

Abstract
Epigenomic processes are believed to play a pivotal role for the effect of environmental exposures in early life to modify disease risk throughout the lifespan. Offspring of women with hypertensive complications of pregnancy (HTNPREG) have an increased risk of developing systemic and pulmonary vascular dysfunction in adulthood. In this preliminary report, we sought to determine whether epigenetic modifications of genes involved in the regulation of vascular function were present in HTNPREG offspring. We contrasted DNA methylation and gene expression patterns of peripheral blood mononuclear cells obtained from young male offspring of HTNPREG (n=5) to those of normotensive controls (n=19). In HTNPREG offspring we identified six differentially methylated regions (DMRs) including three genes (SMOC2, ARID1B and CTRHC1) relevant to vascular function. The transcriptional activity of ARID1B and CTRCH1 was inversely related to methylation status. HTNPREG offspring had higher systolic pulmonary artery pressure (sPPA) vs. controls. Our findings demonstrate that epigenetic marks are altered in offspring of HTNPREG with a modest elevation of sPPA and introduce novel epigenomic targets for further study. On the basis of these findings we speculate that epigenomic mechanisms may be involved in mediating the effect of HTNPREG to raise the risk of vascular disease later in life.
Keywords: epigenetics, preeclampsia, gestational hypertension, developmental programming
Introduction
Intrauterine exposures influence physiological function and disease susceptibility throughout the lifespan, as demonstrated by epidemiologic and experimental animal studies conducted over the past two decades. For instance, maternal hypertensive disorders of pregnancy (HTNPREG) predispose offspring to systemic cardiovascular disease (1, 2) and pulmonary vascular dysfunction (3) in adulthood. Because epigenetic marks are responsive to environmental stimuli (4–6) and mediate gene expression patterns that control embryonic development and organogenesis (7), epigenomic processes are believed to play a central role in the developmental programming of adult disease. In support of this view, DNA methylation changes induced by intrauterine exposures can modify gene expression and physiological function in ways that are of clinical importance (4, 6, 8).
Locus-specific DNA methylation marks, some within genes known to influence vascular development and integrity, have been identified in placental tissue or cord blood derived from HTNPREG (e.g., (9, 10)). Therefore, it is logical to speculate that epigenomic mechanisms may mediate the increased risk of vascular dysfunction in offspring of HTNPREG. To our knowledge no study has queried genome-wide DNA methylation patterns in young adult offspring of HTNPREG to identify the potential role of epigenetics for compromised vascular function in these individuals. Accordingly, in this preliminary report we sought to identify differentially methylated genomic regions (DMRs) in offspring of HTNPREG vs. offspring of normotensive controls.
Understanding the role of epigenetics in mediating the effects of intrauterine exposures to modify long-term disease susceptibility has important clinical applications with respect to the identification of environmental triggers for adult-onset disease and novel therapeutic targets. DNA methylation is a particularly attractive candidate for therapeutic intervention because methylation status is amenable to pharmacologic modification.
Materials and Methods
Study Design
Subjects and sample sizes
Subjects included 24 Andean males between the ages of 18 and 25 living in La Paz-El Alto, Bolivia (3600 – 4100 m). Of these, five were born to women whose pregnancy was complicated by hypertension (HTNPREG), and 19 were born to women having a normotensive pregnancy (controls). Additional exclusion criteria were a history of cardiac or pulmonary disease, evidence of cardiopulmonary disease on clinical exam, chronic bronchitis or frequent smoking (> 4 cigarettes per week). Conducting the overarching study in Bolivia was beneficial because more than 1.5 million persons live in the La Paz-El Alto metropolitan area which is situated at a sufficient altitude to increase the incidence of hypoxia-related pregnancy complications, including HTNPREG (11, 12). An additional advantage of the high-altitude model is that because chronic hypoxia exposes predisposition to pulmonary vascular dysfunction (4) high altitude would be expected to improve the detection of elevated sPPA and its association with methylation status. Physiologic studies and sample collection were performed at the Bolivian Institute of High Altitude Biology (3600 m) in La Paz, Bolivia. On the first visit peripheral blood samples were collected from an antecubital vein by routine venipuncture and the subject participated in a detailed questionnaire to obtain information regarding residential and health history. Cardiopulmonary evaluations were performed on subsequent visits. Medical record reviews and an in-depth interview with the subjects’ mother were conducted to obtain information regarding pregnancy, delivery, and perinatal characteristics. The Colorado Multiple Institutional Review Board and Colegío Medico, its Bolivian counterpart, approved all survey and study procedures.
