

Abstract
Cornelia de Lange Syndrome (CdLS) spectrum disorders are characterized by multiple organ system congenital anomalies that result from mutations in genes encoding core cohesin proteins SMC1A, SMC3, and RAD21, or proteins that regulate cohesin function such as NIPBL and HDAC8. HDAC8 is the Zn2+-dependent SMC3 deacetylase required for cohesin recycling during the cell cycle, and 17 different HDAC8 mutants have been identified to date in children diagnosed with CdLS. As part of our continuing studies focusing on aberrant HDAC8 function in CdLS, we now report the preparation and biophysical evaluation of five human HDAC8 mutants: P91L, G117E, H180R, D233G, and G304R. Additionally, the double mutants D233G-Y306F and P91L-Y306F were prepared to enable cocrystallization of intact enzyme–substrate complexes. X-ray crystal structures of G117E, P91L-Y306F, and D233G-Y306F HDAC8 mutants reveal that each CdLS mutation causes structural changes that compromise catalysis and/or thermostability. For example, the D233G mutation disrupts the D233-K202-S276 hydrogen bond network, which stabilizes key tertiary structure interactions, thereby significantly compromising thermostability. Molecular dynamics simulations of H180R and G304R HDAC8 mutants suggest that the bulky arginine side chain of each mutant protrudes into the substrate binding site and also causes active site residue Y306 to fluctuate away from the position required for substrate activation and catalysis. Significantly, the catalytic activities of most mutants can be partially or fully rescued by the activator N-(phenylcarbamothioyl)-benzamide, suggesting that HDAC8 activators may serve as possible leads in the therapeutic management of CdLS.
Introduction
Cornelia de Lange Syndrome (CdLS) is a disorder of multiple congenital anomalies diagnosed in approximately one in 10,000 births. CdLS is characterized by growth retardation, intellectual disability, distinctive facial features (synophrys, long eyelashes, upturned nose, thin downturned lips), limb malformations, and other organ disorders.1,2 CdLS is characterized by a broad range of phenotypes; while the classical severe phenotype is perhaps most recognizable, moderate and mild cases comprise more subtle phenotypes that can be more challenging to recognize.
CdLS is a cohesinopathy, in that the majority of diagnoses have been linked to genetic defects in proteins that comprise or enable the function of cohesin, the multiprotein complex that encircles sister chromatids during the cell cycle. To date, CdLS has been linked to mutations in genes encoding cohesin subunits SMC1A,3 SMC3,3,4 and RAD21,5,6 the accessory protein NIPBL,7–9 and the zinc-dependent deacetylase HDAC8.10–12 Mutations in NIPBL are predominant in classical CdLS patients and account for about 60% of diagnosed cases, whereas mutations in the other genes are less frequent (5% for SMC1A, and about 5% for HDAC8, SMC3, and RAD21 together) and are seen in patients with more variant phenotypes.
A biological function of the cohesin complex and its mediators is to ensure sister chromatid cohesion during the cell cycle. It is also centrally involved in other important cellular processes, including DNA repair and gene expression.13,14 Four proteins, SMC1A, SMC3, RAD21, and STAGs, assemble to form the cohesin complex with a ring-like quaternary structure.14,15 The SMC1A and SMC3 subunits have similar topology: each contains a long, antiparallel coiled-coil with a “hinge” domain at one end and an ATPase domain at the other end. SMC1A and SMC3 assemble as a heterodimer through hinge-hinge and ATPase-ATPase domain interactions. The resulting cohesin ring is further stablized by the binding of RAD21 to the ATPase domains of SMC1A and SMC3. Finally, STAG proteins binds to RAD21 to complete the complex. Accordingly, SMC1A and SMC3 are analogous to the two halves of a hinged bangle bracelet, and the STAG-RAD21 complex is analogous to the clasp that locks the bracelet shut. The cohesin complex is believed to encircle sister chromatids via its ring-like structure.14–16
The loading of chromatin onto the cohesin complex is promoted by NIPBL during G1 phase.14,16 However, chromatin entrapment inside the cohesin ring is still reversible during this stage of the cell cycle. After DNA replication during S phase, the acetylation of SMC3 is essential for promoting sister chromatid cohesion.17–20 SMC3 acetylation at two conserved lysine residues (K105 and K106 in human SMC3) is catalyzed by the N-acetyltransferases ESCO1 and ESCO2.17 During prophase, most of the cohesin is released from the sister chromatids. The remaining cohesin localized at centromeres is further cleaved by separase during anaphase. This allows for the complete dissolution of the cohesin complex and separation of sister chromatids.14,16,21
In order to recycle the cohesin complex for reuse during the next cell cycle, the acetylated lysines of SMC3 must be deacetylated after dissolution of the cohesin complex. The deacetylation of SMC3 is catalyzed by the Zn2+-dependent deacetylase Hos1 in yeast.22,23 Its human ortholog, histone deacetylase 8 (HDAC8), was recently identified as the SMC3 deacetylase by Deardorff and colleagues.10 Several patients with features overlapping CdLS have been diagnosed with missense or nonsense mutations in HDAC8: to date, 17 missense mutations have been identified that cause partial or complete loss of deacetylase activity (Figure 1).10–12
Figure 1.
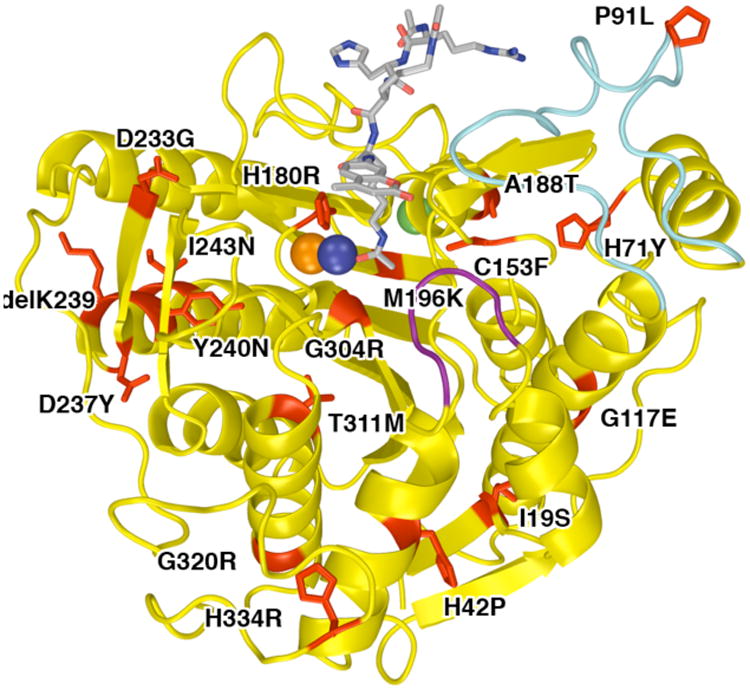
A total of 17 missense mutations in HDAC8 have been identified to date in children diagnosed with Cornelia de Lange Syndrome. Mutations (red) are mapped onto the structure of the Y306F HDAC8–substrate complex (PDB accession code 2V5W; note that the Y240N mutation is accompanied by the deletion of K239). The bound substrate, Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin, is a gray stick-figure, and the active site Zn2+ ion is a dark blue sphere. Monovalent cations required for structural stabilization and regulation of catalytic activity are shown as orange and green spheres. Purple and cyan segments indicate the flexible L1 and L2 loops, respectively, which can undergo conformational changes to accommodate substrate and inhibitor binding.
HDAC8 is a class I metal-dependent histone deacetylase24–26 that catalyzes the hydrolysis of the acetyllysine side chain to form free lysine and acetate; notably, HDAC8 can utilize histone and non-histone substrates.10,27,28 Active site residues important for HDAC8 catalysis include D178, H180, and D267, which coordinate to the catalytically-obligatory Zn2+ ion;29,30 tandem histidine residues H142 and H143, which serve as electrostatic catalyst and general base-general acid, respectively;31,32 and Y306, which donates a hydrogen bond to the scissile carbonyl group in the enzyme–substrate complex, thereby assisting the Zn2+ ion in orienting and polarizing the carbonyl for nucleophilic attack by Zn2+-bound solvent.31,33 A summary of the HDAC8 catalytic mechanism is shown in Figure 2.26,27
Figure 2.
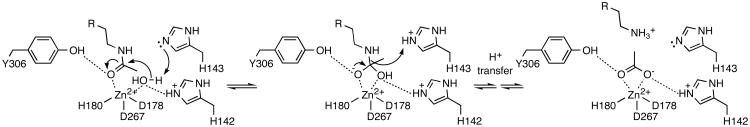
Summary of the HDAC8 mechanism. Coordination to Zn2+ and hydrogen bonding with Y306 activates the scissile carbonyl of acetyllysine for nucleophilic attack by a Zn2+-bound water molecule with the assistance of general base H143. Electrostatic stabilization of the resulting tetrahedral intermediate is achieved by Zn2+ and H142. Proton donation to the leaving amino group by H143 enables collapse of the tetrahedral intermediate to form free lysine and acetate.
