

Abstract
A single molecule of the motor enzyme kinesin-1 keeps a tight grip on its microtubule track, making tens or hundreds of discrete, unidirectional 8 nm steps before dissociating. This high duty ratio processive movement is thought to require a mechanism in which alternating stepping of the two head domains of the kinesin dimer is driven by alternating, overlapped cycles of ATP hydrolysis by the two heads. The R210K point mutation in Drosophila kinesin heavy chain was reported to disrupt the ability of the enzyme active site to catalyze ATP P-O bond cleavage. We expressed R210K homodimers as well as isolated R210K heads and confirmed that both are essentially inactive. We then co-expressed tagged R210K subunits with untagged wild-type subunits and affinity purified R210K/wild-type heterodimers together with the inactive R210K homodimers. In contrast to the R210K head or homodimer, the heterodimer was a highly active (>50% of wild-type) microtubule-stimulated ATPase, and the heterodimer displayed high duty ratio processive movement in single-molecule motility experiments. Thus, dimerization of a subunit containing the inactivating mutation with a functional subunit can complement the mutation; this must occur either by lowering or by bypassing kinetic barriers in the ATPase or mechanical cycles of the mutant head. The observations provide support for kinesin-1 gating mechanisms in which one head stimulates the rate of essential processes in the other.
Kinesin-1 and myosin V are the best characterized examples of motor enzyme molecules that move unidirectionally and continuously along the cytoskeletal filaments (microtubules and F-actin, respectively) that serve as their tracks (1–3). Each enzyme maintains a tight association with its track, undergoing tens or hundreds of catalytic turnovers without detaching from or sliding freely relative to it. Such “processive high duty ratio” movement allows individual isolated enzyme molecules to make forward progress even when opposed by substantial elastic loads. This property may be essential to the biological function of these enzymes as engines that propel subcellular organelles through the viscoelastic cytoplasm. Processive high duty ratio movement is thought to require that the actions of the two identical motor or “head” domains of the enzyme be coordinated so that at least one head is attached to the track at any given instant during movement. This head coordination hypothesis is consistent with observations that one-headed kinesin-1 constructs lack the ability to move processively (4–7) and with high duty ratio (8). The hypothesis is also consistent with demonstrations in two-headed molecules that catalytic turnover of the two heads is not independent, at least under some conditions (9–11).
The combined results from a series of studies (10, 12–17) show that kinesin heads move by an alternating-sites hand-over-hand mechanism, in which the two heads alternately hydrolyze ATP and each head moves forward 16 nm along the microtubule in every other hydrolysis cycle. This results in net movement of the enzyme as a whole by 8 nm per cycle. Curiously, the two heads do not behave quite identically; the data suggest that even- and odd-numbered steps begin with different conformations of the domain that links the heads to the C-terminal coiled-coil and that even and odd cycles have different kcat values under some circumstances (15, 17).
In addition to defining the sequence of domain movements that underlie kinesin motility, the alternating-sites hand-over-hand mechanism provides an attractive hypothesis to explain the head coordination that is thought to be the responsible for the high duty ratio processive property of kinesin movement. In particular, it is widely accepted that such head coordination, and hence processive movement, is a consequence of the alternating ATP hydrolysis between the two heads (9, 18–20). Alternation of the ATPase cycles is enforced by the mechanical linkage of the two subunits through the neck linker domains. Specifically, the linkage is thought to mediate gating processes in which a step in the mechanochemical cycle of one head is slowed or blocked by the presence of the other head in particular states. Such inhibitory gating processes include i) blocking of ADP release by the “tethered” head until nucleoside triphosphate binds to the “bound” head (9, 11, 21), and ii) slowing of ATP binding to the leading head when the neck linker in the trailing head is docked (22, 23). Conversely, acceleration of trailing head detachment by mechanical strain transmitted from the leading head may represent an unusual stimulatory gating process, but there are disagreements concerning the magnitude and mechanistic significance of this process (4, 23, 24).
To investigate possible additional stimulatory gating processes in kinesin, we prepared by co-expression a heterodimeric kinesin derivative in which one head has the wild-type sequence and the other has a single point mutation. Unlike previous studies using heterodimers (4, 10, 25, 26), we chose to focus on an active site mutation known to specifically target the chemical step of ATP hydrolysis. The Drosophila kinesin-1 R210K mutation almost completely blocks the ATP phosphoanhydride bond hydrolysis step while having little or no effect on other head functions (27). We here show that the R210K/wild-type heterodimer has novel properties distinct from those of previously characterized heterodimers. Pairing the inactive R210K subunit with a wild-type subunit produces a dimer with essentially the full mechanochemical function of homodimeric wild-type kinesin.
MATERIALS AND METHODS
Plasmids
Plasmid pWC2 encodes wild-type subunit K401-BIO-H6, which consists of the first 401 residues of Drosophila kinesin heavy chain fused to a biotin acceptor domain (this fusion is K401-BIO of ref. 5) with the C-terminal extension (one-letter amino acid code) LSETSGHHHHHH added to permit Ni2+ chelation affinity purification. pWC2 is identical to pEY4 (ref. 5 and Genbank AY621072) except for the insertion 5′-TATCTGAGACTAGTGG-GCACCACCATCACCATCACTGA-3′, which encodes the extension, at position 1476. pEY4 is identical to pSK4 (28), except that the coding region is that given in Genbank AY621072. Plasmid pTTR210K encoding the mutant subunit K401-BIO-H6 R210K was made from pWC2 by directed mutagenesis (QuikChange, Stratagene) of the R210 CGA codon to AAA. pTTSTOP, which encodes untagged wild-type K401, was generated by directed mutagenesis of codon 402 (CGC) of pEY4 to the stop codon TGA. Co-expression experiments used pTTSTOP2, which was prepared by ligation of the BstBI/HindIII coding region fragment of pTTSTOP with the BsaHI/HindIII replication origin fragment of pACYC184 (29). pTTR210K340, which encodes the tagged mutant kinesin head K340-BIO-H6 R210K, was constructed by ligating the 374 bp PstI/EcoRI fragment of pKA2 (28) to the 3390 bp PstI/EcoRI fragment of pTTR210K. The entire coding regions of pTTSTOP, pTTSTOP2 and pTTR210K were confirmed by sequencing.
