

Abstract
Background and aims
The concept of enteric glia as regulators of intestinal homeostasis is slowly gaining acceptance as a central concept in neurogastroenterology. Yet how glia contribute to intestinal disease is still poorly understood. Purines generated during inflammation drive enteric neuron death by activating neuronal P2X7 purine receptors (P2X7R), triggering ATP release via neuronal pannexin-1 channels that subsequently recruits intracellular calcium ([Ca2+]i) responses in the surrounding enteric glia. We tested the hypothesis that the activation of enteric glia contributes to neuron death during inflammation.
Methods
We studied neuroinflammation in vivo using the 2,4-dinitrobenzenesulfonic acid model of colitis and in situ using whole-mount preparations of human and mouse intestine. Transgenic mice with a targeted deletion of glial connexin-43 (Cx43) [GFAP∷CreERT2+/−/Cx43f/f] were used to specifically disrupt glial signaling pathways. Mice deficient in inducible nitric oxide (NO) synthase (iNOS−/−) were used to study NO production. Protein expression and oxidative stress were measured using immunohistochemistry and in situ Ca2+ and NO imaging were used to monitor glial [Ca2+]i and [NO]i.
Results
Purinergic activation of enteric glia drove [Ca2+]i responses and enteric neuron death through a Cx43-dependent mechanism. Neurotoxic Cx43 activity, driven by NO production from glial iNOS, was required for neuron death. Glial Cx43 opening liberated ATP and Cx43-dependent ATP release was potentiated by NO.
Conclusions
Our results show that the activation of glial cells in the context of neuroinflammation kills enteric neurons. Mediators of inflammation that include ATP and NO activate neurotoxic pathways that converge on glial Cx43 hemichannels. The glial response to inflammatory mediators might contribute to the development of motility disorders.
Keywords: Enteric nervous system, purines, hemichannels, oxidative stress
INTRODUCTION
Reflex behaviors of the intestine, such as peristalsis, are orchestrated by the enteric nervous system (ENS); a complex network of neurons and glia embedded in the gut wall. The basic neural circuitry of the ENS is now well defined and it is generally accepted that the breakdown of ENS control is a major contributing factor to the development of functional bowel disorders1. However, it is only recently that we are beginning to appreciate the potential roles of enteric glial cells in the physiology and pathophysiology of the ENS2. Despite intense interest in enteric glia as regulators of enteric neurons, the precise functions of enteric glia remain poorly defined.
Enteric glia are a unique population of peripheral astroglial cells that surround enteric neurons and are thought to sustain neural signaling and survival. In support, enteric glia secrete neuroprotective factors3 and the selective ablation of glial signaling alters the neural control of motility4. Likewise, in vivo models of glial ablation cause enteric neuron death5,6. Thus, the loss of glial supportive functions is postulated as a potential mechanism that contributes to enteric neuropathy2. However, new data show that chronic astroglial activation, rather than glial cell loss, is responsible for driving neurodegeneration during neuroinflammation in the brain7. Indeed, the conversion of astroglia to reactive astrocytes can promote the secretion of factors that promote neuron death8.
We recently discovered that enteric glia are activated by purines released from enteric neurons prior to neuronal death during colitis9. Specifically, the activation of neuronal P2X7 purine receptors (P2X7Rs) triggers the release of ATP from neurons through pannexin-1 (panx1) channels as a signal to enteric glia. In the brain, neuronal ATP release through panx1 is considered a danger signal that glial cells interpret as a “search and destroy” message; causing glia to execute otherwise healthy neurons. Given that stimulation of P2Y1Rs is a potent stimulus for reactive astrogliosis in the central nervous system10, we hypothesized that the activation of glial purine receptors contributes to neuropathy in the ENS.
We tested our hypothesis using a combination of in vivo models of colitis with inducible and conditional transgenic mice and ex vivo intestinal preparations to address specific mechanisms. Our data show that glial activation is sufficient to cause enteric neuron death via a mechanism that depends on the activation of connexin-43 (Cx43) hemichannels and subsequent ATP release. Surprisingly, our data show that glial-driven neuron death requires the gating of glial Cx43 hemichannels by nitric oxide (NO). In all, our results suggest that the activation of enteric glial cells is a central mechanism in the development of enteric neuropathy.
