable similarities. In PurT an arginine residue and a lysine residue form the 5′-phosphate binding site, and a glutamate residue and main chain hydrogen bonds from the strand-loop-helix motif of the A-domain form the ribose binding site. All of these features are present in PurK and indicate a common mode of substrate binding. In PurD, either a glycine residue or an alanine residue replaces all three of these residues, suggesting different interatomic interactions even though the overall binding cavity is conserved. The most recent ATP-grasp member in the purine biosynthetic pathway, PurP, also shares a similar R5P binding motif. While lacking the C2-domain, PurP utilizes the C1-domain from an adjacent monomer to complete the 5′-phosphate binding site, and the orientation of the nucleotide differs slightly from that of PurT. The overall structural homology and the conservation of the R5P binding motif suggests a common ancestor for these ATP-grasp superfamily members with the most similarity between PurT and PurK, and PurD and PurP being more distantly related. The same R5P binding motif is in part observed in PurN, despite having a different overall fold [75]. Although PurC is also related to the ATP-grasp proteins and binds the catalytic nucleotide similarly, the substrate/product binding site is different, suggesting that this enzyme diverged early on.
Despite PurL and PurM having a very similar fold and a well-conserved ATP-binding pocket, the binding pockets for the FGAR/FGAM substrates do not superimpose well (Figure 21B). If indeed the proposed histidine residues are catalytic in PurM and the sulfate found in the PurM structure marks the binding site of the 5′-phosphate, then the FGAM binding pocket is different from that for FGAR in PurL. This binding mode would still utilize the 5′-phosphate interaction with an N-terminal end of a helix in PurM as also seen for PurL but of a different helix. It remains unclear whether there was any evolutionary pressure necessitating the conservation of the FGAR/FGAM binding site as seen for the second substrate, ATP. Structural characterization of PurM in complex with FGAM or AIR would further shed light on the relationship between the two enzymes.
Two unrelated amidotransferases
Two enzymes in the purine biosynthetic pathway are amidotransferases. PurF, catalyzes the conversion of PRPP to PRA in the first step, and PurLQS, catalyzes the conversion of FGAR to FGAM in the fourth step (Figure 2) [72]. As for all amidotransferase reactions known to date, the source of nitrogen atom for PRA and FGAM formation is ammonia generated via glutamine hydrolysis. Amidotransferases are complex enzymes with the glutamine hydrolysis taking place in an active site spatially separated from the active site where the acceptor molecule receives ammonia [76]. The two active sites are connected by an ammonia channel, and are capable of coupled catalysis with the binding of the acceptor molecule activating the glutaminase activity. While both PurF and PurLQS (large PurL), possess these features common to all amidotransferases, the similarities end here. Not only are the domains responsible for binding of the acceptor different, which is expected considering the structural variation of the acceptor molecules, but so are the glutaminase domains. PurF belongs to the NTN-hydrolase family of glutaminases characterized by a fold of two β-sheets sandwiched together, and PurLQS/large PurL) utilizes a triad glutaminase with one large β-sheet, and the nucleophilic Cys located in a nucleophilic elbow. Therefore, PurF and PurLQS are unrelated structurally and represent a case of convergent evolution where the same function is accomplished in two unrelated ways.
Two unrelated THF-dependent enzymes
Two enzymes in the purine biosynthetic pathway utilize the N10-formylTHF cofactor to transfer a formyl moiety onto the corresponding pathway intermediate thereby installing a new carbon atom of the growing IMP heterocycle. These enzymes are PurN, which catalyzes the third step and introduces the C(8) carbon of IMP, and PurH, which catalyzes the ninth step and introduces the C(2) carbon. Although both enzymes catalyze a formylation reaction, they are not related structurally. The N10-formylTHF binding site in PurN is formed by three loops of a single monomer, and such a binding site is characteristic of other folate-dependent enzymes. The closest structural homologs of PurN are methionyl-tRNAfMet formyltransferase (PDB: 2FMT), N10-formylTHF dehydrogenase (PDB: 1S3I), and the 4-amino-4-deoxy-L-arabinose transformylase domain of a resistance enzyme ArnA (PDB: 1Z7E), and all of these enzymes utilize N10-formylTHF for formyl group transfer [77–79]. Unlike PurN the N10-formylTHF binding site in PurH is located at a dimer interface and is formed by three strand-turn-helix motifs coming from both monomers. While the overall structure of PurH contains no known structural homologs, the fold of the two large subdomains is most closely related to cytidine deaminase (PDB: 1P6O) [80], deoxycytidylate deaminase (PDB: 1VQ2) [81], and an unpublished deaminase involved in riboflavin biosynthesis (PDB: 2B3Z). However, none of these enzymes utilize folate in their corresponding reactions. While PurN and PurH represent an example of convergent evolution, it is interesting to note that the alternative enzymes for steps 3 and 9 (PurT and PurP, respectively) are most likely derived from a common ancestor by divergent evolution.
Variations in gene organization
In E. coli, each step of the purine biosynthesis pathway is catalyzed by a single monofunctional gene product, with the exception of the last two steps, which are catalyzed by the bifunctional enzyme by PurHJ. However, variations of the gene organization have been observed in other organisms (Figure 24).
Figure 24.

Variations in gene organization for the purine biosynthesis pathway. Organizational variations of (A) PurD, PurM, PurN, (B) PurL, PurS, PurQ, and (C) PurK, PurE, PurC from various species are illustrated.