Protein Expression and Purification
K401-BIO-H6 R210K homodimers were expressed from strain BL21(DE3) pLysS pTTR210K and purified as described (5), except that a Ni2+ chelation column (Qiagen Ni-NTA Superflow) was used in place of the avidin column. The resin was incubated with lysate overnight, and then washed for 2 h with 20 mM imidazole-Cl− pH 7.2, 4 mM MgCl2, 10 mM 2-mercaptoethanol. Tagged kinesin was eluted with 500 mM imidazole-Cl− pH 7.2, 4 mM MgCl2, 10 mM 2-mercaptoethanol and dialyzed against 50 mM imidazole-Cl− pH 6.7, 4 mM MgCl2, 10 mM 2-mercaptoethanol prior to ion-exchange chromatography. The same procedure was used to purify heterodimers from BL21(DE3) pTTR210K pTTSTOP2, wild-type homodimers from BL21(DE3) pLysS pWC2 and monomeric mutant heads from BL21(DE3) pLysS pTTR210K340. Untagged wild-type K401 homodimer was expressed using BL21(DE3) pLysS pTTSTOP, and BL21(DE3) pLysS pWC2 was used to express K401-BIO-H6 homodimer.
Sedimentation
Protein diffusion coefficients were measured by fitting (SVEDBERG; John Philo Software) absorbance profiles obtained in a synthetic boundary experiment at low speed (8,000 r.p.m.) in an analytical ultracentrifuge (XL-A with An60Ti rotor; Beckman-Coulter) in 20 mM imidazole-Cl− pH 7.2, 4 mM MgCl2, 0.1 mM tris(2-carboxyethyl)phosphine at 20°C. Absorbance of the sample before centrifugation (OD280 = 1.2; 1.0 cm path length) corresponded closely with that measured in the centrifuge (OD280 = 1.4; 1.2 cm), demonstrating the absence of significant quantities of protein aggregates large enough to be pelleted. Profiles from a conventional meniscus depletion experiment (44,000 r.p.m.) were then analyzed by differential methods and fit to determine the sedimentation coefficient (30). Separate peaks from the heterodimer and mutant homodimer were not resolved in sedimentation experiments because the differences between the molecular weights and shapes of these species are small.
ATPase and Motility Assays
Steady-state ATPase activity was measured as described (28), except that the tubulin concentration was 1.2 mg/mL. Motility assays and bead tracking were conducted essentially as described (5) except that the beads were 150 nm diameter and the ATP regeneration system was omitted. A bead movement was included in the data set only when the complete sequence of bead binding to the microtubule, processive movement, and release was observed, and only movements longer than a minimum length needed for reliable detection (225 and 500 nm for heterodimer and wild-type, respectively) were used for subsequent quantitative analysis. To measure the characteristic run length of a given enzyme species, individual movement length measurements were binned (100 and 500 nm bin width for heterodimer and wild-type, respectively) and normalized, and the resulting histogram was fit with a single exponential probability density function.
RESULTS
Properties of R210K Homodimers and Monomers
In the mutant enzyme studied here, arginine substitutes for lysine at position 210 (R210K) in the well-conserved switch I loop (31) adjacent to the kinesin active site. Despite the conservative nature of this mutation, Klumpp et al. (27) showed that a homodimeric R210K construct is almost completely catalytically inactive; its ATPase kcat of 0.22/dimer/s is ~200-fold reduced from the corresponding wild-type construct. In contrast, microtubule association kinetics and nucleotide affinities of the mutant homodimer were similar to wild-type. Furthermore, nucleotide-stimulated ADP release was unimpaired in the R210K homodimer, evidence that machinery for head-head communication remains functional. To confirm and extend these earlier studies, we expressed and purified a similar mutant kinesin consisting of the N-terminal 401 amino acids of Drosophila kinesin with C-terminal biotin and six-histidine tags (K401-BIO-H6 R210K). K401 constructs lack the complete C-terminus of intact kinesin heavy chain, but contain at least 40 amino acids of the α-helical coiled-coil neck domain, a length sufficient to induce formation of stable dimers (32–34). We also prepared a K340-BIO-H6 R210K construct that lacks this dimerization domain; that protein was homogeneously monomeric as expected (Figure 1). The ATPase specific activities of both the mutant monomer and homodimer constructs were dramatically reduced relative to wild-type dimers (Table 1, lines 4 and 2 vs. line 1). We further tested the ability of the mutant homodimers to move streptavidin-coated 150 nm diameter polystyrene beads along immobilized microtubules, using an experimental design in which wild-type homodimers show robust activity (5). No bead movement was detected at enzyme:bead mole ratios of 1:1, 10:1, or 100:1 (Table 1 and data not shown). At all ratios, bead-labeled mutant homodimers remained bound at fixed positions on microtubules in 1 mM ATP or in controls with no added nucleotide, but did not bind in controls with 1 mM ADP. These findings are consistent with observations in bulk experiments (27) that linkage between nucleotide and microtubule binding is retained in the mutant.
