

Abstract
The benzylindazole derivative 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1) is an allosteric stimulator of soluble guanylate cyclase (sGC) that sensitizes the enzyme to the gaseous ligands carbon monoxide (CO) and nitric oxide (NO). In this study, we examined whether YC-1 also promotes the production of these gaseous monoxides by stimulating the expression of the inducible isoforms of heme oxygenase (HO-1) and NO synthase (iNOS) in vascular smooth muscle cells (SMCs). YC-1 increased HO-1 mRNA, protein, and promoter activity and potentiated cytokine-mediated expression of iNOS protein and NO synthesis by SMCs. The induction of HO-1 by YC-1 was unchanged by the sGC inhibitor, 1H-(1,2,4)oxadiazolo[4,3-α]quinozalin-1-one (ODQ), or by the protein kinase G inhibitors, KT 5823 and DT-2, and was not duplicated by 8-bromo-cGMP or the NO-independent sGC stimulator BAY 41-2272. However, theYC-1-mediated induction of HO-1 was inhibited by the phosphatidylinositol-3-kinase (PI3K) inhibitors, wortmannin and LY294002. In contrast, the enhancement of cytokine-stimulated iNOS expression and NO production by YC-1 was prevented by ODQ and the protein kinase A inhibitor, KT 5720, and mimicked by 8-bromo-cGMP and BAY 41-2272. In conclusion, these studies demonstrate that YC-1 stimulates the expression of HO-1 and iNOS in vascular SMCs via the PI3K and sGC-cGMP-protein kinase A pathway, respectively. The ability of YC-1 to sensitize sGC to gaseous monoxides while simultaneously stimulating their production through the induction of HO-1 and iNOS provides a potent mechanism by which the cGMP-dependent and -independent biological actions of this agent are amplified.
The benzylindazole derivative 3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1) is a nitric oxide (NO)-independent allosteric stimulator of soluble guanylate cyclase (sGC) that elevates intracellular guanosine 3′,5′-cyclic monophosphate (cGMP) levels in diverse tissues, including platelets, blood vessels, and vascular endothelial and smooth muscle cells (SMCs) (Ko et al, 1994; Freibe et al, 1996; Mulsch et al., 1997; Schmidt et al., 2001; Friebe and Koesling, 1998; Tulis et al, 2000; Tulis et al., 2002). YC-1 activates sGC by stabilizing the active configuration of the enzyme and increasing the affinity of sGC for substrate, GTP (Friebe et al., 1996; Freibe and Koesling, 1998; Lee et al., 2000). Activation of sGC by YC-1 is crucially dependent on the presence of the reduced prosthetic heme moiety of the enzyme since its removal or oxidation virtually abolishes YC-1-induced sGC activation (Freibe and Koesling, 1998). Interestingly, YC-1 enhances sGC activation by the ligands NO and carbon monoxide (CO) through stabilization of the active enzyme configuration and by decreasing the dissociation rates of these gases from the activated enzyme (Friebe and Koesling, 1998; Russwurm et al., 2002). Remarkably, YC-1 converts the relatively weak sGC activator CO to a potent activator whose effectiveness is comparable to that of NO (Friebe et al., 1996; Stone and Marletta, 1998).
Vascular SMCs generate CO and NO via the heme oxygenase (HO) and NO synthase (NOS) enzymes, respectively. HO oxidatively degrades heme to yield equimolar amounts of CO, biliverdin and iron. Two distinct isoforms of HO, HO-1 and HO-2, have been identified in vascular SMCs (see Wu and Wang, 2005; Durante et al., 2006). While HO-2 is primarily constitutively expressed, HO-1 is a highly inducible isoform that is upregulated by a number of biochemical and biophysical stimuli. Induction of HO-1 elicits potent antiproliferative, anti-inflammatory, antithrombotic, and antioxidant effects in the circulation via the generation of CO and biliverdin. Alternatively, the predominant source of NO formation by vascular SMCs is via the metabolism of L-arginine by inducible NOS (iNOS). The iNOS isoform is expressed after stimulation with inflammatory mediators and generates large amounts of NO over a prolonged period of time. In addition to reducing vasomotor tone and platelet aggregation, the generation of NO by vascular SMCs limits intimal thickening at sites of vascular injury by blocking SMC proliferation and migration (see Kibbe et al., 1999).
YC-1 represents the prototype of an important novel class of NO-independent stimulators of sGC that exhibit therapeutic potential for the treatment of a range of vascular diseases, including hypertension, thrombosis, erectile dysfunction, and postangioplasty restenosis (see Tulis, 2004; Evgenov et al., 2006). In this respect, YC-1 inhibits platelet aggregation and thrombosis in various experimental models (Teng et al., 1997). In addition, YC-1 lowers blood pressure and dilates blood vessels in normotensive and hypertensive animals (Rothermund et al., 2000). Furthermore, studies from our laboratories and others found that YC-1 inhibits vascular SMC proliferation and neointima formation following balloon-injury of rat carotid arteries (Tulis et al., 2000; Tulis et al, 2002; Liu et al., 2006; Wu et al., 2004). Moreover, YC-1 markedly enhances the antiaggregatory and vasodilator potency of NO and CO (Freibe et al., 1998; Rothermund et al., 2000; McLaughlin et al., 2000; Motterlini et al., 2005).
While YC-1 has been shown to potentiate the biological actions of NO and CO by sensitizing sGC to these gaseous monoxides, the ability of YC-1 to influence the production of these gases has not been fully investigated. In the present study, we investigated the role of YC-1 in regulating HO and iNOS expression in vascular SMCs. In addition, we identified signaling pathways by which YC-1 modulates the expression of these gas-generating enzymes. The ability of YC-1 to coordinately stimulate the expression of enzymes responsible for generating diatomic gases and sensitize sGC to these gaseous ligands may provide a novel mechanism by which this agent is able to amplify increases in tissue cGMP and promote its beneficial effects in the circulation. In addition, the induction of HO-1 and iNOS by YC-1 may contribute to the versatile pharmacological profile of this promising therapeutic compound.
Materials and Methods
Materials
Streptomycin, penicillin SDS, cycloheximide, actinomycin D, trypsin-EDTA, TES, HEPES, collagenase, elastase, glutamine, glycerol, bromophenol blue, 8-bromo-cGMP, BAY 41-2272, DT-2, Tes, 3-isobutyl-1-methylxanthine, naphthylethylenediamine dihydrochloride, curcumin, sulfanilamide, and minimum essential medium were from Sigma-Aldrich (St. Louis, MO); 1H-(1,2,4)oxadiazolo[4,3-α]quinozalin-1-one (ODQ), KT-5823, KT-5270, wortmannin, LY294002, U0125, PD98059, SB203580, and SP600125 were from Calbiochem-Novabiochem (San Diego, CA); interleukin-1β was from R&D Systems (Minneapolis, MN); tumor necrosis factor-α was from Genzyme (Boston, MA); polyclonal HO-1 and HO-2 antibodies were from StressGen Biotechnologies Inc. (Victoria, Canada); a polyclonal iNOS antibody was from BD Biosciences (San Diego, CA); a polyclonal antibody against β-actin was from Santa Cruz Biotechnologies (Santa Cruz, CA); antibodies against phospho-ERK1/2, phospho-p38 MAPK, phospho-JNK, and phospho-Akt were from Cell Signaling (Beverley, MA); [32P]dCTP (3000 Ci/mmol) was from Amersham (Arlington Heights, IL).
Cell Culture
Vascular SMCs were isolated by collagenase and elastase digestion of rat thoracic aortas and characterized by morphological and immunological criteria, as we previously described (Durante et al., 1993). Cells were cultured serially (passage 3 to 12) in minimum essential media supplemented with 10% bovine serum, 2mM L-glutamine, 5mM TES, 5mM HEPES, 100U/ml penicillin and 100U/ml streptomycin, and incubated in an atmosphere of 95% air/5% CO2.