In human and other mammals, PurD (step 2), PurM (step 5), and PurN (step 3) are encoded by the same gene and expressed as a trifunctional enzyme [82–84] (Figure 24A).
In Drosophila, a highly homologous copy of PurM is fused and organized in the order of PurD-PurM-PurM′-PurN. Interestingly, an alternative transcript consisting only of PurD is also found in Drosophila [85]. In yeast and reptiles, only PurD and PurM are fused as a bifunctional enzyme. The structures of the human PurD and PurN components have been determined separately; however, neither the human PurM by itself nor the complete trifunctional enzyme has been structurally characterized. Likewise, no structures are available for the trifunctional enzyme from flies or the bifunctional enzyme from yeast. While these structures await to be determined, the PurD-PurM bifunctional enzyme from yeast S. cerevisiae is known to exist as a dimer [86], which is consistent with PurM forming a dimer in the lower organisms. This also suggests that human PurD-PurM-PurN might also form a dimer, while the Drosophila enzyme probably functions as a monomer with a pseudodimeric PurM-PurM′ core.
PurL (step 4), provides an example of gene fusion during protein evolution (Figure 24B). In Gram-positive bacteria and archaea, three separate gene products PurS, small PurL, and PurQ assemble as a protein complex in order to be functional, while in eukaryotes and Gram-negative bacteria, the three genes are fused into large PurL, which expresses as a multidomain protein consisting of a single polypeptide chain. Not only sharing significant sequence similarities, the structure of the small PurL, PurQ and PurS complex also showed the same spatial organization and conserved active sites as that of the functional domains of the large PurL [42].
The last known variation in gene organization involves steps 6 and 7 (Figure 24C). While most bacteria express PurE (class I) and PurK separately, in yeast and plants a bifunctional fusion protein PurK-PurE is produced [2,87]. In vertebrates, class II PurE, which catalyzes the conversion to CAIR from AIR directly and lacks PurK, is always a C-terminal fusion to PurC [88,89]. Finally, in some methanogenic bacteria gene duplication and fusion, presumably, resulted in PurE-PurE′, which has ~35% sequence identity between the two halves. At the present time, structures are available for PurE and PurC from lower organisms and for the human bifunctional PurCE.
Missing purine biosynthetic enzymes in archaea
Lacking tetrohydrofolate as a cofactor, archaea have evolved differently from most organisms for the last two steps of the purine biosynthesis pathway. The bifunctional enzyme PurHJ is not present in archaea. Instead, PurP and PurO, which have been recently identified from M. jannaschii, catalyze step 9 utilizing an ATP-dependent formate ligation and the step 10 cyclization, respectively. The identification of these new archaeal enzymes raised additional questions. PurP had been thought to be an archaeal signature gene; however, as additional genome sequences become available, it is evident that the purP gene is abundant but not ubiquitous in archaeal organisms. For instance, organisms such as Halobacterium salinarum and Methanoregula boonei do not possess the purP gene, while organisms such as M. jannaschii have a single purP gene and P. furiosus and most other purP containing archaea have multiple copies of purP genes.
Thus far the purP gene has been found in 22 archaeal organisms for which complete genomes are available. Interestingly, 17 of these encode more than one PurP ortholog. While still very little is known about the enzymology of the PurP orthologs, sequence analysis revealed that PurPs can be divided into three groups that share ~30% sequence identity with each other [68]. M. jannaschii PurP is the best biochemically and structurally characterized PurP and belongs to group 1, which consists of five species having a single purP gene. Group 2 PurPs are similar to group 1 PurPs while group 3 PurPs show significant variation at key active site residues and most likely do not catalyze the step 9 reaction. Although a structure of a group 2 PurP has been reported, including an AICAR-complexed structure, this PurP shows no FAICAR synthetase activity and awaits to be classified functionally [68]. No group 3 PurP has been either biochemically or structurally characterized.
Finally, in contrast to PurP, PurO is only present in a few archaeal species such as M. jannaschii and M. thermoautotrophicus. This suggests that the enzyme catalyzing step 10 remains to be discovered in most archaea and further fuels the speculation that other variations of the purine biosynthetic pathway may be discovered as new genome sequences become available.
Implications for pathway evolution
Structural studies of the purine biosynthetic pathway have identified common features that may be related to pathway evolution [75]. The coupling of an amino group and a carboxylate group is required in five steps (2, 3, 6a, 7 and 9) and the ATP-dependent enzymes that catalyze these reactions have common features. Four of the enzymes are members of the ATP-grasp superfamily while PurC has some commonality with these four. Therefore it is possible to hypothesize an ancestral ligase, with broad substrate specificity, that catalyzed all five steps. Over time the individual enzymes evolved to efficiently catalyze a specific step. Figure 25 shows a phylogenetic tree based on comparison of the available sequences for these enzymes. The tree includes the PurP orthologs for which functions have not yet been assigned.
Figure 25.

Evolution of the purine biosynthetic pathway. The phylogenic diagram shows the divergence of the ATP-grasp purine biosynthetic enzymes.
Class I (prokaryotic pathway) and class II (eukaryotic pathway) PurEs are also closely related. Both show a homooctameric arrangement with active sites requiring the participation of three monomers. Class II PurE catalyzes the direct carboxylation of AIR to form CAIR, while class I PurE catalyzes the transfer of the carboxylate group of NCAIR, which was formed in previous step by addition of bicarbonate to the N5 positions of AIR, to the C4 position of the imidazole. While the reactions catalyzed by class I and II PurEs are similar, the activities are specific and cannot be interchanged. This suggests that class I PurE may have evolved first in a carbonate rich environment and that class II PurE evolved later as CO2 became more abundant.