Figure 1.

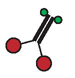
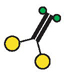
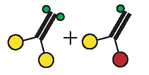
Population distributions (g) of apparent sedimentation coefficients (s*) in sedimentation velocity experiments on the heterodimer (blue) and mutant monomer (purple) samples (left). Circles are measurements; lines are theoretical non-interacting single-species distributions using the sedimentation and diffusion coefficients (right, rows 1–2) determined by fitting (see Materials and Methods). Cartoons show the domain structures of protein species in the samples: red, wild-type head; yellow, R210K mutant head; thick black, neck coiled-coil; green, biotin and six-histidine tags; white, biotin tag. Wild-type data (right, rows 3–4) from (5) are included for comparison.
Table 1.
ATPase Activity and Motility of Kinesin Derivatives
| Enzyme Species a | ATPase Activity b (mean ± S.D.) (s−1) | Movement Velocity c (mean ± S.D.) (μm s−1) | Run Frequency c (mean ± S.E.) (min −1) | Run Length c (± S.E.) (μm) |
|---|---|---|---|---|
|
24.3 ± 0.5 (N = 6) | 0.8 ± 0.2 (N = 79) | 1.0 ± 0.2 (N = 25) | ≥1.5 ± 0.1 d (N = 79) |
|
0.19 ± 0.02 (N = 6) | <0.00010e (N = 8) | <0.04 e | No motility |
|
10.0 ± 0.1 f (N = 3) | 0.09 ± 0.12 (N = 50) | 0.4 ± 0.1 (N = 10) | 0.31 ± 0.06 (N = 50) |
|
|
0.18 ± 0.02 (N = 3) | N.D.g | N.D.g | N.D.g |
Schematic diagrams of enzyme species present, using the same symbols as in Figure 1.
Microtubule-stimulated ATPase specific activity per dimer (rows 1–3) or per monomer (row 4) in 1 mM ATP, 1.2 mg/ml microtubules, 25 °C.
In single-molecule bead movement assay at 1:1 enzyme dimer:bead mole ratio, 1 mM ATP, 22–23 °C.
Data set included some runs where motor ran off the microtubule end.
No motility detected. Value given is the detection limit.
Implies that heterodimer specific activity is ~15 s−1, since the preparation is estimated by gel densitometry to contain ~35% mutant homodimers, which have negligible activity.
Not determined.
Wild-Type/R210K Heterodimers
Having confirmed that R210K inactivates kinesin both in homodimers and in isolated heads, we made preparations containing heterodimers with one wild-type and one mutant subunit. Tagged mutant subunits (K401-BIO-H6 R210K) and untagged wild-type subunits (K401) were co-expressed and proteins that contained at least one tagged subunit were purified by affinity and ion exchange chromatography, yielding mutant/wild-type heterodimers mixed with the inactive mutant homodimers. Denaturing electrophoresis of the purified protein reveals two bands at the expected sizes of the tagged and untagged subunits (Figure 2). The tagged:untagged molar concentration ratio estimated by densitometry was 1.6:1. That is close to the 2:1 ratio expected if the two types of subunits are expressed equally, subunits associate randomly, and untagged wild-type homodimers are fully removed during purification. Application of the purification protocol to control cells expressing only untagged K401 yielded no detectable protein (Figure 2, lane 4), confirming that the untagged homodimers are efficiently removed.
Figure 2.

SDS-polyacrylamide gel electrophoresis of kinesin preparations after purification to remove proteins lacking the six-histidine tag: co-expressed K401 and K401-BIO-H6 (R210K) (lane 1, lower and upper bands, respectively); K401-BIO-H6 (R210K) alone (lane 2); K401-BIO-H6 alone (lane 3); and K401 alone (lane 4). The lane 4 sample was loaded in 30-fold volumetric excess relative to lane 1 to confirm that removal of the untagged homodimer is essentially complete. Diagrams show the protein species expected in each sample using the same symbols as in Figure 1. Scale (left): molecular weight markers.
Sedimentation velocity measurements on the heterodimer sample demonstrate that it is uniformly dimeric, with sedimentation and diffusion coefficients indistinguishable from those of wild-type homodimers (Figure 1). Thus, the R210K mutation does not affect the oligomeric state of the construct nor significantly alter its overall shape, consistent with previous conclusions (27) that R210K causes a local structural alteration at the catalytic site, not a global folding defect.
If the kinesin mechanism requires that the heads alternate catalysis (9, 18–20), a mutation which blocks ATP hydrolysis in one head of a mutant/wild-type heterodimer would be expected to prevent steady-state catalytic turnover of the wild-type head. Despite the fact that R210K heads are essentially inactive in both monomer and homodimer contexts, the heterodimer sample microtubule-stimulated ATPase specific activity is only ~2-fold reduced relative to that of wild-type homodimers (Table 1). The observed activity was not from the mutant homodimers present in the sample; that species is inactive (Table 1). Nor can the activity be explained by postulating contamination of the heterodimer sample by wild-type homodimers, because the wild-type homodimer and heterodimer sample ATPase activities were observed to have quite different salt dependences: the wild-type homodimer ATPase decreased ~58-fold upon addition of 150 mM NaCl whereas heterodimer ATPase decreased by only ~8-fold.
Motility of Heterodimer Sample
Can ATP hydrolysis by the mutant/wild-type heterodimer drive processive movement of a single enzyme molecule along a microtubule? The heterodimer sample was mixed with streptavidin-coated beads at a 1:1 mole ratio of protein dimers to beads; in the motility experiment these beads moved processively and unidirectionally along microtubules, at a velocity ~9-fold slower than that of wild-type homodimers (Table 1, Video S1, and Video S2).