Recently, we reported X-ray crystal structures of 5 missense mutants of HDAC8 identified in children diagnosed with CdLS, showing how individual amino acid substitutions 8–25 Å away from the active site can trigger local conformational changes that impair catalytic activity and thermostability.34 Here, we extend this analysis with the characterization of 5 additional HDAC8 mutants using X-ray crystallography, molecular dynamics (MD), and measurements of catalytic activity and thermostability. Specifically, we report the X-ray crystal structures of G117E HDAC8 complexed with an inhibitor, and the double mutants P91L-Y306F HDAC8 and D233G-Y306F HDAC8 complexed with a substrate (the Y306F mutation deactivates catalysis to enable cocrystallization with an intact acetyllysine substrate). Although HDAC8 mutants H180R and G304R were not amenable to crystallization, the structural consequences of these substitutions were evaluated in MD simulations. Interestingly, compromised catalytic activity in three of these mutants can be partially or fully rescued by a small molecule activator,35 as previously demonstrated for other CdLS HDAC8 mutants,34 suggesting a potential therapeutic strategy for managing CdLS in children diagnosed with HDAC8 mutations.
Methods
Reagents
Most chemicals used for buffers or crystallization were purchased from Fisher or Sigma, unless otherwise specified. The HDAC inhibitors 4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl]benzamide (M344) and trichostatin A (TSA) were obtained from Sigma and Cayman Chemical, respectively. The HDAC8 activator N-(phenylcarbamothioyl)benzamide was purchased from Oakwood Products, Inc. Inhibitor and activator compounds were used without any further purification.
Expression and Purification of HDAC8 Mutants
The P91L, G117E, H180R, D233G, G304R, and Y306F mutations were introduced into the HDAC8-6His-pET20b construct31 using standard protocols outlined in the Quickchange site directed-mutagenesis kit (Agilent Technologies, Inc.). Forward and reverse primers used for PCR mutagenesis are listed in Table S1. Incorporation of desired mutations was confirmed by DNA sequencing. Recombinant HDAC8 mutants P91L, P91L-Y306F, G117E, H180R, D233G, D233G-Y306F, and G304R were expressed in BL21(DE3) Escherichia coli cells according to a previously published procedure,34 with minor modifications. Briefly, 50-mL cultures (Lysogeny Broth (LB) media supplemented with 100 μg/mL ampicillin) were grown overnight and used to inoculate 1-L flasks (minimal media supplemented with 100 μg/mL ampicillin). Typically, 6 liters were expressed for each mutant, except for G117E (12 liters). Cells were grown at 37°C until OD600 reach approximately 0.5, at which point the temperature was lowered to 18°C, except for G117E (16°C). After 30 minutes, cells were induced by the addition of isopropyl-β-D-thiogalactopyranoside (0.4 mM final concentration for each mutant, except for G117E (0.1 mM)) and ZnCl2 (100 μM final concentration), and grown overnight at 18°C, except for G117E (16°C). Cells were pelleted by centrifugation and kept at -80°C until purification. HDAC8 mutants were purified according to a previously described protocol.34 The protein was concentrated to 7–15 mg/mL. Protein concentrations were determined from the absorbance at 280 nm using the calculated extinction coefficient ε = 50,240 M-1cm-1 for all the single mutants, and ε = 48,960 M-1cm-1 for all the double mutants.36
Enzyme Activity Assays
The catalytic activities of HDAC8 mutants was measured using the Fluor-de-Lys tetrapeptide assay substrate Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin (BML-KI178-0005, Enzo Life Sciences). Deacetylation of the substrate by HDAC8 is followed by the cleavage of the amide bond linking the C-terminal 7-amino-4-methylcoumarin to the peptide backbone by a protease developer, resulting in a fluorescence shift. Activity assays were run at 25°C in assay buffer [25 mM Tris (pH 8.2), 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2] and contained 150 μM tetrapeptide substrate with the following enzyme concentrations: 0.5 μM (wild-type), 0.5 μM (P91L), 1.5 μM (G117E), 3.0 μM (H180R), 1.0 μM (D233G), or 3.0 μM (G304R) in a final volume of 50 μL. After 30 min, reactions were quenched by the addition of the same volume of a developing solution containing 200 μM M344 (a known inhibitor of HDAC8) and the commercial Developer II (BML-KI176-1250, Enzo Life Sciences) in assay buffer. After 45 min, reaction solution samples (100 μL) were transferred to a 96-well plate and the fluorescence of the aminomethylcoumarin product was measured using a Fluoroskan plate reader (excitation = 355 nm, emission = 460 nm). All assays were run in triplicate. Results are reported in Table 1.
Table 1. Catalytic Activities and Melting Temperatures of CdLS HDAC8 Mutants.
|
|
|||
|---|---|---|---|
| Melting Temperature (Tm, °C)a | |||
|
| |||
| Activitya (nmol product•μmol enzyme-1•min-1) | no ligand | with M344 | |
| wild type | 1520 ± 90 | 50.1 ± 0.1b | 55.7 ± 0.2b |
| H180R | 0 ± 1 | 44.3 ± 0.3 | 44.1 ± 0.1 |
| G304R | 0 ± 1 | 43.3 ± 0.1 | 43.4 ± 0.1 |
| G117E | 70 ± 10 | 46.2 ± 0.1 | 49.2 ± 0.1 |
| D233G | 740 ± 40 | 43.3 ± 0.1 | 49.7 ± 0.1 |
| P91L | 1310 ± 60 | 49.3 ± 0.1 | 55.2 ± 0.1 |
All measurements made in triplicate and reported as mean ± standard deviation.
From reference 34.
Activity assays with the HDAC8 activator35 N-(phenylcarbamothioyl)benzamide were performed under similar conditions. Briefly, the assay buffer contained 150 μM tetrapeptide substrate, 0, 1, 10 or 100 μM TM-251, 3% DMSO, and the following enzyme concentrations: 0.15 μM (wild-type), 0.25 μM (P91L), 1.5 μM (G117E), 3.0 μM (H180R), 0.40 μM (D233G), or 3.0 μM (G304R). Enzyme was first incubated with the activator for 10 min before starting the reaction with the substrate. Reaction time, developer, developing time, and fluorescence reading were the same as described above. All assays were run in triplicate.
Thermostability assays
The thermostabilities of HDAC8 mutants were determined using a thermal shift assay.37 Assay mixtures contained 5 μM HDAC8 mutant, 0 or 50 μM M344, 25 mM HEPES (pH = 7.5), 150 mM KCl, 500 μM TCEP, and SYPRO orange dye (S6650, Life Technologies) at a 5× final concentration. HDAC8 enzymes were incubated with or without M344 for 45 min at 4°C before the addition of SYPRO orange dye. Since the M344 stock solution was prepared in DMSO, the corresponding amount of DMSO was added to each enzyme for assays in the absence of inhibitor. 20-μL samples of each mixture were transferred to a 96-well plate (MicroAmp fast 96-well reaction plate, Applied Biosystems). The plate was sealed (MicroAmp adhesive film, Applied Biosystems) and incubated in a real-time polymerase chain reaction instrument (StepOnePlus, Applied Biosystems) for 1 min at 20°C followed by a 1°C increase per minute up to 90°C. During the thermal scan, fluorescence was monitored using a predefined filter (ROX). Protein unfolding induces an increase in SYPRO orange fluorescence, which was used to monitor thermal denaturation. Melting temperatures (Tm) were designated as the inflection point by fitting the initial portion of the curve (up to its maximum) with a Boltzmann equation.37 All assays were run in triplicate. Melting temperatures are reported in Table 1.
Crystallization and Data Collection
Crystals of G117E HDAC8 complexed with TSA were prepared by cocrystallization at 21°C in sitting drops using the vapor diffusion method. A 500 nL drop containing G117E HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% glycerol, 1 mM DTT, 2 mM TSA and 0.03 M glycylglycylglycine was added to a 500 nL drop of precipitant solution and equilibrated against a 100-μL reservoir of precipitant solution. The precipitant solution consisted of 100 mM BisTris (pH 6.5), 10% (w/v) PEG 35,000 (Fluka), and 4 mM TCEP. Crystals of double mutants complexed with the tetrapeptide assay substrate Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin (Enzo Life Sciences) were cocrystallized in similar fashion. Briefly, a 500 nL drop containing 4.5 mg/mL HDAC8 double mutant, 2.5 mM substrate, 0.03 M glycylglycylglycine, 50 mM Tris (pH 8.0), 76.4 mM KCl, 68.5 mM NaCl, 2.5% glycerol, 0.5 mM MgCl2, and 0.5 mM DTT was added to a 500 nL drop of precipitant solution and equilibrated against a 100 μL reservoir of precipitant solution. P91L-Y306F HDAC8 was cocrystallized with substrate at 21°C using a precipitant solution of 100 mM Tris (pH 8.0), 10% (w/v) PEG 35,000 (Fluka), and 4 mM TCEP. D233G-Y306F HDAC8 was cocrystallized with substrate at 21°C using a precipitant solution of 100 mM Tris (pH 8.0), 10% (w/v) PEG 3,350 (Hampton Research), and 4 mM TCEP.