Systolic Pulmonary Artery Pressure (sPPA)
Transthoracic echocardiography (GE Vivid I Ultrasound 2007) was used to estimate sPPA as previously described (13). Quantification of tricuspid regurgitation was measured with the use of continuous wave Doppler and averaged from three complete waveforms of good quality. Maximal tricuspid regurgitation jet flow velocity (TRmax) was used to estimate sPPA using a modified Bernoulli equation as previously reported (3).
Medical Records Review and Maternal Interview
Perinatal data were obtained by structured interview of the subject’s mother and a review of medical records from the hospital or clinic where the subject was born. Data collected were: a) maternal health and reproductive history, b) pregnancy or delivery complications, c) fetal complications, and d) newborn characteristics and complications. Hypertensive pregnancy was determined by a diagnosis of preeclampsia or gestational hypertension as noted in the medical records or as obtained from maternal interview. Specific questions regarding HTNPREG included, “Were you told you had preeclampsia or gestational hypertension during your pregnancy?”, “Were you told you had high blood pressure multiple times during your pregnancy?” and, to distinguish between preexisting hypertension and HTNPREG, “Did you have high blood pressure before your pregnancy?”. Gestational hypertension and preeclampsia were grouped into a single HTNPREG category for this preliminary test since both conditions impair fetal growth and influence vascular function in adulthood (2, 14).
Differences between HTNPREG and controls with respect to demographic variables and physiologic characteristics were determined using independent T-tests or chi-squared tests as appropriate. Data are reported as the mean ± SEM or the proportion and 95% CI.
Sampling and Isolation of Genomic DNA (gDNA) and Total RNA
Peripheral blood samples were collected from an antecubital vein using standard phlebotomy, placed into a BD Vacutainer Cell Preparation Tube containing sodium citrate and Ficoll Hypaque density fluid. PBMCs were isolated according to manufacturer guidelines, resuspended in RNAlater (Ambion) solution and stored at −80°C until analysis. gDNA and RNA were isolated using the Qiagen AllPrep DNA/RNA Mini Kit. gDNA was purified using the Qiagen QIAEX II Kit and quantitated with the NanoDrop ND-100 UV-Vis spectrophotometer (NanoDrop Technologies).
DNA Methylation Arrays, Analysis, and Interpretation
DNA methylation was assessed using Illumina’s Infinium HumanMethylation450 BeadChip arrays. This methylation platform permits the interrogation of >485k methylation sites at single-nucleotide resolution and has been validated with respect to technical performance and functional relevance. Methylated cytosines were bisulfite converted using the EZ DNA Methylation Gold Kit (Zymo Research), amplified by WGA and then enzymatically fragmented. 4μl of bisulfite converted DNA at 50ng/ul was hybridized to the BeadChip. Quantitative methylation scores were generated for each of the 485k loci for each sample using Illumina’s GenomeStudio Methylation module. Signal intensities of methylated and unmethylated probes were exported from the GenomeStudio interface, along with detection p-values representing the likelihood of detection relative to background. Individual data points with a p-value outside of detection criteria (p>0.05) were treated as missing data. Data were normalized using the SWAN method (15) within the R package minfi, and M-values were calculated. Methylation differences between HTNPREG and controls were identified using limma (16) and multcomp (17) packages in R. From the resultant p-values, differentially methylated regions (DMRs) between HTNPREG and controls were identified using Comb-p, an analytical tool developed to analyze spatially correlated p-values (18). To define DMRs, Comb-p uses a sliding window approach to identify genomic regions with multiple low p-values after accounting for local dependencies and multiple testing using a false discovery rate cut off for multiple comparisons. DMRs were ranked by p-value, and annotated to the nearest gene. Significance was defined as an α < 5% after correcting for the number of possible DMRs. Because methylation marks can influence the transcriptional activity of adjacent genes, we inspected genes located within 1Mb upstream or downstream of intergenic DMRs for additional vascular-related candidates.