At the 1:1 dimer:bead ratio, most beads had ≤1 enzyme molecule. Nonetheless, the frequency of heterodimer bead movements was roughly similar to that observed with wild-type homodimers (Table 1). This is strong evidence that processive movement can be generated by single heterodimers; it is not restricted to the extremely small subpopulation of beads with >1 enzyme molecule simultaneously interacting with the microtubule (2). The small (~2-fold) reduction in movement frequency in the heterodimer sample relative to wild-type homodimers is explained in large part by the fact that a significant fraction of molecules in the former are inactive mutant homodimers.
In principle, the heterodimer preparation might contain wild-type homodimers due to incomplete removal in the purification, homologous recombination between the plasmids, or by subunit exchange after purification. Two lines of evidence confirm that the observed motility is not from wild-type homodimers. First, no motility was seen in control experiments in which the streptavidin-coated beads were pre-treated with excess biotin before adding enzyme. This implies that the movements observed in the heterodimer sample arise from enzyme molecules specifically attached to the beads by a biotin-streptavidin linkage; the wild-type homodimers should lack the biotin tag. Second, movement in the heterodimer samples has different properties from that driven by wild-type homodimers, suggesting that the two arise from distinct enzyme species. Velocity distributions observed for wild-type homodimers and the heterodimers are nearly disjoint (Figure 3). Also, the characteristic distance moved by each enzyme molecule (“run length”) was significantly lower for heterodimers, and the mean duration of the run (i.e., run length divided by mean velocity) was substantially larger (Table 1).
Figure 3.

Movement velocity distributions for wild-type homodimer (white; N = 79) and wild-type/R210K heterodimer (black; N = 50) at 1:1 enzyme dimer:bead mole ratio, 1 mM ATP, 22–23 °C. The overlap (gray) between the two distributions is minimal.
In control experiments, we examined motility from equimolar mixtures of wild-type K401-BIO-H6 with the mutant/wild-type heterodimer sample or with mutant homodimers. In the former, the bead velocity distribution had two peaks, one at the wild-type velocity (~800 nm/s) and one at the heterodimer velocity (~95 nm/s). The latter had only a single peak at the wild-type velocity. These experiments demonstrate that movement of wild-type homodimers is not slowed by the simultaneous presence of heterodimers or mutant homodimers, confirming that contaminating wild-type homodimers do not cause the slow motility observed in the heterodimer sample.
Nanometer-scale tracking of single molecules (35) was used to determine whether beads driven by the mutant/wild-type heterodimers moved unidirectionally. As is the case with wild-type homodimers, clear backwards displacements were rare (Figure 4). For each molecule tracked, the distribution of bead positions normal to the microtubule axis was narrow and movement was parallel to the axis. These observations demonstrate that the R210K/wild-type heterodimers, like wild-type homodimers, follow microtubule protofilament tracks and therefore must move processively (5, 7). Protofilament tracking is confirmed by observation that in traces with sufficiently low movement velocity, movement occurred in discrete ~8 nm steps (Figure 4B).
Figure 4.

(A) Movement records of five individual bead-labeled wild-type/R210K heterodimer molecules in 1 μM ATP. Samples were prepared at 1:1 enzyme dimer:bead mole ratios and observed at 22–23°C. Traces show bead movements both parallel (top panels) and perpendicular (bottom panels) to the microtubule axis. Positions were recorded at 30 Hz (gray) and smoothed with a 10-point mean filter (black). (B) Expanded plot of a portion of a run taken at 0.5 μM ATP that shows individual ~8 nm steps. Horizontal grid lines are spaced at 8 nm intervals.
A different heterodimer, R14A/wild-type, was shown to exhibit alternating long and short dwells between 8 nm steps (10), consistent with alternating movement by the two heads. Such “limping” (15) is less pronounced in our R210K/wild-type heterodimer, but we did see alternation of long and short dwells in some records (e.g., Figure 4B, 4–10 s). A larger and higher resolution data set would be required to quantitatively evaluate the extent of R210K/wild-type limping because limping measurements on weakly limping motors are subject to statistical bias as described previously (15).
A high duty ratio motor enzyme can be defined as one that does not freely slide along or detach from its track during most or all of its catalytic cycle (8). This is the key feature of kinesin movement that is thought to require coordinated action of the two heads (8): one-headed constructs of kinesin and its homologs can be processive (36) or non-processive (4–6, 8), but all have low duty ratios. Unidirectional processive movement of single wild-type kinesin homodimers implies that the enzyme has a high duty ratio, because any significant time spent sliding or detached would result in reverse movements due to Brownian motion (8, 36). The observation that single R210K/wild-type heterodimers move processively and unidirectionally confirms that they also have a high duty ratio.
DISCUSSION
Association of the inactive R210K subunit with a wild-type subunit produces a dimer with near-wild-type ATPase and motor activity
Kaseda et al. (10) hypothesized that the activity of some kinesin mutant/wild-type heterodimers can be explained by a hand-over-hand mechanism in which the mutation slows a step in every other catalytic cycle, leading to the alternating long and short dwell times that they observed between successive 8 nm steps. In its simplest form, this hypothesis predicts that movement speed and ATPase kcat of a wild-type/mutant heterodimer will be no more than twice those of the corresponding mutant/mutant homodimer, since the slow step occurs in every cycle of the latter and in every other cycle of the former.