METHODS
Animals
All work involving animals was approved by the Institutional Animal Care and Use Committee (IACUC) of Michigan State University. Male mice (8–10 weeks of age) were maintained on a 12-hour light/dark cycle with access to food and water ad libitum. C57Bl/6 mice were purchased from Charles River and inducible nitric oxide synthase (iNOS) null mice (B6.129S2-Nos2tm1MrlN12; Taconic Labs; RRID: MGI_4837857; hereafter referred to as iNOS−/−) from Taconic11. Transgenic mice with an inducible and conditional deletion of Cx43 in GFAP-expressing glia (GFAP∷CreERT2+/−/Cx43f/f; hereafter referred to as Cx43i-cKO) and their Cre negative littermate controls (GFAP∷CreERT2+/+/Cx43f/f) were generated in-house as previously described4 by crossing GFAP∷CreERT2+/− mice (GFAP-cre/ERT2)505Fmv/J; Jackson Labs; RRID: IMSR_JAX:012849) with Cx43f/f mice (B6.129S7-Gja1tm1Dlg/J; Jackson Labs; RRID: IMSR_JAX:008039). Cre recombinase activity was induced by feeding animals tamoxifen citrate in chow (400 mg/kg) for two weeks. Animals were returned to normal chow for 1 week to clear tamoxifen prior to beginning experiments.
Human tissue
Work involving human tissue was approved by the Institutional Review Board of Michigan State University (IRB# 13-945M). Samples of live, full-thickness human jejunum were collected from a 57-year-old female with hypertension and type 2 diabetes who underwent elective laparoscopic bariatric surgery for weight loss, and placed in chilled DMEM/F12 media during transfer to the laboratory. Live longitudinal muscle myenteric plexus whole-mount preparations were prepared by microdissection for Ca2+ imaging.
Whole-mount immunohistochemistry
Whole-mount preparations of mouse colonic longitudinal muscle and myenteric plexus (LMMP) were prepared by microdissection from tissue preserved in Zamboni's fixative. Processing of LMMPs via immunohistochemistry was conducted as described previously4 with the primary and secondary antibodies listed in Tables 1 and 2, respectively. Briefly, dissected LMMP preparations underwent three 10 min washes in 0.1% Triton X-100 in phosphate-buffered saline (PBST) followed by a 45 min block in blocking solution containing 4% normal goat serum, 0.4% Triton X-100 and 1% bovine serum albumin (4% NGS). Preparations were incubated in primary antibodies (listed in Table 1) for 48hours at 4°C and secondary antibodies (listed in Table 2) for 2hours at room temperature before mounting. Antibody specificity was confirmed by preadsorption with the corresponding control peptides or in knockout mice as previously described9. Fluorescent labeling was visualized using the 40X (PlanFluor, 0.75 numerical aperture) objective of an upright epifluorescence microscope (Nikon Eclipse Ni, Melville, NY) with a Retiga 2000R camera (QImaging, Surrey, BC, Canada) controlled by QCapture Pro 7.0 (QImaging) software or by confocal imaging through the Plan-Apochromat 60x oil immersion objective (1.42 numerical aperture) of an inverted Olympus Fluoview FV1000 microscope (Olympus, Center Valley, PA).
Table 1.
Primary antibodies used.
| Antibody | Source | Dilution | Catalog # |
|---|---|---|---|
| Rabbit anti-iNOS | Abcam, Cambridge, MA | 1:200 | ab15323 |
| Biotinylated Mouse Anti-Human Hu C/D | Molecular Probes, Grand Island, NY | 1:200 | A-21272 |
| Chicken anti-GFAP | Abcam, Cambridge, MA | 1:1000 | ab4674 |
| Rabbit anti-Nitrotyrosine | Millipore, Billerica, MA | 1:100 | 06–284 |
| Rabbit anti-P2Y1R | Alomone Labs, Jerusalem, Israel | 1:200 | APR-021 |
Table 2.
Secondary antibodies used.
| Antibody | Source | Dilution | Catalog # |
|---|---|---|---|
| Alexa Fluor 488 Goat anti-rabbit | Invitrogen, Carlsbad, CA | 1:200 | A-11034 |
| Alexa Fluor 488 Goat anti-chicken | Invitrogen, Carlsbad, CA | 1:200 | A-11039 |
| Alexa Fluor 568 Goat anti-chicken | Invitrogen, Carlsbad, CA | 1:200 | A-11041 |
| Alexa Fluor 594-conjugated Streptavidin | Jackson ImmunoResearch, West Grove, PA | 1:200 | 016-580-084 |
Quantification of neuronal thiol oxidation
We quantified neuronal thiol oxidation as a measure of oxidative stress as described previously12. Reduced (-SH) and oxidized (-SS) thiols were labeled in live LMMP preparations with 1 μM Alexa Fluor 680 C2 maleimide and 1 μM Alexa Fluor 546 C5 maleimide, respectively. Fluorescent dyes were dissolved in 4% paraformaldehyde, 0.02% Triton X-100 and 1 mM N-ethylmaleimide (NEM) in PBS. Oxidized thiols were converted to reduced thiols for labeling by washing tissue in 5 mM tris(2-carboxyethyl)phosphine hydrochloride in PBS for 20 minutes. Images were obtained by epifluorescence microscopy as described above and the ratio of 546-maleimide/680-maleimide (SS/SH) was calculated with ImageJ software (NIH).