PurM and PurL catalyze consecutive steps in purine biosynthesis and involve chemically similar reactions (Figure 11). Both are ATP-dependent and the PurL product (FGAM) is the PurM substrate. In addition PurM and PurL are structurally homologous and belong to superfamily that also includes ThiL, HypE and SelD (Figure 22). The similarity of the chemical transformations catalyzed by PurL and PurM and the similarity in structure is remarkable and points to a common ancestor. While PurM is a homodimer, T. maritima PurL consists of a single chain with pseudotwofold symmetry in which each half is structurally similar to the PurM monomer. S. typhimurium PurL is homolgous to T. maritima PurL but is fused at the N-terminus to a PurS-like domain and at the C-terminus to a PurQ-like domain. Each PurM monomer contains one active site while the N-terminal half of PurL contains an active site and the C-terminal half contains an auxillary ADP binding site. Furthermore, PurM is inhibited by ADP and assembly of the PurL/PurQ/PurS complex requires ADP. These observations suggest a possible ancestral enzyme that catalyzed both the PurL and PurM reactions (Figure 21D). When ADP is absent, the ancestral enzyme catalyzes the conversion of FGAM to AIR. When ADP is bound, the PurM activity is inhibited and the PurQ and PurS proteins are recruited to form a complex that catalyzes the conversion of FGAR to FGAM. Over time PurL and PurM evolved and specialized. A PurLQS complex is found in Gram-positive and archaeal organisms, while the genes for PurL, PurQ and PurS fused to create large PurL in Gram-negative bacteria and eukaryotic organisms. In modern day organisms, the regulatory site of PurL is no longer needed, and has become a purely structural remnant.
In higher organisms the ten purine biosynthetic activities are contained in six enzymes. These gene fusions (steps 2,3 and 5; steps 6 and 7; steps 9 and 10) could provide several advantages. Fused proteins localize the activities and provide the possibility of channeling from one active site to the next. Very recently, evidence for a large, multiprotein complex called the purinosome was published [90]. These studies show that the purinosome forms under conditions of purine starvation and is reversible. Other factors such as protein modification, additional small molecules, metabolites or additional protein components may regulate assembly of the purinosome. The highly evolved purinosome results from the evolution of individual protein activities, gene fusions and the emergence of transient protein interactions, likely under the control of multiple factors.
Conclusions
Structures are now available for all of the known purine biosynthetic enzymes. Together with biochemical studies, the structural details provide a basis for understanding mechanistic details of the enzyme reactions. However, many areas remain to be investigated. In some archaea the genes for steps 9 and 10 have not yet been identified and it is possible that other purine biosynthetic enzymes remain to be discovered as new genome sequences become available. Other areas of investigation include the organization of multifunctional proteins. Structures of PurCE and PurHJ are available; however, no structure exists for the PurD-PurM-PurN trifunctional protein (or the bifunctional enzymes in other organisms) and no structure exists for the PurKE bifunctional enzyme. Transient protein-protein interactions that are not well understood must occur given the instability of intermediates such as PRA and NCAIR. Finally, the structural biology of the newly discovered purinosome will present challenges for many years to come.
Supplementary Material
Acknowledgments
We thank Leslie Kinsland for assistance in the preparation of the manuscript. This work was supported by NIH grant GM73220 to SEE. SEE is indebted to the Lucille P. Markey Charitable Trust and the W.M. Keck Foundation.
References
- 1.Buchanan JM, Hartman SC. Enzymic reactions in the synthesis of the purines. Adv Enzymol Relat Areas Mol Biol. 1959;21:199–261. [Google Scholar]
- 2.Mueller EJ, Meyer E, Rudolph J, Davisson VJ, Stubbe J. N5-carboxyaminoimidazole ribonucleotide: evidence for a new intermediate and two new enzymatic activities in the de novo purine biosynthetic pathway of Escherichia coli. Biochemistry. 1994;33:2269–2278. doi: 10.1021/bi00174a038. [DOI] [PubMed] [Google Scholar]
- 3.Marolewski AE, Mattia KM, Warren MS, Benkovic SJ. Formyl phosphate: a proposed intermediate in the reaction catalyzed by Escherichia coli PurT GAR transformylase. Biochemistry. 1997;36:6709–6716. doi: 10.1021/bi962961p. [DOI] [PubMed] [Google Scholar]