The properties of the R210K/wild-type heterodimer do not conform to this prediction. We observed that this species moved rapidly along microtubules, and it hydrolyzed ATP at rates >50-fold faster than the R210K homodimer. The high activity of the heterodimer is a striking contrast to previously reported processive mutant/wild-type kinesin heterodimers, none of which has steady-state activity significantly greater than twice that of the corresponding mutant homodimer (10, 26).1 We found single R210K/wild-type heterodimers to be processive, high duty ratio motors like wild-type homodimers. Additional experiments confirmed that the activities attributed to heterodimers are not due to contamination by wild-type homodimers or mutant homodimers and are not a result of aggregation or higher-order oligomerization of heterodimers.
Intra-dimer complementation of the motility defect in the R210K subunit
High duty ratio processive motility requires two heads; it is absent in single-headed kinesin-1 constructs (4–7). The data presented here show that motility is restored when a wild-type subunit is associated with an R210K subunit that in other contexts (monomer or homodimer) is non-motile and almost completely inactive. Analogous effects have been seen with other motor enzymes: for both myosin II and Rep helicase, dimer-specific functions can be restored when a wild-type subunit associates with a non-functional mutant subunit (38, 39). Since the R210K/wild-type kinesin heterodimer takes 8 nm steps, tracks microtubule protofilaments, and appears to limp, there is a strong likelihood that it moves by the same pattern of alternating hand-over-hand head movements as wild-type homodimers.
In the most widely accepted view of the kinesin catalytic cycle (see refs. 23, 40), the enzyme hydrolyzes ATP in the trailing head in a configuration in which the leading head is empty and both heads are bound to the microtubule (Figure 5A, uppermost species). Klumpp et al. (27) showed that the hydrolysis step is blocked by the R210K mutation. How is this kinetic barrier overcome in the R210K/wild-type heterodimer? In purely formal terms, the presence of the wild-type subunit can complement the kinetic defect in the R210K subunit either by acting directly to lower the kinetic barrier (Figure 5, orange) or by bypassing the barrier through an alternative movement mechanism (for example, Figure 5, violet). In the schemes shown, both possibilities result in hand-over-hand movement coupled to ATP hydrolysis. The key difference is that the former hydrolyzes one ATP per 8 nm step (Figure 5B), like wild-type homodimers, while the latter hydrolyzes one ATP per two steps (Figure 5C). These two possibilities could in principle be distinguished by comparing the ATPase specific activity in bulk with the single-molecule movement velocity measured under the same conditions (35, 41). However, approaches based on specific activity will not be reliable until the heterodimer mechanism is understood in enough detail that active site titatration methods can be used to make accurate measurements of the concentration of active heterodimer molecules (35, 41), and the extent to which heterodimer ATPase might be partially uncoupled from motility is assessed. Without such additional data, both barrier lowering and barrier bypass pathways remain as plausible explanations for the complementation of the R210K kinetic defect by heterodimerization with a wild-type subunit.
Figure 5.

Scheme for the mechanochemical function of R210K/wild-type heterodimers. Microtubule protofilaments with alternating αβ-tubulin dimers (gray/white) are shown with plus ends to the right; T, ATP; D, ADP; Pi, inorganic phosphate. (A) Overcoming the kinetic barrier when the R210K head is in the trailing position. In a single turnover cycle for wild-type kinesin homodimers (analogous to that shown in the green box, but with two identical heads), ATP is hydrolyzed in the trailing head from a state (top) in which both heads are bound to the microtubule. In the R210K/wild-type heterodimer, individual cycles with the mutant head in the leading position (not shown) alternate with individual cycles (green box) in which the wild-type head (red) is leading and the mutant head (yellow) is trailing. In these cycles where the R210K head is trailing, its pairing with a wild-type head in the heterodimer must enable it to evade the kinetic barrier to ATP hydrolysis (red “X”) present in isolated R210K monomers. Evasion could be accomplished by lowering the barrier (orange arrow), allowing the trailing mutant head to hydrolyze ATP much more rapidly in the heterodimer context than in the monomer context, or by bypassing the barrier via a pathway in which the trailing mutant head steps forward without accompanying hydrolysis of ATP. The mechanism of this putative bypass pathway might consist of ATP binding to the leading head, followed by stepping, followed by release of unhydrolyzed ATP from the (now leading) mutant head (violet arrow), but other arrangements of steps are also possible. (A possible alternative is that ATP remains continuously bound to the mutant head, as in the short runs seen in mixtures of ATP and an inhibitor in ref. 47.) In the wild-type kinesin mechanism, the position of ADP-associated heads has been variously reported as microtubule-bound, tethered, or docked to the leading head (see discussion and citations in refs. 40, 50); for simplicity, only the first of these possibilities is illustrated. Docked and undocked neck linker strands in the trailing head are represented with straight and curved lines, respectively. (B and C) Summary of predicted reaction stoichiometries for the barrier lowering (B) and barrier bypass (C) mechanisms. Each box shows the net stoichiometry of substrate binding and product release for two consecutive 8 nm steps; the step with the mutant head (yellow) trailing is shown first, followed by the step with the wild-type head (red) trailing. In these schemes, the first 8 nm step is assumed to proceed by the pathways shown as orange (B) or violet (C) arrows in panel A. The second step is assumed to proceed by the wild-type mechanism since the functions performed by the leading head (binding ATP and stimulating nucleotide release from the other head) are essentially unaffected by the R210K mutation (27).