Compared to associations based on the differential methylation of single CpG sites, DMR analysis has greater specificity and power to detect functionally relevant methylation-phenotype associations (19). For instance, evidence indicates that methylation status may be profoundly affected by sequence content and genetic variation, including single nucleotide polymorphisms (20, 21). Because DMRs require multiple adjacent probes to be differentially methylated, the Comb-p method reduces the influence of sequence-specific bias. Prior investigations have used similar methods to identify clinically relevant methylation-phenotype associations (22).
Influence of DMRs on Gene Expression
RT-PCR was used to determine the influence of the DMRs identified on gene expression. cDNA was generated using random primers with Invitrogen’s Superscript III First-Strand Synthesis System SuperMix. PCR reactions were prepared using TaqMan Fast Advanced Master Mix and TaqMan Gene Expression Assays (Applied Biosystems) as specified by the manufacturer. RT-PCR was performed and analyzed on the ViiA 7 Real-Time PCR System with ViiA 7 1.22 software (Applied Biosystems). The quantification of target gene transcription relative to that of GAPDH was assessed using the 2ΔΔCT method. Unpaired Student’s t tests were used to identify differential expression between HTNPREG and controls using a significance threshold of P < 0.05. Data are expressed as means ± SD.
Results
HTNPREG and controls were equivalent with respect to age, height, weight, resting heart rate and SaO2. When compared to controls, HTNPREG had higher estimated sPPA (p=0.007) (Table 1).
Table 1.
Subject characteristics
| HTNPREG Offspring (n = 5) | Controls (n = 19) | P-value | |
|---|---|---|---|
|
|
|||
| Age, yrs | 21.4 ± 2.5 | 21.8 ± 1.5 | NS |
| Height, cm | 164.8 ± 3.1 | 167.8 ± 6.1 | NS |
| Weight, kg | 63.1 ± 3.4 | 62.8 ± 7.5 | NS |
| Heart rate, bpm | 64 ± 5 | 63 ± 10 | NS |
| SaO2, % | 90.4 ± 3.0 | 91.5 ± 2.4 | NS |
| RVSP, mmHg | 40.6 ± 1.1 | 37.4 ± 4.2 | 0.007 |
| DLCO corr, % predicted | 121 ± 19 | 142 ± 29 | NS |
| FEV1/FVC | 86.8 ± 5.8 | 85.8 ± 7.5 | NS |
Data are reported as the mean ± SD. Abbreviations: arterial oxygen saturation (SaO2), right ventricular systolic pressure (RVSP), pulmonary CO diffusion capacity (DLCO) corrected for altitude and hemoglobin, forced expiratory volume at one second (FEV1) and forced vital capacity (FVC).
Comparing HTNPREG with control subjects, we identified six genome-wide significant DMRs (Figure 1). Each DMR was associated with a unique gene: namely, collagen triple helix repeat containing 1 (CTHRC1), tripartite motif containing 31 (TRIM31), AT rich interactive domain 1B (ARID1B), SPARC related modular calcium binding 2 (SMOC2), leucine-rich repeats and IQ motif containing 3 (LRRIQ3) or long intergenic non-protein coding RNA 226 (LINC00226).
Figure 1.