- 4.Graupner M, Xu H, White RH. New class of IMP cyclohydrolases in Methanococcus jannaschii. J Bacteriol. 2002;184:1471–1473. doi: 10.1128/JB.184.5.1471-1473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waldrop GL, Rayment I, Holden HM. Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase. Biochemistry. 1994;33:10249–10256. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]
- 6.Fan C, Moews PC, Walsh CT, Knox JR. Vancomycin resistance: structure of D-alanine:D-alanine ligase at 2.3 Å resolution. Science. 1994;266:439–443. doi: 10.1126/science.7939684. [DOI] [PubMed] [Google Scholar]
- 7.Galperin MY, Koonin EV. A diverse superfamily of enzymes with ATP-dependent carboxylate-amine/thiol ligase activity. Protein Sci. 1997;6:2639–2643. doi: 10.1002/pro.5560061218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathews II, Kappock TJ, Stubbe J, Ealick SE. Crystal structure of Escherichia coli PurE, an unusual mutase in the purine biosynthetic pathway. Structure. 1999;7:1395–1406. doi: 10.1016/s0969-2126(00)80029-5. [DOI] [PubMed] [Google Scholar]
- 9.Smith JL. Glutamine PRPP amidotransferase: snapshots of an enzyme in action. Curr Opin Struct Biol. 1998;8:686–694. doi: 10.1016/s0959-440x(98)80087-0. [DOI] [PubMed] [Google Scholar]
- 10.Zalkin H. The amidotransferases. Adv Enzymol Relat Areas Mol Biol. 1993;66:203–309. doi: 10.1002/9780470123126.ch5. [DOI] [PubMed] [Google Scholar]
- 11.Messenger LJ, Zalkin H. Glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Purification and properties. J Biol Chem. 1979;254:3382–3392. [PubMed] [Google Scholar]
- 12.Kim JH, Krahn JM, Tomchick DR, Smith JL, Zalkin H. Structure and function of the glutamine phosphoribosylpyrophosphate amidotransferase glutamine site and communication with the phosphoribosylpyrophosphate site. J Biol Chem. 1996;271:15549–15557. doi: 10.1074/jbc.271.26.15549. [DOI] [PubMed] [Google Scholar]
- 13.Krahn JM, Kim JH, Burns MR, Parry RJ, Zalkin H, Smith JL. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry. 1997;36:11061–11068. doi: 10.1021/bi9714114. [DOI] [PubMed] [Google Scholar]
- 14.Chen S, Tomchick DR, Wolle D, Hu P, Smith JL, Switzer RL, Zalkin H. Mechanism of the synergistic end-product regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase by nucleotides. Biochemistry. 1997;36:10718–10726. doi: 10.1021/bi9711893. [DOI] [PubMed] [Google Scholar]
- 15.Zhou G, Smith JL, Zalkin H. Binding of purine nucleotides to two regulatory sites results in synergistic feedback inhibition of glutamine 5-phosphoribosylpyrophosphate amidotransferase. J Biol Chem. 1994;269:6784–6789. [PubMed] [Google Scholar]
- 16.Smith JL, Zaluzec EJ, Wery JP, Niu L, Switzer RL, Zalkin H, Satow Y. Structure of the allosteric regulatory enzyme of purine biosynthesis. Science. 1994;264:1427–1433. doi: 10.1126/science.8197456. [DOI] [PubMed] [Google Scholar]
- 17.Wong JY, Bernlohr DA, Turnbough CL, Switzer RL. Purification and properties of glutamine phosphoribosylpyrophosphate amidotransferase from Bacillus subtilis. Biochemistry. 1981;20:5669–5674. doi: 10.1021/bi00523a005. [DOI] [PubMed] [Google Scholar]
- 18.Muchmore CR, Krahn JM, Kim JH, Zalkin H, Smith JL. Crystal structure of glutamine phosphoribosylpyrophosphate amidotransferase from Escherichia coli. Protein Sci. 1998;7:39–51. doi: 10.1002/pro.5560070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rudolph J, Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry. 1995;34:2241–2250. doi: 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Kappock TJ, Stubbe J, Ealick SE. X-ray crystal structure of glycinamide ribonucleotide synthetase from Escherichia coli. Biochemistry. 1998;37:15647–15662. doi: 10.1021/bi981405n. [DOI] [PubMed] [Google Scholar]
- 21.Almassy RJ, Janson CA, Kan CC, Hostomska Z. Structures of apo and complexed Escherichia coli glycinamide ribonucleotide transformylase. Proc Natl Acad Sci U S A. 1992;89:6114–6118. doi: 10.1073/pnas.89.13.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen P, Schulze-Gahmen U, Stura EA, Inglese J, Johnson DL, Marolewski A, Benkovic SJ, Wilson IA. Crystal structure of glycinamide ribonucleotide transformylase from Escherichia coli at 3.0 Å resolution. A target enzyme for chemotherapy. J Mol Biol. 1992;227:283–292. doi: 10.1016/0022-2836(92)90698-j. [DOI] [PubMed] [Google Scholar]
- 23.Dahms TE, Sainz G, Giroux EL, Caperelli CA, Smith JL. The apo and ternary complex structures of a chemotherapeutic target: human glycinamide ribonucleotide transformylase. Biochemistry. 2005;44:9841–9850. doi: 10.1021/bi050307g. [DOI] [PubMed] [Google Scholar]