Typically, crystals appeared within 1-2 days for each mutant. Crystals were flash-cooled in liquid nitrogen after transfer to a cryoprotectant solution consisting of precipitant solution supplemented with 25-30% glycerol. X-ray diffraction data were collected on beamline X29 at the National Synchrotron Light Source (NSLS, Brookhaven National Laboratory, New York). Data collection statistics are presented in Table 2. Data were indexed, integrated, and scaled using HKL2000.38
Table 2. Data Collection and Refinement Statistics.
|
|
||||
|---|---|---|---|---|
| G117E HDAC8–TSA complex | D233G-Y306FHDAC8–substrate complex | P91L-Y306FHDAC8–substrate complex | ||
| Unit cell | ||||
| space group symmetry | P21 | P212121 | P212121 | |
| a, b, c (Å) | 52.2, 83.0, 98.5 | 83.0, 97.9, 104.7 | 82.3, 98.0, 105.9 | |
| α, β, γ (deg) | 90, 102.8, 90 | 90, 90, 90 | 90, 90, 90 | |
| Data collection | ||||
| wavelength (Å) | 1.075 | 1.075 | 1.075 | |
| resolution limits (Å) | 49.8–2.90 | 44.4–1.42 | 44.5–2.01 | |
| total/unique reflections | 60436/18263 | 2004072/159962 | 548533/57560 | |
| Rmergea,b | 0.145 (0.469) | 0.072 (1.112)c | 0.135 (1.153)c | |
| I/σ(I)a | 8.3 (2.7) | 28.5 (2.5) | 13.2 (2.1) | |
| redundancya | 3.3 (3.3) | 12.5 (11.4) | 9.5 (9.1) | |
| completeness (%)a | 99.9 (99.8) | 99.9 (100) | 99.9 (98.8) | |
| Refinement | ||||
| reflections used in refinement/test set | 18247/934 | 159859/8011 | 57481/2915 | |
| Rcrystd | 0.193 | 0.146 | 0.175 | |
| Rfreee | 0.228 | 0.167 | 0.206 | |
| protein atomsf | 5527 | 6029 | 5676 | |
| water moleculesf | 17 | 819 | 272 | |
| ligand moleculesf | 2 | 2 | 2 | |
| Zn2+ ionsf | 2 | 2 | 2 | |
| K+ ionsf | 4 | 4 | 4 | |
| glycerol moleculesf | - | 1 | - | |
| R.m.s. deviations from ideal geometry | ||||
| bonds (Å) | 0.003 | 0.009 | 0.003 | |
| angles (°) | 0.6 | 1.3 | 0.7 | |
| dihedral angles (°) | 13 | 12 | 11 | |
| Ramachandran plot(%)g | ||||
| allowed | 89.9 | 90.7 | 90.2 | |
| additionally allowed | 9.8 | 9.3 | 9.8 | |
| generously allowed | 0.2 | - | - | |
| disallowed | 0.2 | - | - | |
| PDB accession code | 5D1B | 5D1C | 5D1D | |
Values in parentheses refer to the highest shell of data.
Rmerge= Σ|Ih - 〈I〉h|/ΣIh, where 〈I〉h is the average intensity calculated from replicate reflections.
Given the high redundancy for the outer shells of these data sets, Rpim is a more appropriate measure of the data quality than Rmerge.52 Rpim=0.029 (0.490) and 0.048 (0.376) for D233G-Y306F HDAC8 and P91L-Y306F HDAC8, respectively.
Rcryst=Σ‖Fo| - |Fc||/Σ|Fo| for reflections contained in the working set; |Fo|and|Fc|are the observed and calculated structure factor amplitudes, respectively.
Rfree=Σ‖Fo| - |Fc||/Σ|Fo| for reflections contained in the test set held aside during refinement.
Per asymmetric unit.
Calculated with PROCHECK version 3.4.4.
Phasing, Model Building, and Structure Refinement
Structures were solved by molecular replacement using PHENIX39 with the atomic coordinates of the H143A HDAC8–tetrapeptide substrate complex (PDB accession code 3EWF)31 less substrate, ion, and solvent used as a search probe for rotation and translation function calculations. Each model was refined through iterative cycles of refinement with PHENIX and manual model rebuilding using COOT.40 Solvent molecules and inhibitors were added after several rounds of refinement for each structure. For the D233G-Y306F HDAC8–substrate complex, Translation Libration Screw (TLS) refinement was performed in the late stages of refinement. TLS groups were automatically determined using PHENIX.
Certain segments in each structure (the N-terminus, the C-terminus, and a portion of the L2 loop; side chains of some surface residues) appeared to be disordered and were accordingly excluded from each final model as summarized in the Supporting Information. Also, occasional ambiguous electron density peaks were observed either on the protein surface (e.g., near F336 for the G117E HDAC8–TSA complex; near M54, F189, F225, and V376 for the D233G-Y306F HDAC8–substrate complex; or near F208 and K289 for the P91L-Y306F HDAC8–substrate complex) or in the protein interior (e.g., near W141 for G117E HDAC8–TSA complex). Ambiguous electron density near W141 likely corresponds to alternative conformations. However, since such conformations were not confidently interpretable, the W141 side chain was modeled in only one primary conformation. Other ambiguous electron density peaks on the protein surface were usually elongated and attributed to disordered PEG fragments or other molecules present in the buffer solution used for crystallization. However, since these electron density peaks were not confidently interpretable, they were left unmodeled.
Circular dichroism spectroscopy
The secondary structures of wild-type, H180R, and G304R HDAC8 enzymes were evaluated using circular dichroism (CD) spectroscopy on an Aviv model 425 spectrometer in a quartz cell with a 0.1 cm path length. Prior to each measurement, enzyme was extensively buffer exchanged with 25 mM Tris (pH = 8.0) and 5% (v/v) glycerol. Spectra were recorded at 4°C in the far UV region between 190–260 nm with a 5 μM enzyme concentration.
Molecular dynamics simulations
GROMACS (GROningen MAchine for Chemical Simulation) is a program for calculating MD simulations written in ANSI C that is compatible with the GROMOS, OPLS, AMBER, and CHARMM force fields.41–43 GROMACS is free and open source online. The goal of the GROMACS developers is to enable MD calculations on desktop-grade computing hardware instead of supercomputers to which such calculations had previously been restricted. GROMACS comprises an efficient and high-throughput molecular simulation suite of programs that allow for hardware scaling, both in multiple computer clusters and in single computers using multiple cores and graphics accelerators, using both Message-Passing Interface (MPI) and Open Multi-Processing (OpenMP) parallelization. A computer was built to exploit these features as described in the Supporting Information.
The starting point for each MD simulation was the crystal structure of wild-type HDAC8 as determined an enzyme–inhibitor complex (PDB accession code 3RQD).44 After stripping the coordinate file of all water and inhibitor atoms, amino acid substitutions were made to the protein model in silico to generate atomic coordinate sets for HDAC8 mutants H180R and G304R. The pdb2gmx command was then run to generate a Gromacs-compatible topology, position restraint, and structure file using the AMBER99SB-ILDN force field.45 A cubic or dodecahedral box was then defined with faces ranging 10–17Å away from the protein surface. The box was filled with water molecules, and ions were added so that the net charge of the system was 0. The protein-solvent system was subject to energy minimization to ensure stability and the integrity of subsequent MD runs. Thermal equilibration was run in two phases to bring the system to the correct temperature, pressure, and density, first using the constant Number of particles, Volume, and Temperature (NVT), and then the constant Number of particles, Pressure, and Temperature (NPT) equilibration schemes. Equilibration steps were judged to be successful if the system quickly converged to the selected temperature and if the pressure and density of the system remained stable for a 100 ps run. All MD simulations were run for 10 ns with a step size of 2 fs; frames were generated every 2 ps to sample each trajectory.