Precedents for Overcoming the R210K Kinetic Barrier in Heterodimers
All kinesin and myosin motor domains have an invariant arginine residue in the switch I motif at the position corresponding to R210 in Drosophila kinesin (31, 42). For myosin, there is extensive evidence (reviewed in ref. 42) that the arginine side chain, together with a adjacent conserved glutamic acid side chain to which it forms a salt bridge when the nucleotide binding pocket is closed, play an essential role in nucleoside triphosphate hydrolysis by precisely positioning a water molecule adjacent to the γ-phosphate of ATP. A similar role has been proposed for R210 in kinesin (27). In both families of enzymes, single point mutations at these positions abolish or greatly reduce microtubule- or actin-stimulated ATPase activity of isolated monomer heads (this report and refs. 27, 39, 42–45). Thus, available data suggest that mutations at R210 impair ATPase function primarily by local disruption of the active site geometry needed for catalysis. This view is supported by structural modeling which shows that the conservative R210K substitution is expected merely to subtly alter the network of H-bonds in the ATP-bound active site (27).
If R210K causes only a subtle localized alteration in active site conformation, the barrier lowering pathway is a reasonable explanation for its near wild-type function. Specifically, one might hypothesize that when the mutant head is trailing (yellow head in the topmost state of Figure 5A), mechanical strain from the bound leading head transmitted through the neck linker might be sufficient to rearrange the structure of the active site and restore the hydrolytic activity that is absent in the R210K monomer, which lacks the internal mechanical strain of the two head bound state. There is ample precedent for mechanical strain transmitted through the neck linkers altering the kinetics of chemical processes within the heads; for example, this is thought to be the mechanism that prevents premature binding of ATP to the lead head (22, 46).
Conversely, the essential steps of the barrier bypass pathway shown in Figure 5A are also supported by precedents in the literature. This pathway (violet arrow) postulates detachment of the trailing mutant head (yellow) while it is in the strongly microtubule binding ATP state. This process is expected to be slower than detachment of an ADP bound trailing head as occurs in the wild-type kinesin mechanism (Figure 5A, green box), but inhibitor studies suggest that it can occur at a substantial rate (23, 47). The feasibility of the bypass pathway is supported by a computational model of R210K/wild-type heterodimer function (48). Also, the idea that a kinesin-1 heterodimer can function with only one catalytically active head parallels the recent revelation that the heterodimeric kinesin-14 Kar3/Vik1 functions as a motor despite the absence of catalytic activity in the Vik1 head (49). However, the barrier bypass pathway proposed here, in which the inactive R210K head binds ATP only to release it later in the cycle, is probably not directly applicable to Kar3/Vik1 since the a structure of an isolated Vik1 head shows that the nucleotide binding pocket is absent in this protein (49).
Failure to Overcome the Kinetic Barrier in R210K Homodimers
In the hypothesized scheme for heterodimer function, 8 nm steps occurring through the barrier lowering or bypass pathway alternate with steps occurring by the wild-type pathway (Figure 5B, C). The implication is that since no such alternation can take place in the R210K homodimer, reactions of that enzyme should be restricted to the altered pathway in every step. Thus, the bypass pathway predicts no ATPase activity and no ATP-driven motility, in good agreement with the observed properties of the homodimer (this study and ref. 27). In contrast, the barrier lowering pathway predicts that the homodimer will be functional as long as it can successfully enter the two head bound state shown at the top of Figure 5A. Therefore, to explain the low activity of the homodimer in the barrier lowering scheme, it is necessary to make the additional assumption that the enzyme becomes trapped in a kinetically refractory state (e.g., an off-cycle one-head-bound species) from which it is unable to efficiently enter the catalytic cycle. This scenario is consistent with available data, but there is no independent evidence for such a state.
Implications for the Mechanism of Wild-Type Kinesin-1
High duty ratio processive movement by kinesin-1 requires coordination of the cycles of the two head domains. Most gating processes that have been demonstrated or proposed to enforce this coordination are inhibitory in nature. In both the slowing by internal mechanical strain of lead head ATP binding (22) and in the blockage of ADP release from the tethered head (9), a reaction step that occurs readily in one-headed constructs is inhibited (in some parts of the mechanochemical cycle) as a consequence of the head-head linkage in the dimer. It is only in the hypothesized strain-induced dissociation of the trailing head (4, 24) that the partner head is proposed to have a stimulatory rather than inhibitory effect. The observations that the presence of the wild-type head in the R210K/wild-type heterodimer complements the kinetic defect in the mutant head provides a new example of such stimulatory subunit-subunit interactions and raises the possibility that additional stimulatory gating processes may exist in the mechanism of wild-type kinesin-1.
Acknowledgments
We thank Radhika Subramanian and Feng Wang for their collaboration in developing the purification protocol and Wendy Choy, Han Chen, and Kelsa Teeters for providing DNA constructs. We also thank Susan Gilbert, Chris Miller, and Walter Stafford for advice or comments on an earlier version of the manuscript.
This work was funded by the National Institute of General Medical Sciences, GM43369.
Footnotes
A potential exception is the loop 12/helix 5 microtubule binding site triple mutant Y274A/R278A/K281A. However, this mutant homodimer binds microtubules so weakly that it was not possible to measure the ATPase kcat or single-molecule movement velocity (10, 37). Thus, homo- and heterodimer activities for this species cannot be compared.