- 24.Greasley SE, Marsilje TH, Cai H, Baker S, Benkovic SJ, Boger DL, Wilson IA. Unexpected formation of an epoxide-derived multisubstrate adduct inhibitor on the active site of GAR transformylase. Biochemistry. 2001;40:13538–13547. doi: 10.1021/bi011482+. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Desharnais J, Greasley SE, Beardsley GP, Boger DL, Wilson IA. Crystal structures of human GAR Tfase at low and high pH and with substrate β-GAR. Biochemistry. 2002;41:14206–14215. doi: 10.1021/bi020522m. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Desharnais J, Marsilje TH, Li C, Hedrick MP, Gooljarsingh LT, Tavassoli A, Benkovic SJ, Olson AJ, Boger DL, Wilson IA. Rational design, synthesis, evaluation, and crystal structure of a potent inhibitor of human GAR Tfase: 10-(trifluoroacetyl)-5,10-dideazaacyclic-5,6,7,8-tetrahydrofolic acid. Biochemistry. 2003;42:6043–6056. doi: 10.1021/bi034219c. [DOI] [PubMed] [Google Scholar]
- 27.Greasley SE, Yamashita MM, Cai H, Benkovic SJ, Boger DL, Wilson IA. New insights into inhibitor design from the crystal structure and NMR studies of Escherichia coli GAR transformylase in complex with β-GAR and 10-formyl-5,8,10-trideazafolic acid. Biochemistry. 1999;38:16783–16793. doi: 10.1021/bi991888a. [DOI] [PubMed] [Google Scholar]
- 28.Klein C, Chen P, Arevalo JH, Stura EA, Marolewski A, Warren MS, Benkovic SJ, Wilson IA. Towards structure-based drug design: crystal structure of a multisubstrate adduct complex of glycinamide ribonucleotide transformylase at 1.96 Å resolution. J Mol Biol. 1995;249:153–175. doi: 10.1006/jmbi.1995.0286. [DOI] [PubMed] [Google Scholar]
- 29.Warren MS, Marolewski AE, Benkovic SJ. A rapid screen of active site mutants in glycinamide ribonucleotide transformylase. Biochemistry. 1996;35:8855–8862. doi: 10.1021/bi9528715. [DOI] [PubMed] [Google Scholar]
- 30.Thoden JB, Firestine S, Nixon A, Benkovic SJ, Holden HM. Molecular structure of Escherichia coli PurT-encoded glycinamide ribonucleotide transformylase. Biochemistry. 2000;39:8791–8802. doi: 10.1021/bi000926j. [DOI] [PubMed] [Google Scholar]
- 31.Thoden JB, Firestine SM, Benkovic SJ, Holden HM. PurT-encoded glycinamide ribonucleotide transformylase. Accommodation of adenosine nucleotide analogs within the active site. J Biol Chem. 2002;277:23898–23908. doi: 10.1074/jbc.M202251200. [DOI] [PubMed] [Google Scholar]
- 32.Westheimer FH. Monomeric metaphosphates. Chem Rev. 1981;81:313–326. [Google Scholar]
- 33.Buchanan JM. Covalent reaction of substrates and antimetabolites with formylglycinamide ribonucleotide amidotransferase. Methods Enzymol. 1982;87:76–84. doi: 10.1016/s0076-6879(82)87009-2. [DOI] [PubMed] [Google Scholar]
- 34.Schendel FJ, Mueller E, Stubbe J, Shiau A, Smith JM. Formylglycinamide ribonucleotide synthetase from Escherichia coli: cloning, sequencing, overproduction, isolation, and characterization. Biochemistry. 1989;28:2459–2471. doi: 10.1021/bi00432a017. [DOI] [PubMed] [Google Scholar]
- 35.Anand R, Hoskins AA, Stubbe J, Ealick SE. Domain organization of Salmonella typhimurium formylglycinamide ribonucleotide amidotransferase revealed by X-ray crystallography. Biochemistry. 2004;43:10328–10342. doi: 10.1021/bi0491301. [DOI] [PubMed] [Google Scholar]
- 36.Hoskins AA, Anand R, Ealick SE, Stubbe J. The formylglycinamide ribonucleotide amidotransferase complex from Bacillus subtilis: metabolite-mediated complex formation. Biochemistry. 2004;43:10314–10327. doi: 10.1021/bi049127h. [DOI] [PubMed] [Google Scholar]
- 37.Anand R, Hoskins AA, Bennett EM, Sintchak MD, Stubbe J, Ealick SE. A model for the Bacillus subtilis formylglycinamide ribonucleotide amidotransferase multiprotein complex. Biochemistry. 2004;43:10343–10352. doi: 10.1021/bi0491292. [DOI] [PubMed] [Google Scholar]
- 38.Batra R, Christendat D, Edwards A, Arrowsmith C, Tong L. Crystal structure of MTH169, a crucial component of phosphoribosylformylglycinamidine synthetase. Proteins. 2002;49:285–288. doi: 10.1002/prot.10209. [DOI] [PubMed] [Google Scholar]
- 39.Mathews II, Krishna SS, Schwarzenbacher R, McMullan D, Abdubek P, Ambing E, Canaves JM, Chiu HJ, Deacon AM, DiDonato M, Elsliger MA, Godzik A, Grittini C, Grzechnik SK, Hale J, Hampton E, Han GW, Haugen J, Jaroszewski L, Klock HE, Koesema E, Kreusch A, Kuhn P, Lesley SA, Levin I, Miller MD, Moy K, Nigoghossian E, Paulsen J, Quijano K, Reyes R, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, White A, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA. Crystal structure of phosphoribosylformylglycinamidine synthase II (smPurL) from Thermotoga maritima at 2.15 Å resolution. Proteins. 2006;63:1106–1111. doi: 10.1002/prot.20650. [DOI] [PubMed] [Google Scholar]