Results
H180R HDAC8
The imidazole side chain of H180 is one of three protein ligands to the catalytically-obligatory Zn2+ ion in the active site of HDAC8, so the substitution of H180 by a bulky, positively charged arginine residue disrupts the metal binding site. As a consequence, H180R HDAC8 is completely inactive (Table 1). Among all the CdLS HDAC8 mutations identified to date,10–12 H180R HDAC8 is the only one that involves the substitution of a residue that plays a direct role in catalysis. Since our attempts to crystallize this critical mutant in the presence and absence of ligands were unsuccessful, we investigated the structural consequences of the H180R substitution through MD simulations. Since the CD spectrum of H180R HDAC8 is essentially identical to that of wild-type HDAC8 (Figure S1, Supporting Information), the overall fold of the mutant enzyme is probably not significantly perturbed relative to that of the wild-type enzyme. Therefore, wild-type HDAC8 serves as a suitable basis for modeling and MD simulations of the H180R mutant.
Two separate MD trajectories were calculated for H180R HDAC8: one in which Zn2+ remained in the active site, coordinated by protein ligands D178 and D267, and the other in which Zn2+ was absent from the active site. We reasoned that while the H180R substitution would destabilize Zn2+ binding, it might not completely abrogate Zn2+ binding since the R180 side chain would not necessarily block metal coordination by the remaining protein ligands. The 10-ns MD trajectory calculated for Zn2+-bound H180R HDAC8 reveals that the arginine side chain can fluctuate into the acetyllysine binding groove of the active site, so the bulky R180 side chain may sterically block substrates or inhibitors from binding even if a Zn2+ ion is weakly bound in the active site. While the conformation of R180 occasionally fluctuates so as to minimize this blockage, the guanidinium group generally remains in the region of the acetyllysine Cα atom throughout the simulation (Figure 3a). Additionally, the steric bulk of the R180 side chain triggers fluctuations of Y306 away from the “in” conformation required for catalysis, such that the Y306 OH---Zn2+ separation increases by ca. 2 Å in comparison with the wild-type enzyme (Figure 3b). Such fluctuations would also weaken inhibitor binding, since Y306 typically hydrogen bonds with the Zn2+-bound hydroxamate group of inhibitors.29–31
Figure 3.
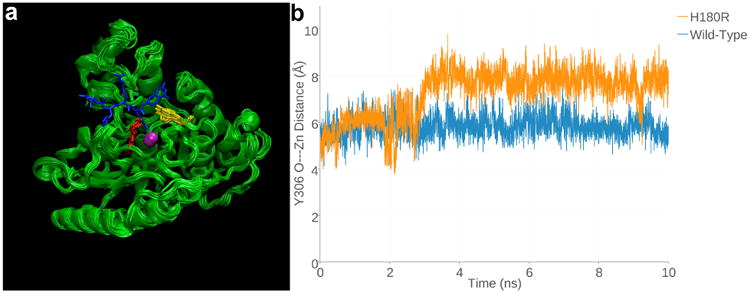
(a) Superimposed 1-ns snapshots from the 10-ns MD simulation of Zn2+-bound H180R HDAC8, with a tetrapeptide assay substrate (blue) superimposed for reference (from the structure of the H143 HDAC8-substrate complex, PDB accession code 3EWF). Zn2+ is a magenta sphere, R180 is red, and Y306 is yellow. (b) Y306 fluctuates ∼2Å away from the “in” conformation required for catalysis in H180R HDAC8 relative to its fluctuations in the wild-type enzyme over the course of the 10-ns MD simulation. The fluctuations of Y306 in the MD simulation of Zn2+-free H180R HDAC8 are just slightly less (Figure S2, Supporting Information).
The 10-ns MD trajectory calculated for Zn2+-free H180R HDAC8 reveals that the arginine side chain protrudes into the acetyllysine binding groove of the active site in a similar fashion as observed for the Zn2+-bound mutant. Fluctuations observed for Y306 in Zn2+-free H180R HDAC8 are slightly less than those observed in the Zn2+-bound mutant (Figure S2, Supporting Information). Even so, it appears that the conformation of Y306 is sensitive to structural changes in the HDAC8 active site, regardless of Zn2+ occupancy.
The H180R substitution causes a significant decrease in thermostability, with ΔTm= -5.8°C; equilibration with the inhibitor M344 does not enhance thermostability, in contrast with wild-type HDAC8 (Table 1). This is consistent with MD simulations indicating that substrate or inhibitor binding could be blocked by the bulky R180 side chain. Since the hydroxamate group of M344 ordinarily coordinates to the active site Zn2+ ion and hydrogen bonds with Y306,31 the weakening of Zn2+ binding, the addition of steric bulk in the acetyllysine binding groove, and the movement of Y306 away from the “in” conformation observed in MD simulations completely disable inhibitor binding. These features are similarly responsible for the complete loss of catalytic activity exhibited by this mutant.
G304R HDAC8
G304 is located in a glycine-rich loop that connects β-strand 8 to helix H1, ∼4Å away from the active site Zn2+ ion, and G304R HDAC8 is completely inactive (Table 1). Of the residues in the glycine-rich loop G302-G303-G304-G305, G304 and G305 are strictly conserved among class I HDACs and HDAC-related enzymes such as polyamine deacetylase.46,47 Importantly, the glycine-rich loop is immediately adjacent to Y306, which must adopt the “in” conformation for catalysis to enable hydrogen bonding with the scissile carbonyl of substrate acetyllysine.31 Possibly, this glycine-rich loop confers flexibility to facilitate conformational transitions between “in” and “out” conformations for residue 306, as observed in crystal structures of polyamine deacetylase,46,47 and MD simulations of HDAC348 and HDAC8 (N. H. Christianson, unpublished results).
Since our attempts to crystallize G304R HDAC8 in the presence and absence of ligands were unsuccessful, we investigated the structural consequences of this amino acid substitution through MD simulations. The CD spectrum of G304R HDAC8 is essentially identical to that of wild-type HDAC8 (Figure S1, Supporting Information), so the overall fold of the mutant enzyme is essentially identical to that of the wild-type enzyme. Thus, the wild-type enzyme structure serves as a valid starting point for modeling and MD simulations of the G304R mutant.
The 10-ns MD trajectory calculated for G304R HDAC8 reveals that the R304 side chain significantly protrudes into the acetyllysine binding groove of the active site, so the bulky side chain will sterically block substrates or inhibitors from binding. The R304 side chain generally remains in this position during the entire course of the 10-ns simulation (Figure 4a). Additionally, the G304R substitution triggers significant fluctuations of Y306 away from the “in” conformation required for catalysis, such that the Y306 OH---Zn2+ separation increases by ca. 2 Å in comparison with the wild-type enzyme (Figure 4b). Thus, even if R304 and the associated glycine-rich loop were to move so as to allow acetyllysine binding, this movement would also take Y306 out of the position normally required for catalysis. These results may suggest that mutations in the glycine-rich loop could influence catalysis by compromising conformational fluctuations required to position Y306 for catalysis.
Figure 4.
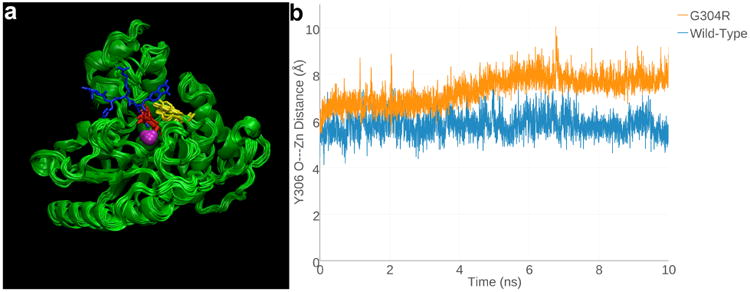
(a) Superimposed 1-ns snapshots from the 10-ns MD simulation of Zn2+-bound G304R HDAC8, with a tetrapeptide assay substrate (blue) superimposed for reference (from the structure of the H143 HDAC8-substrate complex, PDB accession code 3EWF). Zn2+ is a magenta sphere, R304 is red, and Y306 is yellow. (b) Y306 fluctuates ∼2Å away from the “in” conformation required for catalysis in G304R HDAC8 relative to its fluctuations in the wild-type enzyme over the course of the 10-ns MD simulation.
The G304R substitution causes a significant decrease in thermostability, with ΔTm= -6.8°C; equilibration with the inhibitor M344 does not enhance thermostability, in contrast with wild-type HDAC8, indicating that inhibitor binding is completely destabilized (Table 1). Since inhibitors generally hydrogen bond with Y306,29–31 the conformational transition of Y306 away from the “in” conformation required for catalysis or inhibitor binding, as predicted from MD simulations, could sufficiently destabilize binding so as to be unobserved in thermal shift assays. Thus, it appears that both the blockage of the acetyllysine binding site, as well as the movement of Y306 away from the “in” conformation triggered by the nearby G304R substitution, completely disable inhibitor binding, substrate binding, and catalysis.