Western Blotting
Cells were lysed in sample buffer (125 mM Tris [pH 6.8], 12.5% glycerol, 2% SDS, 50 mM sodium fluoride, and trace bromophenol blue) and proteins separated by SDS-PAGE. Following transfer to nitrocellulose membrane, blots were blocked with PBS and nonfat milk (5%) and then incubated with antibodies directed against HO-1 (1:1,500), HO-2 (1:500), iNOS (1:2000), phospho-ERK1/2 (1:1000), phospho-p38 mitogen-activated protein kinase (MAPK) (1:500), phospho-JNK (1:1000), phospho-Akt (1:1000), or β-actin (1:2000). Membranes were washed in PBS, incubated with horseradish peroxidase-conjugated goat anti-rabbit, rabbit anti-mouse, or donkey anti-goat antibody and developed with commercial chemoluminescence reagents (Amersham, Arlington Heights, IL). The expression of HO-1 and HO-2 protein was quantified by scanning densitometry and normalized with respect to β-actin.
HO-1 mRNA Expression
HO-1 mRNA levels were determined by northern blotting. Total RNA (30 μg) was loaded onto 1.2% agarose gels, fractionated by electrophoresis, and blots transferred to Gene Screen Plus membranes (Perkin Elmer Life Sciences). Membranes were prehybridized for 4 hours at 68°C at in rapid hybridization buffer (Amersham) and then incubated overnight at 68°C in hybridization buffer containing [32P]DNA probes (1 × 108 cpm) for HO-1 or 18S rRNA. DNA probes were generated by RT-PCR and labeled with [32P]dCTP using a random priming kit (Amersham, Arlington Heights, IL). Following hybridization, membranes were washed, exposed to X-ray film at -70°C, and HO-1 expression quantified by scanning densitometry and normalized with respect to 18S rRNA.
HO-1 Promoter Analysis
HO-1 promoter activity was determined using a full length murine HO-1 promoter/firefly luciferase construct that was generously provided by Dr. Jawed Alam at the Ochsner Clinic Foundation, New Orleans, LA. The HO-1 promoter (1 μg/ml) and pCMVβgal (1μg/ml) were transfected into SMCs using lipofectamine, cultured for 24 hours and then treated with YC-1. Cells were collected, lysed, and luciferase activity measured using a luciferase assay system and a Glomax luminometer (Promega, Madison, WI). Luciferase activity was normalized with respect to β-glactosidase activity (Liu et al., 2007).
MAPK and PI3K Activation
MAPK and PI3K activity were determined by western blotting using phospho-specific antibodies, as we previously described (Liu et al., 2007).
cGMP Assay
Vascular SMCs were pretreated with 3-isobutyl-1-methylxanthine (0.5mM) for 30 minutes and then treated with YC-1 or Bay 41-2272. In some instances, cells were also pretreated with ODQ (30μM) for 30 minutes prior to the addition of YC-1. Cells were lysed using 0.1 M HCl and cGMP concentrations measured using a commercially available kit (Molecular Devices, Sunnyvale, CA).
NO Synthesis
NO synthesis was assessed by measuring the accumulation of nitrite in the culture media, as we previously described (Durante et al., 1993). Since NO breaks down to both nitrite and nitrate, the levels of nitrite in the culture media were determined after incubation of samples with nitrate reductase to convert nitrate to nitrite. Samples were then mixed with an equal volume of Griess reagent (0.1% naphthylethylenediamine dihydrochloride and 1% sulfanilamide in 3 % H3PO4), incubated at room temperature for 10 minutes, and absorbance measured at 540 nm. Concentrations were determined relative to a standard curve obtained using aqueous solutions of nitrite.
Statistics
Results were expressed as the means ± SEM. Statistical analyses were performed using a Students two-tailed t-test or with an analysis of variance with post-hoc bonferroni testing when multiple groups were compared. P<0.05 was considered statistically significant.
Results
Treatment of vascular SMCs with YC-1 (5-100μM) stimulated a time- and concentration-dependent increase in HO-1 protein. An increase in HO-1 protein was detected after 24 hours with 20μM YC-1 and higher concentrations of YC-1 resulted in a progressive increase in HO-1 protein (Figure 1A). A significant increase in HO-1 protein was first detected 4 hours after YC-1 exposure and levels remained elevated following 24 hours of treatment (Figure 1B). In contrast, YC-1 had no effect on HO-2 protein expression (Figures 1A and B). Interestingly, another structurally similar NO-independent sGC stimulator BAY 41-2272 (5-100μM), failed to stimulate HO-1 protein expression (Figure 1C).
Figure 1.
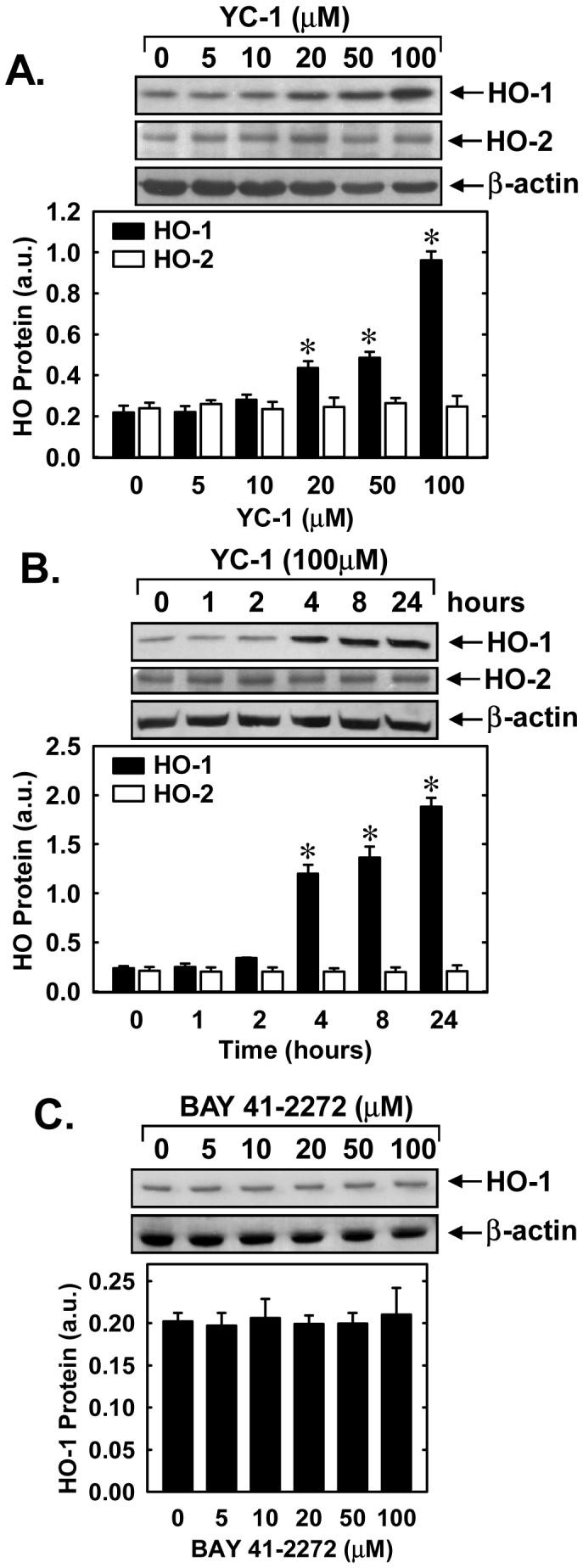
YC-1 stimulates HO-1 protein expression in vascular SMCs. A. Concentration-dependent effect of YC-1 (5-100μM for 24 hours) on HO-1 and HO-2 protein expression. B. Time-course of HO-1 and HO-2 protein expression following the administration of YC-1 (100μM). C. BAY 41-2272 (5-100μM for 24 hours) fails to stimulate HO-1 protein expression. HO-1 and HO-2 protein levels are expressed in arbitrary units (a.u.). Results are means ± SEM of 3-4 experiments. *Statistically significant effect of YC-1.