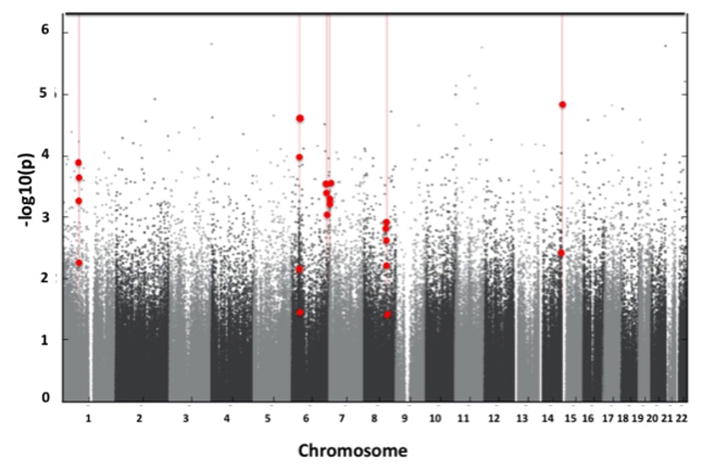
Differentially methylated regions (DMRs) were identified in young male Andean residents of high altitude (18–25 yrs) born to women with hypertensive complications of pregnancy (HTNPREG) vs. women who remained normotensive throughout pregnancy (controls). Shown is a Manhattan plot of the adjusted p-values for the comparison between HTNPREG vs. controls. Each dot represents a p-value for a probe on the Illumina 450k array that has been adjusted by the significance of neighboring probes according to their correlation using Comb-p (18). Probes that comprise the DMRs are identified by red dots.
Of the six DMRs, three were hypomethylated and three were hypermethylated in HTNPREG vs. controls. In each case, the directionality of the differentially methylated sites composing the DMR was consistent. Methylation differences between HTNPREG and controls at each DMR averaged 6.7% (range, 2.6 to 11.2%; Table 2). Three DMRs contained differentially methylated sites within gene bodies (intron, exon, 3’ untranslated region [UTR] and 5’ UTR), and one DMR (CTHRC1) showed differential methylation within 200–1,500 bases from a transcriptional start site (promoter) (Table 2). Several genes with potential relevance for vascular function, including metastasis associated 1 (MTA1) and cytosine-rich protein2 (CRIP2), and thrombospondin 2 (THBS2) are located within 1Mb upstream or downstream of the intergenic LINC00226 and SMOC2 DMRs, respectively. No vascular-related candidate genes were identified within the 1Mb band on either side of the ARID1B DMR.
Table 2.
Differentially methylated regions (DMRs) identified in young male highlanders (18–25 yrs) born to women with hypertensive pregnancy (HTNPREG) vs. normotensive controls
|
DMR Gene Symbol Genomic Location |
Ref Gene Feature(s) | Ref Gene Distance | Slk P-value | Slk-Sidak P-value | Probes | deltaBeta |
|---|---|---|---|---|---|---|
|
CTHRC1 Chr8: 104383714 – 104383798 |
TSS, exon, 5’UTR | 0 | 9.82E-07 | 0.006 | cg04219247 | −0.015 |
| cg20447655 | −0.040 | |||||
| cg20392240 | −0.017 | |||||
| cg11889769 | −0.024 | |||||
| cg21643403 | −0.033 | |||||
|
| ||||||
|
LINC00226
* Chr14: 106806142 – 106806179 |
intergenic | 61176 | 1.97E-06 | 0.025 | cg23404902 | 0.038 |
| cg01842774 | 0.072 | |||||
|
| ||||||
|
TRIM31 Chr6: 30071502 - 30071612 |
intron | 0 | 8.55E-08 | 0.0004 | cg00354641 | −0.034 |
| cg27123071 | −0.028 | |||||
| cg13436843 | −0.086 | |||||
| cg09735274 | −0.087 | |||||
|
| ||||||
|
ARID1B
* Chr6: 159983262 - 156983315 |
intergenic | −115748 | 2.20E-07 | 0.002 | cg05282260 | 0.047 |
| cg08839808 | 0.097 | |||||
| cg00269725 | 0.072 | |||||
|
| ||||||
|
SMOC2
* Chr6: 169284284 – 169284344 |
intergenic | 215610 | 1.81E-08 | 0.001 | cg17720554 | 0.052 |
| cg10213762 | 0.087 | |||||
| cg12550496 | 0.100 | |||||
|
| ||||||
|
LRRIQ3 Chr1: 74663582 - 74663750 |
intron, exon,5’UTR | 0 | 1.81E-08 | 5.11E-05 | cg16356856 | −0.106 |
| cg13900770 | −0.085 | |||||
| cg06034096 | −0.094 | |||||
| cg00402366 | −0.162 | |||||
Denotes hypermethylated genes; the remainder are hypomethlated.