G117E HDAC8
G117 is located in helix B4, ∼20Å away from the active site Zn2+ ion, close to the protein surface and adjacent to the loop connecting β-strand 2 to helix B1. G117E HDAC8 exhibits 5% residual activity relative to the wild-type enzyme (Table 1). The crystal structure of G117E HDAC8 was determined in complex with the hydroxamate inhibitor TSA at 2.9 Å resolution. Although the resolution is somewhat modest, it is clear that the overall structure of G117E HDAC8 is generally similar to that of wild-type HDAC8 in complex with TSA (root mean square (rms) deviation = 0.74 Å for 356 Cα atoms and 0.66 Å for 362 Cα atoms for monomers A and B, respectively). No major structural changes are observed for active site residues important for the chemistry of catalysis.
Figure 5.
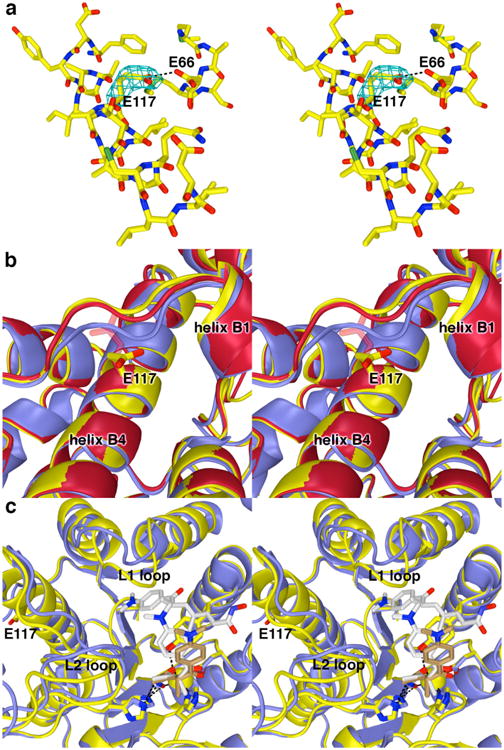
(a) Simulated annealing omit map (contoured at 2.2σ) showing the E117 side chain in the G117E HDAC8-TSA complex (monomer A). Atomic color code are as follows: C = yellow, N = dark blue, O = red, S = green. The side chain of E117 interacts with the side chain of E66; one of these residue is presumably protonated to accommodate this hydrogen bond. (b) Conformational changes induced by the G117E mutation in the nearby loop connecting β-strand 2 to helix B1 in monomers A (yellow) and B (red) of G117E HDAC8 in comparison with the wild-type HDAC8-TSA complex (blue) (PDB accession code 2V5W, monomer A). The side chain of E117 in monomer B is characterized by weak electron density and side chain atoms are not included in the final model after the Cβ atom. However, these unmodeled atoms are included in the figure as transparent sticks. (c) Comparison of the G117E HDAC8-TSA complex (monomer A, yellow; inhibitor, brown carbon atoms) with the wild-type HDAC8-TSA complex (monomer A, blue; inhibitor, white carbon atoms). Two TSA molecules bind to the wild-type enzyme, but only one molecule of TSA binds to G117E HDAC8. Significant conformational differences are observed in the L1 and L2 loops between the two structures.
Electron density for the E117 side chain in helix B4 is oriented toward solvent and is better defined in monomer A than in monomer B (Figure 5a). The substitution of the bulky glutamate side chain for that of the glycine hydrogen atom does not significantly perturb the backbone conformation of residue 117. In monomer A, the E117 carboxylate interacts with the side chain of E66. Presumably, one of these two carboxylate side chains is protonated to accommodate a hydrogen bond interaction (Figure 5a). Additionally, helix B4 shifts ca. 0.5-1.0 Å, and the steric bulk introduced by E117 triggers conformational changes in the nearby loop connecting β-strand 2 to helix B1, with loop Cαatom shifts of up to 2.5-2.6 Å (Figure 5b).
These conformational changes appear to be transmitted through the protein scaffolding to the L1 loop, which slightly constricts the active site cleft and prevents the binding of a second TSA molecule in a pocket adjacent to the active site (as observed for the wild-type HDAC8−TSA complex29). This structural change additionally appears to influence the L2 loop (Figure 5c). The binding of only one molecule of TSA with consequent structural changes is also observed in another CdLS mutant, T311M HDAC8.34
Finally, the G117E substitution causes a modest decrease in thermostability, with ΔTm= 3.9°C; equilibration with the inhibitor M344 restores thermostability with ΔTm= 3.0°C, indicating that inhibitor binding is favorable (Table 1). However, this ΔTm value is just about half that measured for M344 binding to wild-type HDAC8 (ΔTm= 5.6°C), so inhibitor binding appears to be compromised in this mutant.
D233G HDAC8
The side chain of D233 is located on the protein surface in the L6 loop connecting β-strand 6 to helix F, ∼15 Å away from the active site Zn2+ ion, and D233G HDAC8 exhibits 49% residual activity compared with the wild-type enzyme (Table 1). In wild-type HDAC8, D233 makes a hydrogen bonded salt link with K202 at the end of β-strand 5. The side chain of K202 additionally donates a hydrogen bond to S276 in the L7 loop, so the D233-K202-S276 hydrogen bond network appears to be important for the stabilization of loop conformations in the overall tertiary structure of HDAC8.
Cocrystallization of D233G HDAC8 with typical hydroxamate inhibitors such as M344 or TSA yielded only poorly diffracting plate-like crystals unsuitable for diffraction data collection. To circumvent this problem, the Y306F mutation was introduced to inactivate the enzyme and enable cocrystallization with a peptide substrate.33 This strategy recently proved successful in the study of three other CdLS HDAC8 mutants,34 and here it proved similarly succesful for cocrystallization of D233G-Y306F HDAC8 with the tetrapeptide assay substrate Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin. The crystal structure of this enzyme–substrate complex was determined at 1.42 Å resolution and is similar to that of the Y306F HDAC8–substrate complex33 (PDB accession code 2V5W; rms deviation = 0.33 Å for 363 Cα atoms for both monomers A and B). An omit electron density map shows that the tetrapeptide substrate is essentially fully ordered in the active site of D233G-Y306F HDAC8 (Figure 6a) and adopts a conformation nearly identical to that observed in the complex with Y306 HDAC8.33 No major structural changes are observed for active site residues important for the chemistry of catalysis.
Figure 6.
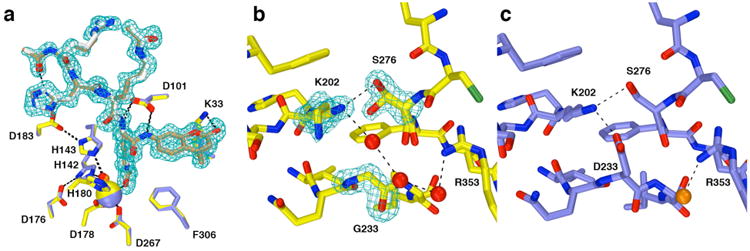
(a) Comparison of substrate binding in the D233G-Y306F HDAC8–substrate complex (C = yellow (protein) or tan (substrate), N = dark blue, O = red, Zn2+= yellow sphere, water = red sphere, monomer B) and the Y306F HDAC8–substrate complex (C = blue (protein) or gray (substrate), N = dark blue, O = red, Zn2+= blue sphere, water = orange sphere, monomer A, PDB accession code 2V5W). Metal coordination and hydrogen bond interactions are shown as solid black and dashed lines, respectively. The simulated annealing omit map (contoured at 3.0σ) shows a nearly fully ordered tetrapeptide substrate bound in the active site of D233G-Y306F HDAC8. (b) Simulated annealing omit maps of the D233G-Y306F HDAC8–substrate complex (monomer B, color coded as in (a)) showing the mutated residue G233 (contoured at 5.0σ) and the side chains of K202 and S276 (contoured at 3.0σ), each of which adopt two conformations. An ordered water molecule fills the void created by the D233G mutation and hydrogen bonds with K202 and a second water molecule. (c) Structure of the Y306F HDAC8–substrate complex (monomer A, color coded as in (a), PDB accession code 2V5W). Comparison with (b) illustrates structural changes resulting from the D233G mutation.
Although the D233G mutation triggers only minor changes in the protein structure, these changes significantly compromise thermostability with ΔTm= -6.8°C (Table 1). The crystal structure of D233G-Y306F reveals that two water molecules occupy the void created by the deletion of the bulky D233 side chain (Figure 6b). Additionally, the D233-K202-S276 hydrogen bond network is disrupted, and the remaining K202-S276 hydrogen bond is weakened based on the appearance of alternative, partially occupied conformers of these side chains that exceed normal hydrogen bonding separations. Thus, the most significant consequence of the D233G substitution appears to be compromised thermostability due to perturbation of a hydrogen bond network that stabilizes key tertiary structural interactions involving the L6 loop, β-strand 5, and the L7 loop. MD simulations of wild-type HDAC8 and D233G-Y306F HDAC8 indicate that the rms fluctuation of K202 nearly doubles from 0.576 Å to 1.003 Å in the absence of the D233 hydrogen bond; additional fluctuations are also triggered in residues adjacent to K202, up to and including F208 (Figure S3, Supporting Information). Increased atomic fluctuations triggered by the D233G mutation may be consistent with the significantly decreased thermostability measured for this mutant.