Correspondingly, the induction of HO-1 protein by YC-1 was associated with a time- and concentration-dependent rise in HO-1 mRNA that preceded the increase in HO-1 protein (Figures 2A and B). In addition, incubation of vascular SMCs with the transcriptional inhibitor, actinomycin D (2μg/ml), completely blocked the induction of HO-1 mRNA while the protein synthesis inhibitor, cycloheximide (5μg/ml) only partially inhibited the rise in HO-1 message (Figure 2C). In the absence of YC-1, actinomycin D or cycloheximide minimally affected HO-1 mRNA expression (data not shown). Furthermore, treatment of vascular SMCs with YC-1 (10-100μM) for 24 hours stimulated a concentration-dependent increase in HO-1 promoter activity (Figure 2D). To evaluate whether stabilization of HO-1 mRNA or protein also contributed to the induction of HO-1, YC-1 (100μM)-pretreated SMCs were incubated with actinomycin D (2μg/ml) or cycloheximide (5μg/ml) either in the presence or absence of YC-1 (100μM). However, YC-1 had no effect on the degradation of HO-1 mRNA or protein (Figures 3A and B).
Figure 2.

YC-1 stimulates HO-1 mRNA expression and promoter activity in vascular SMCs. A. Time-course of HO-1 mRNA expression following the administration of YC-1 (50μM). B. Concentration-dependent increase in HO-1 mRNA following the administration of YC-1 (5-100μM for 24 hours). C. Effect of actinomycin D (ActD; 2μg/ml) and cycloheximide (CX; 5μg/ml) on YC-1 (50μM for 8 hours)-mediated increase in HO-1 mRNA. D. Effect of YC-1 (10-100μM for 24 hours) on HO-1 promoter activity. HO-1 mRNA expression is expressed in arbitrary units (a.u.). Results are means ± SEM of 3-5 experiments. *Statistically significant effect of YC-1.
Figure 3.

YC-1 has no effect on HO-1 mRNA or protein stability in vascular SMCs. A. Effect of YC-1 on HO-1 mRNA stability. SMCs were pretreated with YC-1 (100μM for 24 hours), washed, and then exposed to actinomycin D (ActD; 2μg/ml) in the presence or absence of YC-1 (100μM) and the decay of HO-1 mRNA followed over 8 hours. B. Effect of YC-1 on HO-1 protein stability. SMCs were pretreated with YC-1 (100μM for 24 hours), washed, and then exposed to cycloheximide (CX; 5μg/ml) in the presence or absence of YC-1 (100μM) and the decay of HO-1 protein followed over 24 hours. Results are means ± SEM of 3-4 experiments.
In subsequent experiments we determined the upstream signaling molecules that mediate the induction of HO-1 by YC-1. Since YC-1 activates sGC, we first examined whether cGMP was responsible for triggering HO-1 expression. Treatment of vascular SMCs with YC-1 stimulated the intracellular accumulation of cGMP, and this was completely blocked by the sGC inhibitor, ODQ (30μM) (Figure 4A). Bay 41-2272 (5μM) also resulted in a substantial 7-fold increase in intracellular cGMP, raising levels from 12±4 to 88±10 fmol/well. However, incubation of SMCs with ODQ or the protein kinase G (PKG) inhibitors, KT5823 (5μM) or DT-2 (5μM), failed to prevent the YC-1-mediated induction of HO-1 protein or mRNA (Figures 4B, C and D). Moreover, exposure of SMCs to the cGMP lipophilic analogue, 8-Br-cGMP (5-500μM), had no effect on HO-1 protein expression (Figure 4E).
Figure 4.

YC-1 stimulates HO-1 gene expression in a soluble guanylate cyclase (sGC) and protein kinase G (PKG)-independent manner. A. Effect of the sGC inhibitor ODQ (30μM) on YC-1 (10-100μM for 10 minutes)-mediated increases in intracellular cGMP concentration. B. Effect of the sGC inhibitor ODQ (30μM) or the PKG inhibitor KT5823 (5μM) on YC-1 (100μM for 24 hours)-mediated increases in HO-1 protein. C. Effect of the sGC inhibitor ODQ (30μM) or the PKG inhibitor KT5823 (5μM) on YC-1 (100μM for 24 hours)-mediated increases in HO-1 mRNA. D. Effect of the sGC inhibitor DT-2 (5μM) on YC-1 (100μM for 24 hours)-mediated increases in HO-1 protein. E. 8-Br-cGMP (0-500μM for 24 hours) fails to stimulate HO-1 protein expression. HO-1 protein and mRNA are expressed in arbitrary units (a.u.). Results are means ± SEM of 3-4 experiments. *Statistically significant effect of YC-1.
Treatment of vascular SMCs with YC-1 resulted in the rapid activation of ERK1/2, JNK1, p38 MAPK, and PI3K, as reflected by the phosphorylation of Akt (Figure 5A). Activation of these proteins occurred within 5 minutes and persisted for 30 minutes following YC-1 exposure. Interestingly, the sGC inhibitor, ODQ (30μM), potentiated the YC-1-mediated induction of ERK1/2 and JNK1 but had no effect on the activation of p38 MAPK (Figure 5B), suggesting that YC-1 activates the three arms of MAPK via different signaling pathways. In addition, pretreatment of vascular SMCs with the ERK inhibitors, U0126 (20μM) or PD98059 (30μM), the p38 MAPK inhibitor, SB203580 (30μM), or the JNK inhibitor, SP600125 (20μM), had no effect on the induction of HO-1 protein by YC-1 (Figure 5C). In contrast, the PI3K inhibitors, wortmannin (100nM) and LY294002 (10μM), blocked the induction of HO-1 protein and mRNA (Figures 5D and E). Control experiments confirmed the effectiveness of each inhibitor for its respective enzyme (data not shown). Finally, we also examined the interaction between YC-1 and curcumin, another known inducer of PI3K activation and HO-1 gene expression (Pugazhenthi et al., 2007). While YC-1 and curcumin were both able to stimulate PI3K activation and HO-1 protein expression, the combined addition of these two agents did not lead to further increases in either PI3K activation or HO-1 protein (Figures 6A and B).
Figure 5.

YC-1 stimulates HO-1 gene expression in a phosphatidylinositol-3-kinase (PI3K)-dependent manner. A. Effect of YC-1 (50μM for 0-30 minutes)-treatment on ERK1/2, p38 MAPK, JNK, and Akt activity. B. Effect of the sGC inhibitor ODQ on YC-1-mediated increases in MAPK. Cells were pretreated with ODQ (30μM) for 30 minutes and then treated with YC-1 (50μM for 5 minutes). C. Effect of MAPK inhibitors on YC-1-mediated increases in HO-1 protein. Cells were pretreated with UO125 (UO; 20μM), PD98059 (PD; 30μM), SB203580 (SB; 30μM), or SP600125 (SP; 20μM) for one hour and then treated with YC-1 (50μM for 24 hours). D. Effect of PI3K inhibitors on YC-1-mediated increases in HO-1 protein. Cells were pretreated with LY294002 (LY; 10μM) or wortmannin (W; 100nM) for one hour and then treated with YC-1 (50μM for 24 hours). E. Effect of MAPK or PI3K inhibition on YC-1-mediated increases in HO-1 mRNA. Cells were pretreated with UO125 (UO; 20μM), PD98059 (PD; 30μM), SB203580 (SB; 30μM), SP600125 (SP; 20μM), or LY294002 (LY; 10μM) for one hour and then treated with YC-1 (50μM for 24 hours). HO-1 protein and mRNA are expressed in arbitrary units (a.u.). Results are means ± SEM of 3-5 experiments. *Statistically significant effect of YC-1. †Statistically significant effect of LY.