Genomic regions were defined according to the UCSC RefGene group. Intergenic = site not annotated in a gene, TSS = transcription start site at 200–1500 bp, 5′ region = 5′UTR and 1st exon, Intragenic = gene body including introns and exons and, 3′ region = 3′UTR. UTR – untranslated region. Individual sites that compose the differentially methylated regions and their percent methylation change are shown in the Probes and deltaBeta columns, respectively.
We then evaluated the association between DMRs and the expression of genes with a biologically plausible relationship to vascular function (ARID1B, CTHRC1 and SMOC2). Compared to controls ARID1B gene expression was impaired in HTNPREG offspring (P=0.025), while CTHRC1 tended to be higher (P=0.08) and SMOC2 was equivalent between groups (P=NS). Notably, ARID1B and CTHRC1 expression were inversely related to methylation status. Based on existing literature, the remaining DMRs either do not have a clear relationship to vascular function (TRIM31) or have not been well described (LRRIQ3 and LINC00226) and were therefore not considered for expression studies.
Discussion
Epigenomic processes are believed to play a pivotal role for mediating the effect of environmental exposures in early life on disease risk later in life. Numerous studies have shown that HTNPREG is associated with systemic or pulmonary vascular abnormalities in adulthood (1–3). Here we identified locus-specific DMRs in offspring of HTNPREG with elevated sPPA. Two of the DMRs identified in vascular-related genes, ARID1B and CTHRC1, were also differentially expressed, suggesting the potential functional relevance of the epigenetic modifications identified. Our findings support the hypothesis that epigenetic mechanisms may be involved in mediating the effect of HTNPREG to increase transgenerational vascular disease risk and introduce novel epigenomic targets for further experimental study.
ARID1B is the largest component of the switching defective/sucrose non-fermenting (SWI/SNF) ATP-dependent chromatin-modeling complex. Because SWI/SNF chromatin-remodeling complexes interact with numerous transcription factors to regulate gene expression and are required for the hypoxic induction of several genes including erythropoietin and vascular endothelial growth factor (VEGF) (e.g., (23)), we consider that this gene may be of particular relevance for the vascular dysfunction observed in offspring of HTNPREG. Indeed, the SWI/SNF complex is essential for cellular proliferation and differentiation in multiple cardiac cell lines, cardiomyocyte development, vasculogenesis and vascular smooth muscle proliferation (reviewed in (24)). For instance, components of the SWI/SNF complex (Brg1/Brm) contribute to the pathogenesis of endothelial dysfunction and atherosclerosis via the transcriptional regulation of inflammation-induced cell adhesion molecules (25). Consistent with the inverse methylation-expression relationship observed here, ARID1B is subject to methylation-induced transcriptional silencing. Specifically, hypermethylation upstream of the ARID1B initiation codon impairs ARID1B expression in pancreatic cancer cells (26).
Two of the DMRs identified in HTNPREG offspring, CTHRC1 and SMOC2, are important for vascular remodeling and angiogenesis in the context of vascular injury. Specifically, CTHRC1 is transiently overexpressed by adventitial fibroblasts and smooth muscle of the neointima in the carotid artery after balloon-induced injury (27). CTHRC1 overexpression in fibroblasts and smooth muscle cells enhances migratory ability, reduces collagen matrix deposition, and temporally coincides with extensive adventital remodeling, suggesting its potential involvement in the regulation of vascular remodeling (27). The expression of SMOC2, a novel member of the SPARC family of matricellular proteins that regulate interactions between cells and the extracellular matrix, is upregulated in response to vascular injury (28). SMOC2 overexpression in HUVECS also reduces the expression of thrombospondin 1 (THBS1), an angiogenic inhibitor, and enhances mitogenesis and cellular proliferation in the presence of angiogenic growth factors including vascular endothelial growth factor (VEGF) (29). THBS2 neighbors the SMOC2 intergenic DMR and, similar to THBS1, has potent antiangiogenic actions by antagonizing VEGF activity and via its direct influence on endothelial cell proliferation, survival and migration [reviewed in (30)]. This raises the possibility that the intergenic SMOC2 DMR identified may be acting at a distance on other genes important for vascular function. Further supporting the angiogenic potential of SMOC2 and its potential biological importance for vascular disease, in vitro Matrigel plug invasion assays in which HUVECS were transduced with Ad-SMOC2 showed more projections and greater cell-network area compared to Ad-GFP transduced cells (29).