- 40.Mathews II, Krishna SS, Schwarzenbacher R, McMullan D, Jaroszewski L, Miller MD, Abdubek P, Agarwalla S, Ambing E, Axelrod HL, Canaves JM, Carlton D, Chiu HJ, Clayton T, DiDonato M, Duan L, Elsliger MA, Grzechnik SK, Hale J, Hampton E, Haugen J, Jin KK, Klock HE, Koesema E, Kovarik JS, Kreusch A, Kuhn P, Levin I, Morse AT, Nigoghossian E, Okach L, Oommachen S, Paulsen J, Quijano K, Reyes R, Rife CL, Spraggon G, Stevens RC, van den Bedem H, White A, Wolf G, Xu Q, Hodgson KO, Wooley J, Deacon AM, Godzik A, Lesley SA, Wilson IA. Crystal structure of phosphoribosylformyl-glycinamidine synthase II, PurS subunit (TM1244) from Thermotoga maritima at 1.90 Å resolution. Proteins. 2006;65:249–254. doi: 10.1002/prot.21024. [DOI] [PubMed] [Google Scholar]
- 41.Morar M, Anand R, Hoskins AA, Stubbe J, Ealick SE. Complexed structures of formylglycinamide ribonucleotide amidotransferase from Thermotoga maritima describe a novel ATP binding protein superfamily. Biochemistry. 2006;45:14880–14895. doi: 10.1021/bi061591u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morar M, Hoskins AA, Stubbe J, Ealick SE. Formylglycinamide Ribonucleotide Amidotransferase from Thermotoga maritima: Structural Insights into Complex Formation. Biochemistry. 2008:xxx–yyy. doi: 10.1021/bi800329p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrimsher JL, Schendel FJ, Stubbe J, Smith JM. Purification and characterization of aminoimidazole ribonucleotide synthetase from Escherichia coli. Biochemistry. 1986;25:4366–4371. doi: 10.1021/bi00363a028. [DOI] [PubMed] [Google Scholar]
- 44.Daubner SC, Schrimsher JL, Schendel FJ, Young M, Henikoff S, Patterson D, Stubbe J, Benkovic SJ. A multifunctional protein possessing glycinamide ribonucleotide synthetase, glycinamide ribonucleotide transformylase, and aminoimidazole ribonucleotide synthetase activities in de novo purine biosynthesis. Biochemistry. 1985;24:7059–7062. doi: 10.1021/bi00346a006. [DOI] [PubMed] [Google Scholar]
- 45.Li C, Kappock TJ, Stubbe J, Weaver TM, Ealick SE. X-ray crystal structure of aminoimidazole ribonucleotide synthetase (PurM), from the Escherichia coli purine biosynthetic pathway at 2.5 Å resolution. Structure. 1999;7:1155–1166. doi: 10.1016/s0969-2126(99)80182-8. [DOI] [PubMed] [Google Scholar]
- 46.Thoden JB, Kappock TJ, Stubbe J, Holden HM. Three-dimensional structure of N5-carboxyaminoimidazole ribonucleotide synthetase: a member of the ATP grasp protein superfamily. Biochemistry. 1999;38:15480–15492. doi: 10.1021/bi991618s. [DOI] [PubMed] [Google Scholar]
- 47.Li SX, Tong YP, Xie XC, Wang QH, Zhou HN, Han Y, Zhang ZY, Gao W, Li SG, Zhang XC, Bi RC. Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J Mol Biol. 2007;366:1603–1614. doi: 10.1016/j.jmb.2006.12.027. [DOI] [PubMed] [Google Scholar]
- 48.Hoskins AA, Morar M, Kappock TJ, Mathews II, Zaugg JB, Barder TE, Peng P, Okamoto A, Ealick SE, Stubbe J. N5-CAIR mutase: role of a CO2 binding site and substrate movement in catalysis. Biochemistry. 2007;46:2842–2855. doi: 10.1021/bi602436g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Constantine CZ, Starks CM, Mill CP, Ransome AE, Karpowicz SJ, Francois JA, Goodman RA, Kappock TJ. Biochemical and structural studies of N5-carboxyaminoimidazole ribonucleotide mutase from the acidophilic bacterium Acetobacter aceti. Biochemistry. 2006;45:8193–8208. doi: 10.1021/bi060465n. [DOI] [PubMed] [Google Scholar]
- 50.Settembre EC, Chittuluru JR, Mill CP, Kappock TJ, Ealick SE. Acidophilic adaptations in the structure of Acetobacter aceti N5-carboxyaminoimidazole ribonucleotide mutase (PurE) Acta Crystallogr D. 2004;60:1753–1760. doi: 10.1107/S090744490401858X. [DOI] [PubMed] [Google Scholar]
- 51.Levdikov VM, Barynin VV, Grebenko AI, Melik-Adamyan WR, Lamzin VS, Wilson KS. The structure of SAICAR synthase: an enzyme in the de novo pathway of purine nucleotide biosynthesis. Structure. 1998;6:363–376. doi: 10.1016/s0969-2126(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 52.Zhang R, Skarina T, Evdokimova E, Edwards A, Savchenko A, Laskowski R, Cuff ME, Joachimiak A. Structure of SAICAR synthase from Thermotoga maritima at 2.2 Å reveals an unusual covalent dimer. Acta Crystallogr F. 2006;62:335–339. doi: 10.1107/S1744309106009651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ginder ND, Binkowski DJ, Fromm HJ, Honzatko RB. Nucleotide complexes of Escherichia coli phosphoribosylaminoimidazole succinocarboxamide synthetase. J Biol Chem. 2006;281:20680–20688. doi: 10.1074/jbc.M602109200. [DOI] [PubMed] [Google Scholar]
- 54.Antopyuk SV, Grebenko AI, Levdikov VM, Urusova DV, Melik-Adamyan VR, Lamzin VS, Wilson KS. X-ray study of SAICAR synthase complexes with adenosinetriphosphate. Kristallografiya. 2001;46:687–691. [Google Scholar]
- 55.Toth EA, Yeates TO. The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway. Structure. 2000;8:163–174. doi: 10.1016/s0969-2126(00)00092-7. [DOI] [PubMed] [Google Scholar]