P91L HDAC8
P91 is located in the L2 loop, which flanks one side of the active site and exhibits significant flexibility in the binding of inhibitors and substrates.29–31,34,44,49 Accordingly, P91 is approximately 27–29 Å away from the active site Zn2+ ion, depending on the conformation of the L2 loop. With a residual catalytic activity of 86% compared with the wild-type enzyme (Table 1), P91L HDAC8 is the least functionally compromised of the mutants described herein.
As for D233G HDAC8, cocrystallization attempts with P91L HDAC8 and hydroxamate inhibitors yielded only poorly diffracting plate-like crystals. Accordingly, we prepared and cocrystallized the inactive double mutant P91L-Y306F HDAC8 with the tetrapeptide assay substrate Ac-Arg-His-Lys(Ac)-Lys(Ac)-aminomethylcoumarin. The crystal structure of the D233G-Y306F HDAC8–substrate complex was determined at 2.01 Å resolution and is quite similar overall to that of the Y306F HDAC8–substrate complex33 (PDB accession code 2V5W; rms deviation = 0.30 Å for 363 Cα atoms and 0.36 Å for 356 Cα atoms for monomers A and B, respectively). No major structural changes are observed for active site residues important for the chemistry of catalysis. However, the P91L mutation causes local structural changes that appear to increase the flexibility of the L2 loop; even so, these changes only slightly compromise enzyme thermostability (ΔTm= -0.8°C; Table 1).
In HDAC8 crystal structures, higher thermal B factors are usually observed for residues contained in the L2 loop. Additionally, short segments in the L2 loop are usually characterized by broken electron density and presumed disordered in structures of HDAC8–inhibitor complexes, but these segments are usually not disordered in inactive HDAC8 mutant–substrate complexes.31,33,34 The only enzyme–substrate complex in which a disordered L2 loop segment is observed is I243N-Y306F HDAC8 complexed with the tetrapeptide assay substrate (PDB accession code 4QA3):34 in monomer B only, D88 and D89 are disordered and accordingly not modeled. The electron density map of the P91L-Y306F HDAC8–substrate complex reveals that the D87-P91 segment in monomer B flanking L91 is characterized by poor electron density and is presumed disordered. In monomer A, although the main chain atoms of this segment are characterized by nearly fully connected electron density, these atoms are characterized by high thermal B factors, and the side chains of Q84, E85, D88, D89, L91, D92, I94, and E95 are characterized by broken or missing electron density (Figure 7). Thus, the P91L mutation appears to increase the flexibility of the L2 loop. However, this does not hinder substrate binding in the active site as indicated by clear electron density for the tetrapeptide substrate (although the N-terminal arginine residue of the substrate is partially disordered (Figure S4)), nor does it hinder inhibitor binding in view of the ΔTm of 5.9°C for M344 binding, which is comparable to that of 5.6°C measured for the wild-type enzyme.
Figure 7.
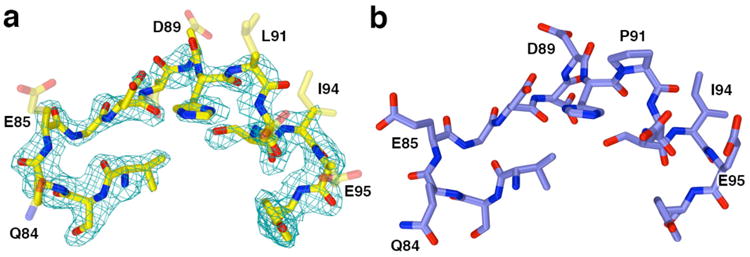
(a) Simulated annealing omit map of the L2 loop segment flanking residue 91 in the P91L-Y306F HDAC8–substrate complex (monomer A, contoured at 2.7σ) indicates that the side chains of residues Q84, E85, D88, D89, L91, D92, I94, and E95 are partially or completely disordered (these side chains are represented as transparent sticks). For comparison, the structure of the Y306F HDAC8–substrate complex (monomer A, PDB accession code 2V5W) is shown in (b). Color codes are as follows: C = yellow (P91L-Y306F HDAC8) or blue (Y306F HDAC8), N = dark blue, O = red.
Catalytic rescue by a small molecule activator
Recently, Singh and colleagues35 reported that N-acylthiourea derivatives can serve as selective activators of HDAC8, and we demonstrated that one of these activators, N-(phenylcarbamothioyl)benzamide (designated “TM251”), was capable of partially or completely rescuing the catalytic activity of certain CdLS HDAC8 mutants in vitro.34 Here, we assessed the ability of TM251 to rescue catalytic activity in the current set of five CdLS mutants (Table 3).
Table 3. Catalytic Activity of CdLS HDAC8 Mutants in the Presence and Absence of Activator TM251.
|
|
||||||
|---|---|---|---|---|---|---|
| Activity (nmol product•umol enzyme-1•min-1) | ||||||
|
| ||||||
| TM251 concentration (μM) | Wild-type HDAC8 | H180R HDAC8 | G304R HDAC8 | G117E HDAC8 | D233G HDAC8 | P91L HDAC8 |
| 0 | 1140 ± 50 | 0 ± 1 | 0 ± 1 | 67 ± 4 | 470 ± 30 | 950 ± 50 |
| 1 | 1500 ± 40 | 0 ± 1 | 0 ± 1 | 101 ± 6 | 620 ± 40 | 1120 ± 50 |
| 10 | 2030 ± 30 | 0 ± 1 | 0 ± 1 | 150 ± 10 | 850 ± 50 | 1420 ± 60 |
| 100 | 2400 ± 100 | 0 ± 1 | 0 ± 1 | 200 ± 10 | 1070 ± 50 | 1900 ± 100 |
As for wild-type HDAC8, the catalytic activities of G117E, D233G, and P91L HDAC8 mutants are enhanced in dose-dependent fashion by TM251. Specifically, we observe a 2–3-fold activation for these mutants, and the catalytic activity of P91L HDAC8 and D233G HDAC8 could be restored to a level comparable to that of the wild-type enzyme. However, the catalytic activity of the completely inactive H180R and G304R mutants could not be rescued by TM251. As previously observed in the evaluation of other CdLS HDAC8 mutants,34 mutants that are completely inactive cannot be rescued by TM251; mutants that exhibit residual activity can be rescued partially or completely by TM251.
Discussion
To date, nearly all of the mutations identified in HDAC8 in subjects with CdLS-like disorders (Figure 1) map onto regions of the protein structure that ordinarily might not be selected for mutagenesis to probe enzyme structure-function relationships. Protein residues corresponding to missense mutations are distributed randomly throughout the protein structure, both near and far from the active site. Surprisingly, some of these distant mutations significantly affect catalysis. Thus, the study of disease-linked mutations provides intriguing new clues regarding long-range effects on the chemistry of catalysis in the enzyme active site.
Some of the amino acid substitutions presented herein exert their influence over distances ranging 15–25 Å or greater, e.g., as observed for G117E HDAC8, D233G HDAC8, or P91L HDAC8. A common theme that emerges from the study of these mutants, which exhibit a wide range of residual activities (5–86%), taken together with our previously reported structure-function relationships for HDAC8 mutants C153F, A188T, I243N, T311M, and H334R,34 is that HDAC8 activity is highly sensitive to the conformation of the L1 and L2 loops flanking the active site, or structural elements that abut these loops. The L1 loop and especially the L2 loop adopt variable conformations in the binding of inhibitors and substrates,29–31,34,44,49 and can fluctuate significantly in MD calculations.50 Thus, the conformational flexibility of these loops required for optimal catalytic function can be compromised by amino acid substitutions within the loops themselves, as observed for P91L HDAC8, or by amino acid substitutions elsewhere that trigger structural changes propagating through the protein scaffolding to these loop(s), e.g., as observed for G117E HDAC8.
Other CdLS-related HDAC8 mutations exert a more direct effect on catalysis, such as the H180R mutation that removes a protein ligand to the catalytically obligatory Zn2+ ion. While MD simulations indicate that metal binding is not sterically precluded in this mutant, these simulations indicate that R180 can protrude into the substrate binding site and hinder the binding of acetyllysine. While the conformation of R180 fluctuates in MD simulations, it does not adopt a conformation that would allow ready access for substrate or inhibitor binding. The blockage of the substrate binding groove, along with compromised Zn2+ binding, are consistent with the lack of inhibitor binding observed suggested by the insignificant ΔTm in the presence and absence of inhibitor M344 (Table 1).