SUPPORTING INFORMATION AVAILABLE: Differential interference contrast microscopy video recording of polystyrene bead-labeled wild-type kinesin homodimers (Video S1) or wild-type/R210K heterodimers (Video S2) moving along stationary microtubules at 1 mM ATP. The microtubule was near vertical in S1 and was diagonal (upper right to the lower left) in S2. Frame width ~2μm in both S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Howard J, Hudspeth AJ, Vale RD. Movement of microtubules by single kinesin molecules. Nature. 1989;342:154–158. doi: 10.1038/342154a0. [DOI] [PubMed] [Google Scholar]
- 2.Block SM, Goldstein LS, Schnapp BJ. Bead movement by single kinesin molecules studied with optical tweezers. Nature. 1990;348:348–352. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 3.Mehta AD, Rock RS, Rief M, Spudich JA, Mooseker MS, Cheney RE. Myosin-V is a processive actin-based motor. Nature. 1999;400:590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- 4.Hancock WO, Howard J. Processivity of the motor protein kinesin requires two heads. J Cell Biol. 1998;140:1395–1405. doi: 10.1083/jcb.140.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berliner E, Young EC, Anderson K, Mahtani HK, Gelles J. Failure of a single-headed kinesin to track parallel to microtubule protofilaments. Nature. 1995;373:718–721. doi: 10.1038/373718a0. [DOI] [PubMed] [Google Scholar]
- 6.Vale RD, Funatsu T, Pierce DW, Romberg L, Harada Y, Yanagida T. Direct observation of single kinesin molecules moving along microtubules. Nature. 1996;380:451–453. doi: 10.1038/380451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelles J, Berliner E, Young EC, Mahtani HK, Perez-Ramirez B, Anderson K. Structural and functional features of one- and two-headed biotinated kinesin derivatives. Biophys J. 1995;68:276S–282S. [PMC free article] [PubMed] [Google Scholar]
- 8.Young EC, Mahtani HK, Gelles J. One–headed kinesin derivatives move by a nonprocessive, low-duty ratio mechanism unlike that of two-headed kinesin. Biochemistry. 1998;37:3467–3479. doi: 10.1021/bi972172n. [DOI] [PubMed] [Google Scholar]
- 9.Hackney DD. Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc Natl Acad Sci USA. 1994;91:6865–6869. doi: 10.1073/pnas.91.15.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaseda K, Higuchi H, Hirose K. Alternate fast and slow stepping of a heterodimeric kinesin molecule. Nat Cell Biol. 2003;5:1079–1082. doi: 10.1038/ncb1067. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert SP, Moyer ML, Johnson KA. Alternating site mechanism of the kinesin ATPase. Biochemistry. 1998;37:792–799. doi: 10.1021/bi971117b. [DOI] [PubMed] [Google Scholar]
- 12.Hoenger A, Thormahlen M, Diaz-Avalos R, Doerhoefer M, Goldie KN, Muller J, Mandelkow E. A new look at the microtubule binding patterns of dimeric kinesins. J Mol Biol. 2000;297:1087–1103. doi: 10.1006/jmbi.2000.3627. [DOI] [PubMed] [Google Scholar]
- 13.Hua W, Chung J, Gelles J. Distinguishing inchworm and hand-over-hand processive kinesin movement by neck rotation measurements. Science. 2002;295:844–848. doi: 10.1126/science.1063089. [DOI] [PubMed] [Google Scholar]
- 14.Yildiz A, Tomishige M, Vale RD, Selvin PR. Kinesin walks hand-over-hand. Science. 2004;303:676–678. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]
- 15.Asbury CL, Fehr AN, Block SM. Kinesin moves by an asymmetric hand-over-hand mechanism. Science. 2003;302:2130–2134. doi: 10.1126/science.1092985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schief WR, Clark RH, Crevenna AH, Howard J. Inhibition of kinesin motility by ADP and phosphate supports a hand-over-hand mechanism. Proc Natl Acad Sci U S A. 2004;101:1183–1188. doi: 10.1073/pnas.0304369101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higuchi H, Bronner CE, Park HW, Endow SA. Rapid double 8-nm steps by a kinesin mutant. Embo J. 2004;23:2993–2999. doi: 10.1038/sj.emboj.7600306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howard J. The movement of kinesin along microtubules. Annu Rev Physiol. 1996;58:703–729. doi: 10.1146/annurev.ph.58.030196.003415. [DOI] [PubMed] [Google Scholar]
- 19.Sablin EP, Fletterick RJ. Coordination between motor domains in processive kinesins. J Biol Chem. 2004;279:15707–15710. doi: 10.1074/jbc.R300036200. [DOI] [PubMed] [Google Scholar]
- 20.Skowronek KJ, Kocik E, Kasprzak AA. Subunits interactions in kinesin motors. European Journal of Cell Biology. 2007;86:559–568. doi: 10.1016/j.ejcb.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Ma YZ, Taylor EW. Interacting Head Mechanism of Microtubule-Kinesin ATPase. J Biol Chem. 1997;272:724–730. doi: 10.1074/jbc.272.2.724. [DOI] [PubMed] [Google Scholar]
- 22.Rosenfeld SS, Fordyce PM, Jefferson GM, King PH, Block SM. Stepping and stretching. How kinesin uses internal strain to walk processively. J Biol Chem. 2003;278:18550–18556. doi: 10.1074/jbc.M300849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guydosh NR, Block SM. Backsteps induced by nucleotide analogs suggest the front head of kinesin is gated by strain. Proc Natl Acad Sci U S A. 2006;103:8054–8059. doi: 10.1073/pnas.0600931103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crevel IM, Nyitrai M, Alonso MC, Weiss S, Geeves MA, Cross RA. What kinesin does at roadblocks: the coordination mechanism for molecular walking. EMBO J. 2004;23:23–32. doi: 10.1038/sj.emboj.7600042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skowronek K, Kasprzak AA. A two-plasmid system for independent genetic manipulation of subunits of homodimeric proteins and selective isolation of chimeric dimers. Anal Biochem. 2002;300:185–191. doi: 10.1006/abio.2001.5456. [DOI] [PubMed] [Google Scholar]