Figure 6.

Interaction between YC-1 and curcumin (Curc) in stimulating PI3K activity and HO-1 protein expression in vascular SMCs. A. Effect of YC-1 and/or Curc on PI3K activity. Cells were treated with YC-1 (50μM) and/or Curc (20μM) for 5 minutes. B. Effect of YC-1 and/or Curc on HO-1 protein expression. Cells were treated with YC-1 (50μM) and/or Curc (20μM) for 24 hours. Results are means ± SEM of 3 experiments. Akt phosphorylation and HO-1 protein are expressed in arbitrary units (a.u.). *Statistically significant from control.
Subsequently, we determined whether YC-1 also regulates the expression of a NO-generating enzyme. Control, untreated SMCs produce minimal amounts of nitrite, however, treatment of cells with a cytokine mixture consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) resulted in a significant accumulation of nitrite in the culture medium (Figure 7A). Interestingly, the simultaneous administration of YC-1 (10-50μM) potentiated the cytokine-mediated production of NO from vascular SMCs (Figure 7A). The cytokine-mediated production of nitrite was accompanied by the induction of iNOS protein, and iNOS protein levels were further increased in the presence of YC-1 (Figure 7B). In the absence of inflammatory cytokines, YC-1 failed to stimulate nitrite production or iNOS expression (Figures 7A and B). The ability of YC-1 to enhance cytokine-mediated nitrite synthesis and iNOS protein expression was dependent on sGC activity since it was prevented by ODQ (Figures 8A and B). In addition, the lipophilic analogue of cGMP, 8-bromo-cGMP (100-500μM), and BAY 41-2272 (5-20μM) potentiated the cytokine-mediated increase in nitrite production (Figures 9A and B). Moreover, 8-bromo-cGMP and BAY 41-2272 augmented the induction of iNOS protein by cytokines (Figure 9C). In the absence of cytokines, 8-bromo-cGMP or BAY 41-2272 minimally affected nitrite production (Figures 9A and B) and failed to induce iNOS protein (data not shown).
Figure 7.
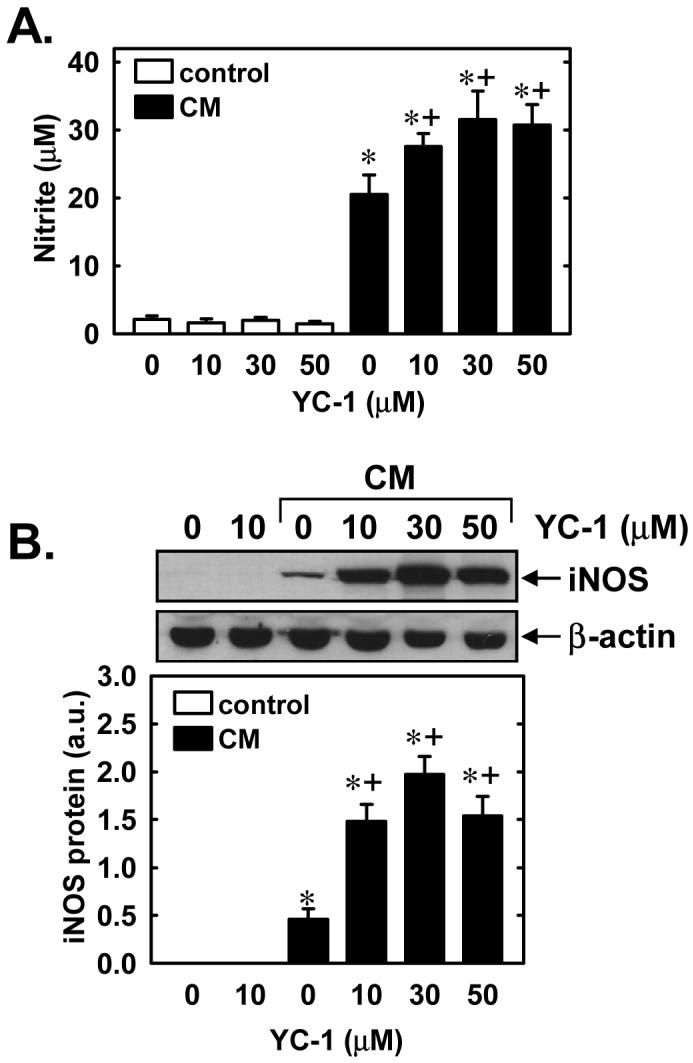
YC-1 enhances cytokine-mediated NO synthesis and iNOS protein expression in vascular SMCs. A. Effect of YC-1 (10-50μM) on nitrite accumulation following the treatment of cells with a cytokine mixture (CM) consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) for 24 hours. B. Effect of YC-1 (10-50μM) on iNOS protein expression following treatment of cells with a CM consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) for 24 hours. iNOS protein is expressed in arbitrary units (a.u.). Results are means ± SEM of 4-6 experiments. *Statistically significant effect of CM. †Statistically significant effect of YC-1.
Figure 8.
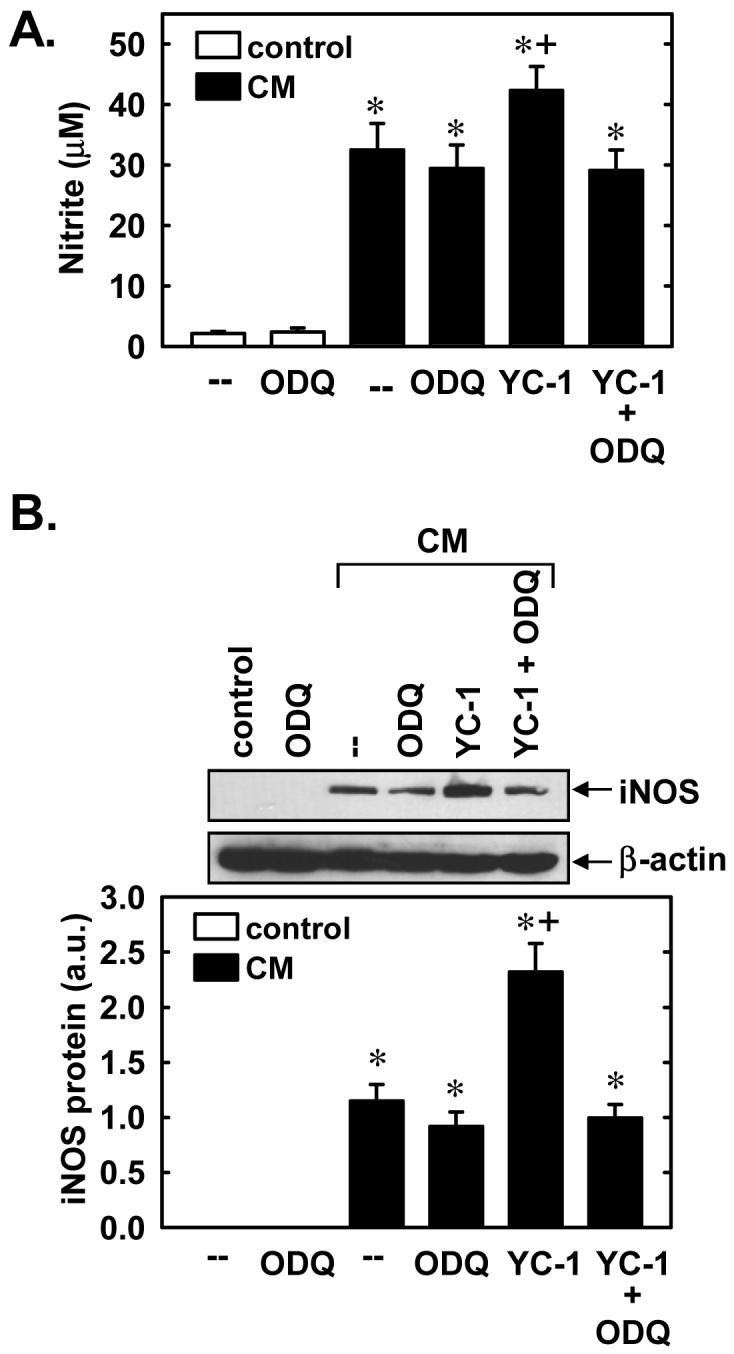
YC-1 enhances cytokine-mediated NO synthesis and iNOS protein expression in a soluble guanylate cyclase (sGC)-dependent manner. A. Effect of the sGC inhibitor, ODQ, (30 μM) on the stimulatory effect of YC-1 (30μM) on nitrite production following the treatment of cells with a cytokine mixture (CM) consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) for 24 hours. B. Effect of the sGC inhibitor, ODQ, (30 μM) on the stimulatory effect of YC-1 (30μM) on iNOS protein expression following the treatment of cells with a CM consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) for 24 hours. iNOS protein is expressed in arbitrary units (a.u.). Results are means ± SEM of 4-6 experiments. *Statistically significant effect of CM. †Statistically significant effect of YC-1.