The deleterious effects on catalysis resulting from the G304R mutation similarly result from partial blockage of the acetyllysine binding site, even though the R304 side chain is capable of fluctuations that could minimize steric interactions with the substrate. The MD simulations provide additional insight on compromised catalysis in both G304R HDAC8 and H180R HDAC8 that is perhaps more subtle, in that both amino acid substitutions cause the conformation of Y306 to fluctuate away from its required position for catalysis (Figures 3 and 4). Since Y306F HDAC8 is essentially devoid of catalytic activity,33 mutations that cause Y306 to move away from the “in” conformation similarly will be catalytically incompetent. Interestingly, that G304R HDAC8 is completely devoid of catalysis suggests a potential dynamical function for the flexible glycine-rich segment G302-G303-G304-G305 adjacent to Y306, which may facilitate the transition of Y306 between “out” and “in” conformations in a possible induced-fit substrate binding model.
While the direct correlation of clinical severity with catalytic activity or thermostability of CdLS HDAC8 mutations is challenging, it is clear that the clinical phenotype is very sensitive to mutations that even slightly compromise activity or thermostability. For example, P91L HDAC8 exhibits 86% residual activity and just a slightly lower melting temperature (ΔTm= -0.8°C), yet the male diagnosed with this mutation exhibits significant deformities in the upper limbs.11 In contrast, however, one male and one female diagnosed with H334R HDAC8, which exhibits 91% residual activity and a significantly lower melting temperature (ΔTm= -7.0°C),34 each present with a relatively moderate CdLS phenotype.11 It is likely that disease severity is related to total cellular HDAC8 activity.
However, two main complicating factors in determining genotype-phenotype correlations are the relatively few mutations identified to date, and the X-linked nature of the HDAC8 clinical presentation. As an inactivated X-linked gene, random differential expression of wild-type and mutant alleles in various tissues and peripheral blood is seen in female patients.11 Thus, expression of the wild-type allele likely alleviates the severity of the defective HDAC8 encoded by the mutant allele due to the location and extent of inactivation of the mutant allele. As expected for an X-linked disorder, the range of CdLS phenotypes outlined by Kaiser and colleagues11 shows that males are typically more affected. In situations where a male presents with CdLS symptoms and a mutant HDAC8 is diagnosed, e.g., as for P91L HDAC8, the clinical presentation can tend to be more severe even though catalytic activity and thermostability are less severely compromised.
Significantly, most CdLS HDAC8 mutants respond in vitro to the small molecule activator TM251. Indeed, the catalytic activities of 5 of the 10 missense mutants studied to date can be restored to wild-type levels (Figure 8). Of these 10 missense mutants, only H180R HDAC8 and G304R HDAC8 – each diagnosed in a female patient – are catalytically inactive and cannot be rescued by TM251 (while C153F HDAC8 exhibits 2% the activity of the wild-type enzyme, it can only be slightly reactivated by TM25134). The structural basis for the lack of reactivation in each of these mutants is the steric blockage of substrate or product binding: in C153F HDAC8, the product acetate release channel is blocked,34 and in H180R HDAC8 and G304R HDAC8, the substrate binding groove is partially or fully blocked (Figures 3 and 4). Additional factors include loss of Zn2+ in H180R HDAC8 and the fluctuations of Y306 away from the “in” conformation required for catalysis in H180R HDAC8 and G304R HDAC8. These results suggest that unless a CdLS mutation directly affects the conformation or chemical function of a catalytic residue, the catalytic activity of a CdLS HDAC8 mutant may be partially or fully rescued by a small molecule activator such as TM251.
Figure 8.
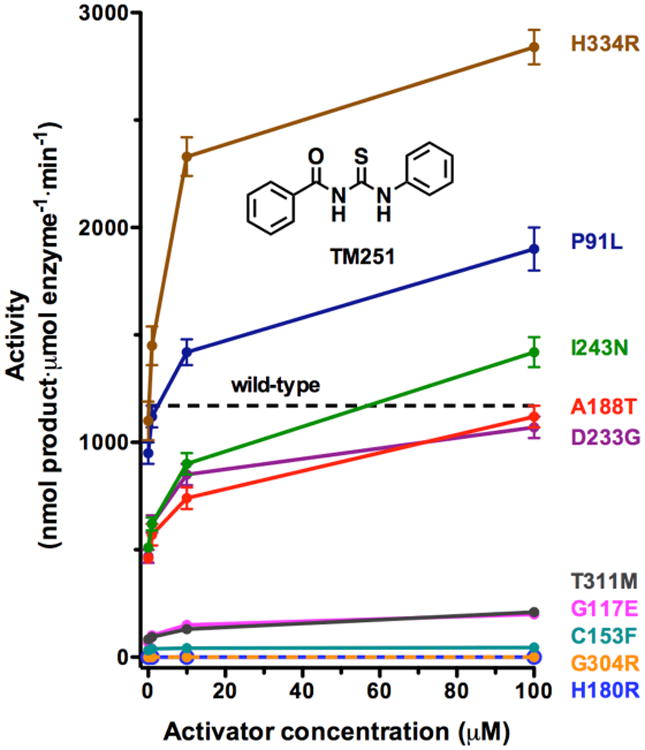
Summary of dose-dependent activation of mutant HDAC8 activity by TM251. Data for the P91L, H180R, D233G, G117E, and G304 mutants are recorded in Table 3; data for the C153F, A188T, I243N, T311M, and H334R mutants are reported by Decroos and colleagues.34 The activity level for wild-type HDAC8 in the absence of activator is indicated by a dashed line. Catalytic activity for several mutants can be restored to wild-type level or better by TM251, and all but two mutants (H180R and G304R) exhibit at least some activation by TM251.
Despite the intriguing results acquired with TM251 as an activator of CdLS HDAC8 mutants, we have as yet been unsuccessful in preparing crystalline ternary complexes with activator, substrate, and CdLS HDAC8 mutants. However, Srivastava and colleagues have utilized enzymological, biophysical, and molecular modeling approaches to understand the molecular basis of HDAC8 activation by TM251.51 Interestingly, these investigators find that TM251 binds to HDAC8 at two sites in a cooperative manner; additionally, TM251 modestly enhances inhibitor binding affinity by approximately two-fold. From analysis of the enzyme structure and molecular modeling of enzyme-activator complexes, these investigators conclude that TM251 binds near the active site, potentially stabilizing active site loops that are important for the binding of substrates and inhibitors.
In summary, the current study demonstrates that TM251 or similar small molecule activators35 may potentially be useful in the clinical management of CdLS in patients diagnosed with HDAC8 mutations, as long as these mutations do not directly affect catalytic residues in the active site. Moreover, it is interesting that the study of disease-linked mutations in HDAC8 elucidates new structure-activity relationships that ordinarily might not be explored in classical structure-based enzyme mutagenesis approaches. Specifically, since CdLS mutations occur randomly throughout the protein structure, and since these mutations affect catalysis as indicated by the disease phenotype, the locations of these mutations both near and far from the active site illuminate new long-range relationships between the structure of the protein scaffolding and the chemistry of catalysis in the active site. Future studies will continue to probe these structural relationships, as well as the ability of small molecule activators to rescue aberrant HDAC8 function.
Supplementary Material
Acknowledgments
We thank the NSLS for access to beamline X29 for X-ray crystallographic data collection. Additionally, D.W.C. thanks the Radcliffe Institute for Advanced Study for the Elizabeth S. and Richard M. Cashin Fellowship.
Funding: We thank the Vagelos Program in Molecular Life Sciences and the NIH for grant GM49758 in support of this research.
Abbreviations
- CD
circular dichroism
- CdLS
Cornelia de Lange Syndrome
- HDAC
histone deacetylase
- M344
4-(dimethylamino)-N-[7-(hydroxyamino)-7-oxoheptyl]benzamide
- MD
molecular dynamics
- NSLS
National Synchrotron Light Source
- PDB
Protein Data Bank
- PEG
polyethylene glycol
- rms
root mean square
- TLS
Translation Libration Screw
- TM251
N-(phenylcarbamothioyl)-benzamide
- TSA
trichostatin A
Footnotes
Supporting Information: Table of primers used to generate CdLS HDAC8 mutants; CD spectra of wild-type HDAC8, H180R HDAC8, and G304R HDAC8; time-dependent fluctutations of the Y306 side chain in Zn2+-bound and Zn2+-free H180R HDAC8 calculated in MD simulations; rms fluctuations in MD simulations of wild-type HDAC8 and D233G HDAC8; and an electron density map showing substrate binding to P91L-Y306F HDAC8. This information is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes: The atomic coordinates and crystallographic structure factors of HDAC8 mutants G117E, D233G-Y306F, and P91L-Y306F have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 5D1B, 5D1C, and 5D1D, respectively.