- 26.Kaseda K, Higuchi H, Hirose K. Coordination of kinesin’s two heads studied with mutant heterodimers. Proc Natl Acad Sci U S A. 2002;99:16058–16063. doi: 10.1073/pnas.252409199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klumpp LM, Mackey AT, Farrell CM, Rosenberg JM, Gilbert SP. A kinesin switch I arginine to lysine mutation rescues microtubule function. J Biol Chem. 2003;278:39059–39067. doi: 10.1074/jbc.M304250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berliner E, Mahtani HK, Karki S, Chu LF, Cronan JE, Jr, Gelles J. Microtubule movement by a biotinated kinesin bound to streptavidin-coated surface. J Biol Chem. 1994;269:8610–8615. [PubMed] [Google Scholar]
- 29.Rose RE. The nucleotide sequence of pACYC184. Nucleic Acids Res. 1988;16:355. doi: 10.1093/nar/16.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philo JS. A method for directly fitting the time derivative of sedimentation velocity data and an alternative algorithm for calculating sedimentation coefficient distribution functions. Anal Biochem. 2000;279:151–163. doi: 10.1006/abio.2000.4480. [DOI] [PubMed] [Google Scholar]
- 31.Vale RD. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozielski F, Sack S, Marx A, Thormahlen M, Schonbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 33.Correia JJ, Gilbert SP, Moyer ML, Johnson KA. Sedimentation studies on the kinesin motor domain constructs K401, K366, and K341. Biochemistry. 1995;34:4898–4907. doi: 10.1021/bi00014a047. [DOI] [PubMed] [Google Scholar]
- 34.Huang TG, Suhan J, Hackney DD. Drosophila kinesin motor domain extending to amino acid position 392 is dimeric when expressed in Escherichia coli. J Biol Chem. 1994;269:16502–16507. [PubMed] [Google Scholar]
- 35.Hua W, Young EC, Fleming ML, Gelles J. Coupling of kinesin steps to ATP hydrolysis. Nature. 1997;388:390–393. doi: 10.1038/41118. [DOI] [PubMed] [Google Scholar]
- 36.Okada Y, Hirokawa N. A processive single-headed motor: kinesin superfamily protein KIF1A. Science. 1999;283:1152–1157. doi: 10.1126/science.283.5405.1152. [DOI] [PubMed] [Google Scholar]
- 37.Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD. Microtubule interaction site of the kinesin motor. Cell. 1997;90:207–216. doi: 10.1016/s0092-8674(00)80329-3. [DOI] [PubMed] [Google Scholar]
- 38.Hsieh C-TJ. DNA unwinding and DNA-stimulated ATPase activities by the Escherichia coli Rep helicase. Washington University; Saint Louis, MO: 2002. Transient kinetic studies on the mechanisms of DNA binding; p. xiv. 306 leaves. [Google Scholar]
- 39.Kad NM, Rovner AS, Fagnant PM, Joel PB, Kennedy GG, Patlak JB, Warshaw DM, Trybus KM. A mutant heterodimeric myosin with one inactive head generates maximal displacement. J Cell Biol. 2003;162:481–488. doi: 10.1083/jcb.200304023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mori T, Vale RD, Tomishige M. How kinesin waits between steps. Nature. 2007;450:750–754. doi: 10.1038/nature06346. [DOI] [PubMed] [Google Scholar]
- 41.Coy DL, Wagenbach M, Howard J. Kinesin takes one 8-nm step for each ATP that it hydrolyzes. J Biol Chem. 1999;274:3667–3671. doi: 10.1074/jbc.274.6.3667. [DOI] [PubMed] [Google Scholar]
- 42.Onishi H, Mochizuki N, Morales MF. On the Myosin catalysis of ATP hydrolysis. Biochemistry. 2004;43:3757–3763. doi: 10.1021/bi040002m. [DOI] [PubMed] [Google Scholar]
- 43.Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 44.Yun M, Zhang X, Park CG, Park HW, Endow SA. A structural pathway for activation of the kinesin motor ATPase. Embo J. 2001;20:2611–2618. doi: 10.1093/emboj/20.11.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki N, Shimada T, Sutoh K. Mutational analysis of the switch II loop of Dictyostelium myosin II. J Biol Chem. 1998;273:20334–20340. doi: 10.1074/jbc.273.32.20334. [DOI] [PubMed] [Google Scholar]
- 46.Auerbach SD, Johnson KA. Alternating site ATPase pathway of rat conventional kinesin. J Biol Chem. 2005;280:37048–37060. doi: 10.1074/jbc.M502984200. [DOI] [PubMed] [Google Scholar]
- 47.Subramanian R, Gelles J. Two distinct modes of processive kinesin movement in mixtures of ATP and AMP-PNP. J Gen Physiol. 2007;130:445–455. doi: 10.1085/jgp.200709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao Q, Gao YQ. Asymmetry in kinesin walking. Biochemistry. 2007;46:9098–9106. doi: 10.1021/bi602382w. [DOI] [PubMed] [Google Scholar]
- 49.Allingham JS, Sproul LR, Rayment I, Gilbert SP. Vik1 modulates microtubule-Kar3 interactions through a motor domain that lacks an active site. Cell. 2007;128:1161–1172. doi: 10.1016/j.cell.2006.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hackney DD. Biochemistry. Processive motor movement. Science. 2007;316:58–59. doi: 10.1126/science.1141549. [DOI] [PubMed] [Google Scholar]