Figure 9.
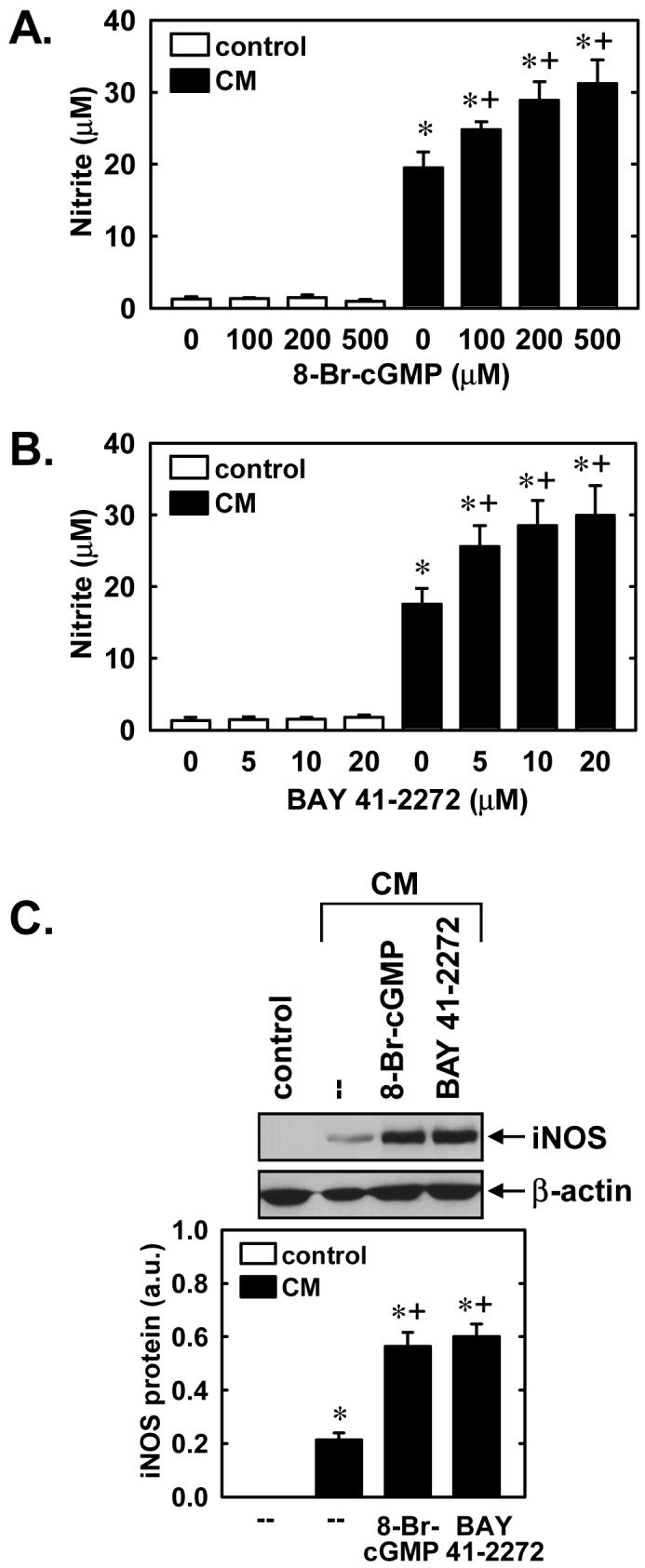
8-Br-cGMP and BAY 41-2272 enhance cytokine-mediated NO synthesis and iNOS protein expression in vascular SMCs. A. Effect of 8-Br-cGMP (100-500μM) on nitrite accumulation following the treatment of cells with a cytokine mixture (CM) consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) for 24 hours. B. Effect of BAY 41-2272 (5-20 μM) on nitrite accumulation following the treatment of cells with a CM consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (10ng/ml) for 24 hours. C. Effect of 8-Br-cGMP (200μM) or BAY 41-2272 (20μM) on iNOS protein expression following treatment of cells with a CM consisting of interleukin-1β (5ng/ml) and tumor necrosis factor-α (20ng/ml) for 24 hours. iNOS protein is expressed in arbitrary units (a.u.). Results are means ± SEM of 4-6 experiments. *Statistically significant effect of CM. †Statistically significant effect of 8-Br-cGMP or BAY41-2272.
In a final series of experiments, we examined the role of downstream protein kinases in mediating the augmentation of nitrite production by YC-1. Treatment of SMCs with the PKG inhibitor, KT5823 (5μM) or DT (5μM), had no effect on the ability of cytokines to stimulate nitrite formation and did not prevent the increase in nitrite synthesis by YC-1 (Figure 10A and B). In contrast, the protein kinase A (PKA) inhibitor, KT5720 (5μM) blunted the cytokine-mediated rise in nitrite formation and reversed the ability of YC-1 to elevate cytokine-mediated nitrite production (Figure 10C). In the absence of cytokines, KT5838, DT-2, or KT5720 failed to influence basal nitrite synthesis.
Figure 10.