Notes: The authors declare no competing conflicts of interest.
References
- 1.Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet. 2009;76:303–314. doi: 10.1111/j.1399-0004.2009.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dorsett D, Krantz ID. On the molecular etiology of Cornelia de Lange syndrome. Ann N Y Acad Sci. 2009;1151:22–37. doi: 10.1111/j.1749-6632.2008.03450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, Gil-Rodríguez C, Arnedo M, Loeys B, Kline AD, Wilson M, Lillquist K, Siu V, Ramos FJ, Musio A, Jackson LS, Dorsett D, Krantz ID. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gil-Rodríguez C, Deardorff MA, Ansari M, Tan CA, Parenti I, Baquero-Montoya C, Ousager LB, Puisac B, Hernández-Marcos M, Teresa-Rodrigo ME, Marcos-Alcade I, Wesselink JJ, Lusa-Bernal S, Bijlsma EK, Braunholz D, Bueno-Martinez I, Clark D, Cooper NS, Curry CJ, Fisher R, Fryer A, Ganesh J, Gervasini C, Gillessen-Kaesbach G, Guo Y, Hakonarson H, Hopkin RJ, Kaur M, Keating BJ, Kibaek M, Kinning E, Kleefstra T, Kline AD, Kuchinskaya E, Larizza L, Li YR, Liu X, Mariani M, Picker JD, Pié A, Pozojevic J, Queralt E, Richer J, Roeder E, Sinha A, Scott RH, So J, Wusik KA, Wilson L, Zhang J, Gómez-Puertas P, Casale CH, Ström L, Selicorni A, Ramos FJ, Jackson LG, Krantz ID, Das S, Hennekam RCM, Kaiser FJ, FitzPatrick DR, Pié J. De novo heterozygous mutations in SMC3 cause a range of Cornelia de Lange syndrome-overlapping phenotypes. Hum Mutat. 2015;36:454–462. doi: 10.1002/humu.22761. [DOI] [PubMed] [Google Scholar]
- 5.Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Mönnich M, Yan Y, Xu W, Gil-Rodríguez MC, Clark D, Hakonarson H, Halbach S, Michelis LD, Rampuria A, Rossier E, Spranger S, Van Maldergem L, Lynch SA, Gillessen-Kaesbach G, Lüdecke HJ, Ramsay RG, McKay MJ, Krantz ID, Xu H, Horsfield JA, Kaiser FJ. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet. 2012;90:1014–1027. doi: 10.1016/j.ajhg.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minor A, Shinawi M, Hogue JS, Vineyard M, Hamlin DR, Tan C, Donato K, Wysinger L, Botes S, Das S, del Gaudio D. Two novel Rad21 mutations in patients with mild Cornelia de Lange syndrome-like presentation and report of the first familial case. Gene. 2014;537:279–284. doi: 10.1016/j.gene.2013.12.045. [DOI] [PubMed] [Google Scholar]
- 7.Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36:636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 9.Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A, Kline AD, Li HH, Devoto M, Jackson LG, Krantz ID. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet. 2004;75:610–623. doi: 10.1086/424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaiser FJ, Ansari M, Braunholz D, Gil-Rodríguez MC, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, Di Donato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillén-Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, McKee S, Mehta SG, Micule I, Care4Rare Canada Consortium. Mohammed S, Moran E, Mortier GR, Moser JAS, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Pérez-Aytés A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheuerle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Wilson LC, University of Washington Center for Mendelian Genomics. Hakonarson H, Quintero-Rivera F, Wierzba J, Musio A, Gillessen-Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pié J, Christianson DW, Krantz ID, Fitzpatrick DR, Deardorff MA. Loss of function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange Syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014;23:2888–2900. doi: 10.1093/hmg/ddu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng L, Zhou D, Zhang Z, Liu Y, Yang Y. Exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J Hum Genet. 2014;59:536–539. doi: 10.1038/jhg.2014.60. [DOI] [PubMed] [Google Scholar]
- 13.Losada A. The regulation of sister chromatid cohesion. Biochim Biophys Acta. 2008;1786:41–48. doi: 10.1016/j.bbcan.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 14.Nasmyth K, Haering CH. Cohesin: its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 15.Gruber S, Haering CH, Nasmyth K. Chromosomal cohesin forms a ring. Cell. 2003;112:765–777. doi: 10.1016/s0092-8674(03)00162-4. [DOI] [PubMed] [Google Scholar]
- 16.Remeseiro S, Losada A. Cohesin, a chromatin engagement ring. Curr Opin Cell Biol. 2013;25:63–71. doi: 10.1016/j.ceb.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, Zhang P, Kim ST, Pan X, Qin J. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008;31:143–151. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Ben-Shahar TR, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 2008;321:563–566. doi: 10.1126/science.1157774. [DOI] [PubMed] [Google Scholar]
- 19.Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008;321:566–569. doi: 10.1126/science.1157880. [DOI] [PubMed] [Google Scholar]
- 20.Beckouët F, Hu B, Roig MB, Sutani T, Komata M, Uluocak P, Katis VL, Shirahige K, Nasmyth K. An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell. 2010;39:689–699. doi: 10.1016/j.molcel.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400:37–42. doi: 10.1038/21831. [DOI] [PubMed] [Google Scholar]
- 22.Xiong B, Lu S, Gerton JL. Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr Biol. 2010;20:1660–1665. doi: 10.1016/j.cub.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 23.Borges V, Lehane C, Lopez-Serra L, Flynn H, Skehel M, Ben-Shahar TR, Uhlmann F. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol Cell. 2010;39:677–688. doi: 10.1016/j.molcel.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 24.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: histones and beyond. Biopolymers. 2013;99:112–126. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, Svinkina T, Natoli T, Lu X, Paulk J, McCarren P, Wagner FF, Barker D, Howe E, Lazzaro F, Gale JP, Zhang YL, Subramanian A, Fierke CA, Carr SA, Holson EB. An unbiased approach to identify endogeneous substrates of “histone”deacetylase 8. ACS Chem Biol. 2014;9:2210–2216. doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 30.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gantt SL, Joseph CG, Fierke CA. Activation and inhibition of histone deacetylase 8 by monovalent cations. J Biol Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Decroos C, Bowman CM, Moser JAS, Christianson KE, Deardorff MA, Christianson DW. Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem Biol. 2014;9:2157–2164. doi: 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh RK, Mandal T, Balsubramanian N, Viaene T, Leedahl T, Sule N, Cook G, Srivastava DK. Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate. Bioorg Med Chem Lett. 2011;21:5920–5923. doi: 10.1016/j.bmcl.2011.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 37.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A message passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 42.van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 43.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a despsipeptide inhibitor of the histone deacetylases. J Am Chem Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct, Funct, Bioinf. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lombardi PM, Angell HD, Whittington DA, Flynn EF, Rajashankar KR, Christianson DW. Structure of a prokaryotic polyamine deacetylase reveals evolutionary functional relationships with eukaryotic histone deacetylases. Biochemistry. 2011;50:1808–1817. doi: 10.1021/bi101859k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decroos C, Christianson DW. Design, synthesis and evaluation of polyamine deacetylase inhibitors, and high-resolution crystal structures of their complexes with acetylpolyamine amidohydrolase. Biochemistry. 2015;54:4692–4703. doi: 10.1021/acs.biochem.5b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arrar M, Turnham R, Pierce L, de Oliveira CAF, McCammon JA. Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Protein Sci. 2013;22:83–92. doi: 10.1002/pro.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Decroos C, Clausen DJ, Haines BE, Wiest O, Williams RM, Christianson DW. Variable active site loop conformations accommodate the binding of macrocyclic largazole analogues to HDAC8. Biochemistry. 2015;54:2126–2135. doi: 10.1021/acs.biochem.5b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kunze MBA, Wright DW, Werbeck ND, Kirkpatrick J, Coveney PV, Hansen DF. Loop interactions and dynamics tune the enzymatic activity of the human histone deacetylase 8. J Am Chem Soc. 2013;135:17862–17868. doi: 10.1021/ja408184x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh RK, Cho K, Padi SKR, Yu J, Haldar M, Mandal T, Yan C, Cook G, Guo B, Mallik S, Srivastava DK. Mechanism of N-acylthiourea-mediated activation of human histone deacetylase 8 (HDAC8) at molecular and cellular levels. J Biol Chem. 2015;290:6607–6619. doi: 10.1074/jbc.M114.600627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weiss MS. Global indicators of X-ray data quality. J Appl Cryst. 2001;34:130–135. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.