- 56.Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. J Mol Biol. 2000;301:433–450. doi: 10.1006/jmbi.2000.3970. [DOI] [PubMed] [Google Scholar]
- 57.Vedadi M, Lew J, Artz J, Amani M, Zhao Y, Dong A, Wasney GA, Gao M, Hills T, Brokx S, Qiu W, Sharma S, Diassiti A, Alam Z, Melone M, Mulichak A, Wernimont A, Bray J, Loppnau P, Plotnikova O, Newberry K, Sundararajan E, Houston S, Walker J, Tempel W, Bochkarev A, Kozieradzki I, Edwards A, Arrowsmith C, Roos D, Kain K, Hui R. Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol. 2007;151:100–110. doi: 10.1016/j.molbiopara.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 58.Tsai M, Koo J, Yip P, Colman RF, Segall ML, Howell PL. Substrate and product complexes of Escherichia coli adenylosuccinate lyase provide new insights into the enzymatic mechanism. J Mol Biol. 2007;370:541–554. doi: 10.1016/j.jmb.2007.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rayl EA, Moroson BA, Beardsley GP. The human purH gene product, 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase. Cloning, sequencing, expression, purification, kinetic analysis, and domain mapping. J Biol Chem. 1996;271:2225–2233. doi: 10.1074/jbc.271.4.2225. [DOI] [PubMed] [Google Scholar]
- 60.Xu L, Chong Y, Hwang I, D’Onofrio A, Amore K, Beardsley GP, Li C, Olson AJ, Boger DL, Wilson IA. Structure-based design, synthesis, evaluation, and crystal structures of transition state analogue inhibitors of inosine monophosphate cyclohydrolase. J Biol Chem. 2007;282:13033–13046. doi: 10.1074/jbc.M607293200. [DOI] [PubMed] [Google Scholar]
- 61.Cheong CG, Wolan DW, Greasley SE, Horton PA, Beardsley GP, Wilson IA. Crystal structures of human bifunctional enzyme aminoimidazole-4-carboxamide ribonucleotide transformylase/IMP cyclohydrolase in complex with potent sulfonyl-containing antifolates. J Biol Chem. 2004;279:18034–18045. doi: 10.1074/jbc.M313691200. [DOI] [PubMed] [Google Scholar]
- 62.Wolan DW, Greasley SE, Wall MJ, Benkovic SJ, Wilson IA. Structure of avian AICAR transformylase with a multisubstrate adduct inhibitor β-DADF identifies the folate binding site. Biochemistry. 2003;42:10904–10914. doi: 10.1021/bi030106h. [DOI] [PubMed] [Google Scholar]
- 63.Greasley SE, Horton P, Ramcharan J, Beardsley GP, Benkovic SJ, Wilson IA. Crystal structure of a bifunctional transformylase and cyclohydrolase enzyme in purine biosynthesis. Nat Struct Biol. 2001;8:402–406. doi: 10.1038/87555. [DOI] [PubMed] [Google Scholar]
- 64.Wolan DW, Greasley SE, Beardsley GP, Wilson IA. Structural insights into the avian AICAR transformylase mechanism. Biochemistry. 2002;41:15505–15513. doi: 10.1021/bi020505x. [DOI] [PubMed] [Google Scholar]
- 65.Xu L, Li C, Olson AJ, Wilson IA. Crystal structure of avian aminoimidazole-4-carboxamide ribonucleotide transformylase in complex with a novel non-folate inhibitor identified by virtual ligand screening. J Biol Chem. 2004;279:50555–50565. doi: 10.1074/jbc.M406801200. [DOI] [PubMed] [Google Scholar]
- 66.Wolan DW, Cheong CG, Greasley SE, Wilson IA. Structural insights into the human and avian IMP cyclohydrolase mechanism via crystal structures with the bound XMP inhibitor. Biochemistry. 2004;43:1171–1183. doi: 10.1021/bi030162i. [DOI] [PubMed] [Google Scholar]
- 67.White RH. Purine biosynthesis in the domain Archaea without folates or modified folates. J Bacteriol. 1997;179:3374–3377. doi: 10.1128/jb.179.10.3374-3377.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, White RH, Ealick SE. Crystal structure and function of 5-formaminoimidazole-4-carboxamide ribonucleotide synthetase from Methanocaldococcus jannaschii. Biochemistry. 2008;47:205–217. doi: 10.1021/bi701406g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang YN, Tran A, White RH, Ealick SE. A novel function for the N-terminal nucleophile hydrolase fold demonstrated by the structure of an archaeal inosine monophosphate cyclohydrolase. Biochemistry. 2007;46:5050–5062. doi: 10.1021/bi061637j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saridakis V, Christendat D, Thygesen A, Arrowsmith CH, Edwards AM, Pai EF. Crystal structure of Methanobacterium thermoautotrophicum conserved protein MTH1020 reveals an NTN-hydrolase fold. Proteins. 2002;48:141–143. doi: 10.1002/prot.10147. [DOI] [PubMed] [Google Scholar]
- 71.Cowtan PEaK. Coot: Model-Building Tools fo Molecular Graphics. Acta Crystallograph D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 72.Flaks JG, Lukens LN. The enzymes of purine nucleotide synthesis de novo. Methods Enzymol. 1963;6:52–95. [Google Scholar]