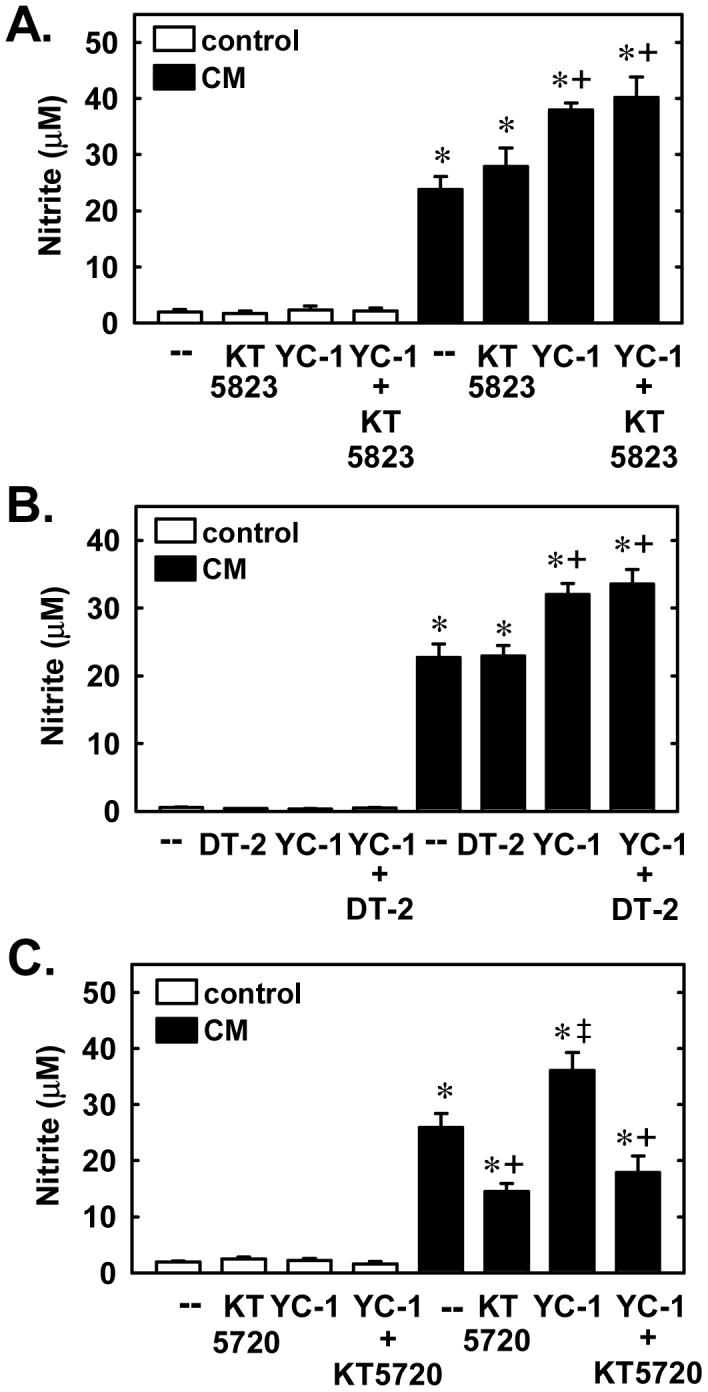
YC-1 enhances cytokine-mediated NO synthesis in a protein kinase A (PKA)-dependent manner. A. Effect of the protein kinase G (PKG) inhibitor KT 5823 (5μM) on the stimulatory effect of YC-1 (30μM) on nitrite production following the treatment of cells with a cytokine mixture (CM) consisting of interleukin-1β (5 ng/ml) and tumor necrosis factor-α (20 ng/ml) for 24 hours. B. Effect of the PKG inhibitor DT-2 (5μM) on the stimulatory effect of YC-1 (30μM) on nitrite production following the treatment of cells with the CM for 24 hours. C. Effect of the PKA inhibitor KT 5720 (5μM) on the stimulatory effect of YC-1 (30μM) on nitrite production following the treatment of cells with the CM for 24 hours. Results are means ± SEM of 5-7 experiments. *Statistically significant effect of CM. †Statistically significant effect of KT5720. ‡Statistically significant effect of YC-1.
Discussion
In the present study, we identified YC-1 as a novel inducer of HO-1 and iNOS protein expression in vascular SMCs. Interestingly, induction of these two enzymes by YC-1 occurs via distinct signaling pathways. While YC-1 stimulates the expression of HO-1 via the PI3K signaling pathway, the augmentation of iNOS expression occurs through the activation of the sGC-cGMP-PKA pathway. This newly discovered capacity of YC-1 to stimulate expression of the enzymes that generate CO and NO in addition to its documented sGC-sensitizing ability may provide an important amplification mechanism by which YC-1 is able to elevate cellular cGMP and regulate SMC function. Moreover, induction of the NO and CO systems by YC-1 may also contribute to its cGMP-independent actions in vascular tissues.
Treatment of vascular SMCs with YC-1 results in time- and concentration-dependent increases in HO-1 mRNA and protein. The induction of HO-1 by YC-1 does not involve mRNA or protein stabilization and is likely due to the transcriptional activation of the gene since actinomycin D completely blocks HO-1 transcript expression. Moreover, transient luciferase reporter assays demonstrate that YC-1 directly stimulates HO-1 promoter activity. Interestingly, the degree of the YC-1-mediated induction of HO-1 mRNA or protein is greater than the increase in HO-1 promoter activity. Although the cause for this discrepancy is not known, its important to note that promoter activity was monitored using a murine HO-1 promoter construct while HO-1 expression was assessed in rat vascular SMCs. Thus, the variation in magnitude between YC-1-mediated increases in HO-1 promoter activity and expression may reflect differences in HO-1 promoter regulation between species (Sikorski et al., 2004).
Since YC-1 stimulates sGC activity in vascular SMCs (Tulis et al., 2000; Tulis et al., 2002; Pan et al., 2004), we initially determined the involvement of this enzyme in mediating the induction of HO-1. Although treatment of vascular SMCs with the sGC inhibitor ODQ blocks the YC-1-mediated increase in cGMP production, ODQ does not prevent the induction of HO-1. Similarly, the PKG inhibitor KT5823 fails to prevent the induction of HO-1 by YC-1. Since KT5823 may not be effective in intact cells (Burkhardt et al., 2000), results obtained with this compound were corroborated using the cell-permeable PKG inhibitory peptide, DT-2. However, DT-2 also failed to block YC-1-mediated HO-1 expression. In addition, administration of the cGMP analogue, 8-bromo-cGMP, or the NO-independent sGC stimulator, BAY 41-2272, had no effect on HO-1 expression. These findings are consistent with previous work from our laboratory showing that cGMP does not alter HO-1 gene transcription in vascular SMCs (Liu et al., 2007). Moreover, these results indicate that the induction of HO-1 is not a common property of allosteric sGC activators and may be a unique property of YC-1.
Since MAPK and PI3K have been implicated in the induction of HO-1 gene expression (Alam and Cook, 2007), the role of these kinases in mediating the induction of HO-1 by YC-1 was investigated. Although YC-1 activates all three arms of the MAPK pathway, pharmacological inhibition of ERK, p38 MAPK, or JNK fails to block the induction of HO-1 by YC-1. Instead, we found that PI3K activity is necessary for the YC-1-mediated increase in HO-1 expression. YC-1 markedly increases PI3K activity in vascular SMCs, and pharmacological inhibition of PI3K activity blocks the YC-1-mediated induction of HO-1. This finding is in agreement with other studies demonstrating a key role for PI3K in stimulating the expression of HO-1 in several cell types, including vascular SMCs (Lee et al., 2004). Interestingly, YC-1 failed to augment PI3K activation or HO-1 protein expression by curcumin, suggesting that these two compounds may utilize a common signaling pathway in stimulating PI3K activity and HO-1 expression.
Aside from stimulating HO-1 expression, YC-1 influences the expression of a NO-generating enzyme in vascular SMCs. In particular, while YC-1 alone does not directly stimulate iNOS protein expression and NO synthesis, it significantly enhances the induction of iNOS protein and NO production by a mixture of inflammatory cytokines. In this case, the potentiation of iNOS expression and NO synthesis by YC-1 is mediated through the sGC-cGMP pathway since inhibition of sGC activity by ODQ abolishes the increase in both iNOS protein and NO synthesis. Moreover, a lipophilic analogue of cGMP or sGC activation by BAY 41-2272 duplicates the stimulatory effects of YC-1 on cytokine-stimulated iNOS expression and NO formation. Surprisingly, the YC-1-mediated increases in iNOS expression and NO synthesis are not mediated by PKG since inhibition of this kinase by KT5823 or DT-2 fails to block the stimulatory effect of YC-1 on NO production. Since YC-1 and cGMP can also activate PKA (Tsong-Long et al., 2003; Cornwell et al., 1994), the role of this kinase in promoting NO synthesis was also examined. Indeed, we found that the PKA inhibitor, KT5720, suppresses the YC-1-mediated increase in NO production in cytokine-treated cells. These findings are consistent with an earlier report showing that PKA is responsible for the enhancement of cytokine-mediated NO production by atrial natriuretic peptide in vascular SMCs (Iimura et al., 1998). Moreover, our finding that KT5720 diminishes NO synthesis following cytokine stimulation is in accordance with previous work demonstrating a key role for PKA in mediating the induction of iNOS by cytokines (Boese et al., 1997). Our finding that YC-1 promotes the induction of iNOS and NO synthesis in vascular SMCs, complements a previous study showing that YC-1 stimulates the release of NO by eNOS in endothelial cells (Wohlfart et al., 1999), further emphasizing the ability of YC-1 to upregulate NO production in the vasculature. However, it contrasts with recent reports showing that YC-1 attenuates lipopolysaccharide-mediated iNOS expression in macrophages and microglia (Hsiao et al., 2004; Lu et al., 2007). The cause for these divergent results may reflect differences in cell type, culture conditions, and/or the nature of the inflammatory stimuli.