Although LINC00226 itself is not well-described several genes within 1Mb of the intergenic LINC00226 are involved in the regulation of vascular function. CRIP2 is a cytoskeletal protein that influences the phenotypic modulation of vascular smooth muscle cells (VSMCs) in response to vascular injury (31). For instance, CRIP2 prevents excessive VSMC migration and proliferation that are associated with increased myocardial infarction risk (31). MTA1 is associated with increased metastatic potential and angiogenesis in numerous cancers, including malignant esophageal, lung, breast and prostate tumors, but also acts as a proangiogenic agent in normal, healthy tissue due, in part, to its ability to upregulate the expression of VEGF and its membrane-bound receptor (Flt-1) [reviewed in (32)].
There are several limitations to our study. A recurrent challenge in epigenomics is the inability to distinguish whether differential methylation patterns are a cause or consequence of the phenotype of interest, or whether their effect is neutral. Additionally, the DMRs we identified may have been influenced by other exposures occurring during development or inherent intra-individual differences. Despite this challenge, our findings offspring provide novel insight with respect to the potential importance of epigenomic dysregulation in individuals exposed to maternal hypertension during gestation and can be used to guide future research in this area. Similarly, the elevated sPPA we observed in HTNPREG offspring is genetic in nature or may result from developmental exposures. In support of the latter possibility, offspring born to preeclamptic mothers have impaired pulmonary and systemic vessel function whereas their siblings born after a normotensive pregnancy do not (3). Another consideration is that we relied primarily on maternal recall of HTNPREG or a clinical diagnosis as noted in the medical record, and lacked repeated blood pressure and proteinuria measurements. Maternal recall of HTNPREG has recently been validated with respect to its concordance with medical records, and the ability to accurately discriminate between gestational hypertension and preeclampsia (33). It is also worth noting that we used PBMCs as a proxy to explore biological processes occurring in or influencing the vasculature. There are several advantages to the use of PBMCs. First, PBMCs are the most transcriptionally active cells in the blood and their gene expression patterns influence systemic processes including the regulation of vascular function. Second, because PBMCs can be obtained using minimally invasive methods the potential for their use in the clinic or for prospective epigenomic studies beginning in early life is high. Finally, epigenomic profiles of PBMCs have successfully identified pathological processes important for various diseases such as asthma (22).
In summary, our observations raise the possibility that intrauterine exposure to HTNPREG modifies epigenetic marks in ways that may be of pathological importance for the development of vascular dysfunction in later life. While further human and experimental animal studies are required to evaluate the role of epigenetics for the effect of HTNPREG to modify transgenerational vascular disease risk, the present study has identified several targets meriting further research.
Acknowledgments
We would like to acknowledge all the individuals who participated in this project as research subjects and the physicians and technical staff at the Bolivian Institute of High Altitude Biology. Support for data collection and analysis was obtained from grants NIH-HL079647 (L.G.M.), R03TW007957 from the Fogarty International Center (L.G.M. and C.G.J.), NIH-HL095393 (D.A.S.), NIH-HL101715, (D.A.S.), NIH-ES18181 (D.A.S.) and the NIH/NCATS Colorado CTSA UL1 TR001082 (CMH-Pilot, C.G.J.). Dr. Julian is supported by the NIH BIRCWH program (5 K12HD057022-07).