- 73.McCulloch KM, Kinsland C, Begley TP, Ealick SE. Structural studies of thiamin monophosphate kinase in complex with substrates and products. Biochemistry. 2008;47:3810–3821. doi: 10.1021/bi800041h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shomura Y, Komori H, Miyabe N, Tomiyama M, Shibata N, Higuchi Y. Crystal structures of hydrogenase maturation protein HypE in the Apo and ATP-bound forms. J Mol Biol. 2007;372:1045–1054. doi: 10.1016/j.jmb.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 75.Kappock TJ, Ealick SE, Stubbe J. Modular evolution of the purine biosynthetic pathway. Curr Opin Chem Biol. 2000;4:567–572. doi: 10.1016/s1367-5931(00)00133-2. [DOI] [PubMed] [Google Scholar]
- 76.Mouilleron S, Golinelli-Pimpaneau Conformational changes in ammonia-channeling glutamine amidotransferases. Curr Opin Struct Biol. 2007;17:653–664. doi: 10.1016/j.sbi.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 77.Chumanevich AA, Krupenko SA, Davies C. The crystal structure of the hydrolase domain of 10-formyltetrahydrofolate dehydrogenase: mechanism of hydrolysis and its interplay with the dehydrogenase domain. J Biol Chem. 2004;279:14355–14364. doi: 10.1074/jbc.M313934200. [DOI] [PubMed] [Google Scholar]
- 78.Gatzeva-Topalova PZ, May AP, Sousa MC. Structure and mechanism of ArnA: conformational change implies ordered dehydrogenase mechanism in key enzyme for polymyxin resistance. Structure. 2005;13:929–942. doi: 10.1016/j.str.2005.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schmitt E, Blanquet S, Mechulam Y. Structure of crystalline Escherichia coli methionyl-tRNAfMet formyltransferase: comparison with glycinamide ribonucleotide formyltransferase. EMBO J. 1996;15:4749–4758. [PMC free article] [PubMed] [Google Scholar]
- 80.Ireton GC, Black ME, Stoddard BL. The 1.14 Å crystal structure of yeast cytosine deaminase: evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure. 2003;11:961–972. doi: 10.1016/s0969-2126(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 81.Almog R, Maley F, Maley GF, Maccoll R, Van Roey P. Three-dimensional structure of the R115E mutant of T4-bacteriophage 2′-deoxycytidylate deaminase. Biochemistry. 2004;43:13715–13723. doi: 10.1021/bi048928h. [DOI] [PubMed] [Google Scholar]
- 82.Aimi J, Qiu H, Williams J, Zalkin H, Dixon JE. De novo purine nucleotide biosynthesis: cloning of human and avian cDNAs encoding the trifunctional glycinamide ribonucleotide synthetase-aminoimidazole ribonucleotide synthetase-glycinamide ribonucleotide transformylase by functional complementation in E. coli. Nucleic Acids Res. 1990;18:6665–6672. doi: 10.1093/nar/18.22.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Daubner SC, Young M, Sammons RD, Courtney LF, Benkovic SJ. Structural and mechanistic studies on the HeLa and chicken liver proteins that catalyze glycinamide ribonucleotide synthesis and formylation and aminoimidazole ribonucleotide synthesis. Biochemistry. 1986;25:2951–2957. doi: 10.1021/bi00358a033. [DOI] [PubMed] [Google Scholar]
- 84.Schild D, Brake AJ, Kiefer MC, Young D, Barr PJ. Cloning of three human multifunctional de novo purine biosynthetic genes by functional complementation of yeast mutations. Proc Natl Acad Sci U S A. 1990;87:2916–2920. doi: 10.1073/pnas.87.8.2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Henikoff S, Keene MA, Sloan JS, Bleskan J, Hards R, Patterson D. Multiple purine pathway enzyme activities are encoded at a single genetic locus in Drosophila. Proc Natl Acad Sci U S A. 1986;83:720–724. doi: 10.1073/pnas.83.3.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tretyakov OY, Ryzhova TA, Velichytina IV, Kostikova TR, Myasnikov AN, Smirnov MN, Domkin VD. Glycinamide ribonucleotide synthetase [EC 6.3.4.13]-aminoimidazole ribonucleotide synthetase [EC 6.3.3.1] of the yeast Saccharomyces cerevisiae. Biokhimiya (Moscow) 1995;60:2011–2021. [PubMed] [Google Scholar]
- 87.Firestine SM, Misialek S, Toffaletti DL, Klem TJ, Perfect JR, Davisson VJ. Biochemical role of the Cryptococcus neoformans ADE2 protein in fungal de novo purine biosynthesis. Arch Biochem Biophys. 1998;351:123–134. doi: 10.1006/abbi.1997.0512. [DOI] [PubMed] [Google Scholar]
- 88.Chen ZD, Dixon JE, Zalkin H. Cloning of a chicken liver cDNA encoding 5-aminoimidazole ribonucleotide carboxylase and 5-aminoimidazole-4-N-succinocarboxamide ribonucleotide synthetase by functional complementation of Escherichia coli pur mutants. Proc Natl Acad Sci U S A. 1990;87:3097–3101. doi: 10.1073/pnas.87.8.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Firestine SM, Poon SW, Mueller EJ, Stubbe J, Davisson VJ. Reactions catalyzed by 5-aminoimidazole ribonucleotide carboxylases from Escherichia coli and Gallus gallus: a case for divergent catalytic mechanisms. Biochemistry. 1994;33:11927–11934. doi: 10.1021/bi00205a031. [DOI] [PubMed] [Google Scholar]
- 90.An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.