The ability of YC-1 to stimulate the production of gaseous monoxides through the induction of HO-1 and iNOS may be of pharmacological and therapeutic significance. Specifically, upregulation of CO and NO synthesis would provide an important long-term mechanism by which YC-1 elevates tissue cGMP levels. Thus, the induction of HO-1 and iNOS may amplify the many cGMP-dependent actions of YC-1, including its antihypertensive, antiaggregatory, antiproliferative, and antiapoptotic properties. (Teng et al., 1997; Rothermund et al., 2000; Tulis et al., 2000; Tulis et al., 2002; Pan et al., 2004; see Tulis, 2004). Interestingly, the induction of HO-1 and iNOS by YC-1 may also underlie the versatile pharmacological profile of this agent. In this respect, YC-1 has recently been shown to elicit an antioxidant effect by inhibiting neutrophil superoxide production through a cGMP-independent pathway (Wang et al., 2002). Furthermore, YC-1 has been reported to exert a cGMP-independent, anti-inflammatory action in leukocytes and in endotoxemic mice (Pan et al., 2005). Given that HO-1 evokes potent antioxidant and anti-inflammatory effects through the generation of biliverdin and bilirubin (Wu and Wang, 2005; Durante et al., 2006), it is plausible that YC-1 may suppress both oxidative stress and inflammation through the induction of HO-1. Moreover, the ability of YC-1 to enhance iNOS-mediated NO synthesis may further intensify its anti-inflammatory actions, since the production of NO by iNOS has been shown to promote the nitrosylation and inactivation of NF-κB, a key transcription factor involved in the inflammatory cascade (Marshall et al., 2001; Reynaert et al., 2004).
In conclusion, these studies demonstrate that YC-1 stimulates the expression of HO-1 and iNOS in vascular SMCs via the PI3K and sGC-cGMP-PKA pathways, respectively. The ability of YC-1 to stimulate CO and NO production through the induction of HO-1 and iNOS, while simultaneously sensitizing sGC to these diatomic gases, may serve to amplify the cGMP-dependent biological actions of this indazole derivative. In addition, the induction of HO-1 and iNOS by YC-1 may contribute to the pleiotropic beneficial effects of this agent in the circulation.
Acknowledgments
This work was supported by NIH Grants HL59976, HL74966, HL82774, and HL81720.
Abbreviations
- YC-1
3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole
- sGC
soluble guanylate cyclase
- NO
nitric oxide
- cGMP
guanosine 3′,5′-cyclic monophosphate
- CO
carbon monoxide
- HO
heme oxygenase
- NOS
nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- SMCs
smooth muscle cells
- SDS
sodium dodecyl sulfate
- EDTA
ethylene diamine tetraacetic acid
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- TES
N-Tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid
- dCTP
deoxycytidine triphosphate
- BAY 41-2272
5-cyclopropyl-2[1-(2-fluorobenzyl)-1H-pyrazolo [3,4-b] pyridine-3-yl] pyrimidin-4-ylamine
- ODQ
1H-(1,2,4)oxadiazolo[4,3-α]quinozalin-1-one
- PKG
protein kinase G
- PKA
protein kinase A
- 8-Br-cGMP
8-bromo- guanosine 3′,5′-cyclic monophosphate
- KT5823
(8R,9S,11S)-(-)-2-methyl-9-methoxyl-9-methoxycarbonyl-8-methyl-2,3,9,10-tetrahydro-8,11-epoxy-1H,8H,11H-2,7b,11a-triazadibenzo(a,g)cyclocta9(cde)trinen-1-one
- KT 5720
(9S,10S,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo(1,2,3-fg:3′,2′,1′-kl)pyrrolo(3,4-i)(1,6)benzodiazocine-10-carboxylic acid hexyl ester
- DT-2 peptide
YGRKKRRQRRRPPLRKKKKKH-amide
- PI3K
phosphatidylinositol-3-kinase
- MAPK
mitogen-activated protein kinase
References
- Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell. Mol. Biol. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- Boese M, Busse R, Mulsch A, Schini-Kerth V. Effect of cyclic GMP-dependent vasodilators on the expression of inducible nitric oxide synthase in vascular smooth muscle cells: role of cyclic AMP. Br. J. Pharmacol. 1996;119:707–715. doi: 10.1111/j.1476-5381.1996.tb15730.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt M, Glazova M, Gambaryan S, Vollkommer T, Butt E, Bader B, Heermeier K, Lincoln TM, Walter U, Palmetshofer A. KT5823 inhibits cGMP-dependent protein kinase activity in vitro but not in intact human platelets and rat mesangial cells. J. Biol. Chem. 2000;275:33536–33541. doi: 10.1074/jbc.M005670200. [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Arnold E, Boerth NJ, Lincoln TM. Inhibition of smooth muscle cell growth by nitric oxide and activation of cAMP-dependent protein kinase by cGMP. Am. J. Physiol. 1994;267:C1405–1413. doi: 10.1152/ajpcell.1994.267.5.C1405. [DOI] [PubMed] [Google Scholar]
- Durante W, Johnson FK, Johnson RA. Role of carbon monoxide in cardiovascular function. J. Cell. Mol. Med. 2006;10:672–686. doi: 10.1111/j.1582-4934.2006.tb00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante W, Schini VB, Catovsky S, Kroll MH, Vanhoutte PM, Schafer AI. Plasmin potentiates induction of nitric oxide synthase in vascular smooth muscle cells. Am. J. Physiol. 1993;264:H617–H624. doi: 10.1152/ajpheart.1993.264.2.H617. [DOI] [PubMed] [Google Scholar]
- Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HHHW, Stasch J-P. NOindependent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat. Rev. 2006;5:755–768. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibe A, Koesling D. Mechanism of YC-1-induced activation of soluble guanylate cyclase. Mol. Pharmacol. 1998;53:123–127. doi: 10.1124/mol.53.1.123. [DOI] [PubMed] [Google Scholar]
- Freibe A, Schultz G, Koesling D. Sensitizing soluble guanylate cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- Hsiao G, Huang H-Y, Fong T-H, Shen M-Y, Lin C-H, Teng C-M, Sheu J-R. Inhibitory mechanisms of YC-1 and PMC in the induction of iNOS expression by lipoteichoic acid in RAW 264.7 macrophages. Biochem. Pharmacol. 2004;67:1411–1419. doi: 10.1016/j.bcp.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Iimura, Kusano E, Homma S, Takeda S, Ikeda U, Shimada K, Asano Y. Atrial natriuretic peptide enhances IL-1β-stimulated nitric oxide production in cultured rat vascular smooth muscle cells. Kidney Blood Press. Res. 1998;21:36–41. doi: 10.1159/000025841. [DOI] [PubMed] [Google Scholar]
- Kibbe M, Billiar T, Tzeng E. Inducible nitric oxide synthase and vascular injury. Cardiovasc. Res. 1999;43:650–657. doi: 10.1016/s0008-6363(99)00130-3. [DOI] [PubMed] [Google Scholar]
- Ko FN, Wu CC, Kuo SC, Lee FY, Teng CM. YC-1, a novel activator of platelet guanylate cyclase. Blood. 1994;84:4226–4233. [PubMed] [Google Scholar]
- Lee TS, Chang CC, Zhu Y, Shyy JY. Simvastatin induces heme oxygenase-1: a novel mechanism of vessel protection. Circulation. 2004;110:1296–1302. doi: 10.1161/01.CIR.0000140694.67251.9C. [DOI] [PubMed] [Google Scholar]