References
- 1.Kajantie E, Eriksson JG, Osmond C, Thornburg K, Barker DJ. Pre-eclampsia is associated with increased risk of stroke in the adult offspring: the Helsinki birth cohort study. Stroke. 2009;40(4):1176–80. doi: 10.1161/STROKEAHA.108.538025. [DOI] [PubMed] [Google Scholar]
- 2.Geelhoed JJ, Fraser A, Tilling K, Benfield L, Davey Smith G, Sattar N, Nelson SM, Lawlor DA. Preeclampsia and gestational hypertension are associated with childhood blood pressure independently of family adiposity measures: the Avon Longitudinal Study of Parents and Children. Circulation. 2010;122(12):1192–9. doi: 10.1161/CIRCULATIONAHA.110.936674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jayet PY, Rimoldi SF, Stuber T, Salmon CS, Hutter D, Rexhaj E, Thalmann S, Schwab M, Turini P, Sartori-Cucchia C, Nicod P, Villena M, Allemann Y, Scherrer U, Sartori C. Pulmonary and systemic vascular dysfunction in young offspring of mothers with preeclampsia. Circulation. 2010;122(5):488–94. doi: 10.1161/CIRCULATIONAHA.110.941203. [DOI] [PubMed] [Google Scholar]
- 4.Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U, Sartori C. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. American journal of physiology Heart and circulatory physiology. 2011;301(1):H247–52. doi: 10.1152/ajpheart.01309.2010. [DOI] [PubMed] [Google Scholar]
- 5.Watson JA, Watson CJ, McCann A, Baugh J. Epigenetics, the epicenter of the hypoxic response. Epigenetics : official journal of the DNA Methylation Society. 2010;5(4):293–6. doi: 10.4161/epi.5.4.11684. [DOI] [PubMed] [Google Scholar]
- 6.Hollingsworth JW, Maruoka S, Boon K, Garantziotis S, Li Z, Tomfohr J, Bailey N, Potts EN, Whitehead G, Brass DM, Schwartz DA. In utero supplementation with methyl donors enhances allergic airway disease in mice. J Clin Invest. 2008;118(10):3462–9. doi: 10.1172/JCI34378. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nature reviews Genetics. 2002;3(9):662–73. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 8.Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang L. Hypoxia-derived oxidative stress mediates epigenetic repression of PKCepsilon gene in foetal rat hearts. Cardiovasc Res. 2012;93(2):302–10. doi: 10.1093/cvr/cvr322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blair JD, Yuen RK, Lim BK, McFadden DE, von Dadelszen P, Robinson WP. Widespread DNA hypomethylation at gene enhancer regions in placentas associated with early-onset pre-eclampsia. Mol Hum Reprod. 2013;19(10):697–708. doi: 10.1093/molehr/gat044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ching T, Ha J, Song MA, Tiirikainen M, Molnar J, Berry MJ, Towner D, Garmire LX. Genome-scale hypomethylation in the cord blood DNAs associated with early onset preeclampsia. Clin Epigenetics. 2015;7(1):21. doi: 10.1186/s13148-015-0052-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Julian CG, Vargas E, Armaza JF, Wilson MJ, Niermeyer S, Villena M, Moore LG. High-altitude ancestry protects against hypoxia-associated reductions in fetal growth. Arch Dis Child Fetal Neonatal Ed. 2007;92(5):F372–7. doi: 10.1136/adc.2006.109579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keyes LE, Armaza JF, Niermeyer S, Vargas E, Young D, Villena M, Moore LG. Intrauterine growth restriction, preeclampsia and intrauterine mortality at high altitude in Bolivia. Pediatr Res. 2003;54(1):20–5. doi: 10.1203/01.PDR.0000069846.64389.DC. [DOI] [PubMed] [Google Scholar]
- 13.Julian CG, Gonzales M, Rodriguez A, Bellido D, Salinas Salmon C, Ladenburger A, Reardon L, Vargas E, Moore LG. Perinatal hypoxia increases susceptibility to high-altitude polycythemia and attendant pulmonary vascular dysfunction. American journal of physiology Heart and circulatory physiology. 2015:ajpheart 00296. doi: 10.1152/ajpheart.00296.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hypertension in pregnancy. . Report of the American College of Obstetricians and Gynecologists' Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122(5):1122–31. doi: 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