- Lee YC, Martin E, Murad F. Human recombinant soluble guanylate cyclase: expression, purification, and regulation. Proc. Natl. Acad. Sci. USA. 2000;97:10763–10763. doi: 10.1073/pnas.190333697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XM, Peyton KJ, Ensenat D, Wang H, Hannink M, Alam J, Durante W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc. Res. 2007;75:381–389. doi: 10.1016/j.cardiores.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-N, Pan S-L, Peng C-Y, Guh J-H, Huang D-M, Chang Y-L, Lin C-H, Pai H-C, Kuo S-C, Lee F-Y, Teng C-M. YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole] inhibits neointima formation in balloon-injured rat carotid through suppression of expressions of activities of matrix metalloproteinases 2 and 9. J. Pharmacol. Exp. Ther. 2006;316:35–41. doi: 10.1124/jpet.105.090563. [DOI] [PubMed] [Google Scholar]
- Lu D-Y, Tang C-H, Liou H-C, Teng C-M, Jeng K-CG, Kuo S-C, Lee F-Y, Fu W-M. YC-1 attenuates LPS-induced proinflammatory responses and activation of nuclear factor-κB. Br. J. Pharmacol. 2007;151:396–405. doi: 10.1038/sj.bjp.0707187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- McLaughlin BE, Chretien ML, Choi C, Brien JF, Makatsu K, Marks GS. Potentiation of carbon monoxide-induced relaxation of rat aorta by YC-1 [3-(5′-hydroxymethyl-2′-furyl)-1-benzyl indazole] Can. J. Physiol. Pharmacol. 2000;78:343–349. [PubMed] [Google Scholar]
- Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, Green CJ. CORMA1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284–286. doi: 10.1096/fj.04-2169fje. [DOI] [PubMed] [Google Scholar]
- Mulsch A, Bauersachs J, Schafer A, Stasch JP, Kast R, Busse R. Effect of YC-1, an NO-independent, superoxide sensitive stimulator of soluble guanylate cyclase, on smooth muscle cell responsiveness to nitrovasodilators. Br. J. Pharmacol. 1997;120:681–689. doi: 10.1038/sj.bjp.0700982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan SL, Guh JH, Chang YL, Kuo SC, Lee FY, Teng CM. YC-1 prevents sodium nitroprusside-mediated apoptosis in vascular smooth muscle cells. Cardiovasc. Res. 2004;61:152–158. doi: 10.1016/j.cardiores.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Pan SL, Guh JH, Peng CY, Chang YL, Cheng FC, Chang JH, Kuo SC, Lee FY, Teng CM. A potential role of YC-1 on the inhibition of cytokine release in peripheral blood mononuclear leukocytes and endotoxemic mouse models. Thromb. Haemost. 2005;93:940–948. doi: 10.1160/TH04-03-0195. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Akhov L, Selvaraj G, Wang M, Alam J. Regulation of heme oxygenase-1 expression by demethoxy curcuminoids through Nrf2 by PI3-kinase/Akt mediated pathway in mouse beta-cells. Am. J. Physiol. Endocrinol. Metab. 2007;293:E645–E655. doi: 10.1152/ajpendo.00111.2007. [DOI] [PubMed] [Google Scholar]
- Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory κB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermund L, Friebe A, Koesling PM, Kreutz R. Acute blood pressure effects of YC-1-induced activation of soluble guanylate cyclase in normotensive and hypertensive rats. Br. J. Pharmacol. 2000;130:205–208. doi: 10.1038/sj.bjp.0703320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Mergia E, Mullershausen F, Koesling D. Inhibition of deactivation of NO-sensitive guanylyl cyclase accounts for the sensitizing effect of YC-1. J. Biol. Chem. 2002;277:24833–24888. doi: 10.1074/jbc.M110570200. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Schrammel A, Koesling D, Mayer B. Molecular mechanisms involved in the synergistic activation of soluble guanylyl cyclase by YC-1 and nitric oxide in endothelial cells. Mol. Pharmacol. 2001;59:220–224. doi: 10.1124/mol.59.2.220. [DOI] [PubMed] [Google Scholar]
- Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: molecular regulation of the heme oxygenase-1 gene in renal injury. Am. J. Physiol. Renal Physiol. 2004;286:F425–F441. doi: 10.1152/ajprenal.00297.2003. [DOI] [PubMed] [Google Scholar]
- Stone JR, Marletta MA. Synergystic activation of soluble guanylate cyclase by YC-1 and carbon monoxide: implications for the role of cleavage of the iron-histidine bond during activation by nitric oxide. Chem. Biol. 1998;5:255–261. doi: 10.1016/s1074-5521(98)90618-4. [DOI] [PubMed] [Google Scholar]
- Teng CM, Wu CC, Ko FN, Lee FY, Kuo SC. YC-1, a nitric oxide-independent activator of soluble guanylate cyclase, inhibits platelet-rich thrombosis in mice. Eur. J. Pharmacol. 1997;320:161–166. doi: 10.1016/s0014-2999(96)00911-9. [DOI] [PubMed] [Google Scholar]
- Tsong-Long H, Hung H-W, Kao S-H, Teng C-M, Wu C-C, Cheng SJ-S. Soluble guanylate cyclase activator YC-1 inhibits human neutrophil functions through a cGMPindependent but cAMP-dependent pathway. Mol. Pharmacol. 2003;64:1419–1427. doi: 10.1124/mol.64.6.1419. 2003. [DOI] [PubMed] [Google Scholar]
- Tulis DA. Salutary properties of YC-1 in the cardiovascular and hematological systems. Curr. Med. Chem.-Cardiovascular & Hematological Agents. 2004;2:343–359. doi: 10.2174/1568016043356200. [DOI] [PubMed] [Google Scholar]
- Tulis DA, Bohl Masters KS, Lipke EA, Schiesser RL, Evans AJ, Peyton KJ, Durante W, West JL, Schafer AI. YC-1-mediated vascular protection through inhibition of smooth muscle cell proliferation and platelet function. Biochem. Biophys. Res. Commun. 2002;291:1014–1021. doi: 10.1006/bbrc.2002.6552. [DOI] [PubMed] [Google Scholar]
- Tulis DA, Durante W, Peyton KJ, Chapman GB, Evan AJ, Schafer AI. YC-1, a benzyl indazole derivative, stimulates vascular cGMP and inhibits neointima formation. Biochem. Biophys. Res. Commun. 2000;279:646–652. doi: 10.1006/bbrc.2000.3942. [DOI] [PubMed] [Google Scholar]
- Wang JP, Chang LC, Raung SL, Hsu MF, Huang LJ, Kuo SC. Inhibition of superoxide anion generation by YC-1 in rat neutrophils through cGMP-dependent and -independent mechanisms. Biochem. Pharmacol. 2002;63:577–585. doi: 10.1016/s0006-2952(01)00882-6. [DOI] [PubMed] [Google Scholar]
- Wohlfart P, Malinski T, Ruetten H, Schindler U, Linz W, Schoenafinger K, Strobel H, Wiemer G. Release of nitric oxide from endothelial cells stimulated by YC-1, an activator of soluble guanylate cyclase. Br. J. Pharmacol. 1999;128:1316–1322. doi: 10.1038/sj.bjp.0702921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-H, Chang W-C, Chang G-Y, Kuo S-C, Teng C-M. The inhibitory mechanism of YC-1, a benzyl indazole, on smooth muscle cell proliferation: an in vitro and in vivo study. J Pharmacol. Sci. 2004;94:252–260. doi: 10.1254/jphs.94.252. [DOI] [PubMed] [Google Scholar]
- Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol. Rev. 2005;57:585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]