- 15.Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS computational biology. 2010;6(5):e1000770. doi: 10.1371/journal.pcbi.1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 17.Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biometrical journal Biometrische Zeitschrift. 2008;50(3):346–63. doi: 10.1002/bimj.200810425. [DOI] [PubMed] [Google Scholar]
- 18.Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28(22):2986–8. doi: 10.1093/bioinformatics/bts545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, Irizarry RA. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41(1):200–9. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Pique-Regi R, Degner JF, Gilad Y, Pritchard JK. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome biology. 2011;12(1):R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, Morris M, Haghighi F, Tycko B. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40(7):904–8. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 22.Yang IV, Pedersen BS, Liu A, O'Connor GT, Teach SJ, Kattan M, Misiak RT, Gruchalla R, Steinbach SF, Szefler SJ, Gill MA, Calatroni A, David G, Hennessy CE, Davidson EJ, Zhang W, Gergen P, Togias A, Busse WW, Schwartz DA. DNA methylation and childhood asthma in the inner city. J Allergy Clin Immunol. 2015 doi: 10.1016/j.jaci.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sena JA, Wang L, Hu CJ. BRG1 and BRM chromatin-remodeling complexes regulate the hypoxia response by acting as coactivators for a subset of hypoxia-inducible transcription factor target genes. Mol Cell Biol. 2013;33(19):3849–63. doi: 10.1128/MCB.00731-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bevilacqua A, Willis MS, Bultman SJ. SWI/SNF chromatin-remodeling complexes in cardiovascular development and disease. Cardiovasc Pathol. 2014;23(2):85–91. doi: 10.1016/j.carpath.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang F, Chen D, Yu L, Dai X, Yang Y, Tian W, Cheng X, Xu H, Weng X, Fang M, Zhou J, Gao Y, Chen Q, Xu Y. Proinflammatory stimuli engage Brahma related gene 1 and Brahma in endothelial injury. Circ Res. 2013;113(8):986–96. doi: 10.1161/CIRCRESAHA.113.301296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khursheed M, Kolla JN, Kotapalli V, Gupta N, Gowrishankar S, Uppin SG, Sastry RA, Koganti S, Sundaram C, Pollack JR, Bashyam MD. ARID1B, a member of the human SWI/SNF chromatin remodeling complex, exhibits tumour-suppressor activities in pancreatic cancer cell lines. Br J Cancer. 2013;108(10):2056–62. doi: 10.1038/bjc.2013.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pyagay P, Heroult M, Wang Q, Lehnert W, Belden J, Liaw L, Friesel RE, Lindner V. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ Res. 2005;96(2):261–8. doi: 10.1161/01.RES.0000154262.07264.12. [DOI] [PubMed] [Google Scholar]
- 28.Nishimoto S, Hamajima Y, Toda Y, Toyoda H, Kitamura K, Komurasaki T. Identification of a novel smooth muscle associated protein, smap2, upregulated during neointima formation in a rat carotid endarterectomy model. Biochim Biophys Acta. 2002;1576(1–2):225–30. doi: 10.1016/s0167-4781(02)00345-7. [DOI] [PubMed] [Google Scholar]
- 29.Rocnik EF, Liu P, Sato K, Walsh K, Vaziri C. The novel SPARC family member SMOC-2 potentiates angiogenic growth factor activity. J Biol Chem. 2006;281(32):22855–64. doi: 10.1074/jbc.M513463200. [DOI] [PubMed] [Google Scholar]
- 30.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harb Perspect Med. 2012;2(5):a006627. doi: 10.1101/cshperspect.a006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen CH, Ho HH, Wu ML, Layne MD, Yet SF. Modulation of cysteine-rich protein 2 expression in vascular injury and atherosclerosis. Mol Biol Rep. 2014;41(11):7033–41. doi: 10.1007/s11033-014-3591-x. [DOI] [PubMed] [Google Scholar]
- 32.Nagaraj SR, Shilpa P, Rachaiah K, Salimath BP. Crosstalk between VEGF and MTA1 signaling pathways contribute to aggressiveness of breast carcinoma. Mol Carcinog. 2015;54(5):333–50. doi: 10.1002/mc.22104. [DOI] [PubMed] [Google Scholar]
- 33.Carter EB, Stuart JJ, Farland LV, Rich-Edwards J, Zera C, McElrath TF, Seely EW. [118-POS]: Validation of a maternal recall questionnaire for pregnancy complications associated with increased future risk for cardiovascular disease. Pregnancy Hypertens. 2015;5(1):63. doi: 10.1089/jwh.2014.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]