Dihydroethidium (DHE) staining
Superoxide levels were measured by quantifying fluorescence of the superoxide marker DHE13 in live LMMP ganglia after a 1hour incubation with 2 μM DHE (Life Technologies, Carlsbad, CA) dissolved in DMEM at 37°C.
In situ model of neuroinflammation
Enteric neuron death was driven, as previously described9, by incubating live LMMP preparations with the P2X7R agonist BzATP (300 μM) for 2hours in 95% air: 5% CO2 at 37°C. Following this incubation, LMMP preparations were rinsed with fresh buffer, incubated for an additional 2hours in Kreb's buffer only, and fixed in Zamboni's fixative overnight.
Ca2+ and NO imaging
Live LMMPs were loaded with 4 μM Fluo-4-AM or DAF-FM (Life Technologies) for 45 min at 37°C as described previously4 to measure intracellular Ca2+ and NO, respectively. Loaded tissue was continuously perfused with pre-warmed buffer (34°C; 3 mL/min) and drugs were bath applied. Images were acquired every 1–5 seconds with a Neo sCMOS digital camera (Andor, South Windsor, CT) through the 40X water-immersion objective of an upright Olympus BX51W1 fixed-stage microscope (Olympus, Center Valley, PA) controlled by Andor IQ3 software.
Determination of cell viability
Cell viability was determined using Calcein-AM; a hydrophobic compound that is converted to hydrophilic, fluorescent Calcein in live, membrane-intact cells14. The Calcein-AM cell viability assay is a simple, rapid and accurate method to measure cytotoxicity that is not dependent upon the mode of cell death and is a true end-point assay for cell viability. Non-fluorescent, hydrophobic Calcein-AM easily permeates live cells where it is acted upon by esterases, producing the strongly fluorescent hydrophilic compound Calcien, which is retained in the cell cytoplasm. A loss of membrane integrity in dying cells permits the dye to escape from the cell and a loss of cellular fluorescence is indicative of neuron death. Live LMMPs were loaded with 4 μM Calcien-AM for 30 min at 37°C following in situ induction of neuroinflammation with BzATP (300 μM) or ADP (100 μM) or incubation with buffer control. Static images of 10 ganglia per whole mount were acquired as described for Ca2+ imaging and neuronal density calculated based on the number of live (fluorescent) cells per ganglionic area.
Detection of ATP release
ATP release was measured using an in-house-fabricated, selective ATP sensor15,16. LMMPs were continuously perfused with buffer (34°C; 6 mL/min) and ATP release was stimulated by bath application of either BzATP (300 μM) or ADPβS (100 μM) in the presence or absence of various pharmacological inhibitors. The ATP-selective sensor was placed directly on a ganglion and a superfusion pipette was located within 100 μm of the sensor. Traces were analyzed by measuring the area under the curve and converting this to the concentration of ATP using pre-obtained calibration curves15.
Induction of colitis
Colitis was induced in mice via an enema of dinitrobenzene sulfonic acid (DNBS, 5.5 mg/mouse in 0.1 mL ethanol/saline administered via a gavage needle inserted 3 cm into the colon) as previously described9. Animal weight was recorded daily and tissue harvested 6hours and 48hours following DNBS treatment. Upon tissue collection, macroscopic damage was assessed to quantify acute inflammation17. Macroscopic damage assessment scored for the presence of colonic ulcers, hemorrhaging, fecal blood, diarrhea, increased colon wall thickness and adhesion of the colon to the peritoneal cavity and/or other organs. Inflammation was classified as mild if damage scores were <1, moderate if scores were >1 but <5 and severe if > 5.
Chemicals/ Drugs
2'(3')-O-(4-Benzoylbenzoyl)adenosine 5'-triphosphate triethylammonium salt (BzATP), Adenosine 5'-diphosphate monopotassium salt dihydrate (ADP), Adenosine 5'-[β-thio]diphosphate trilithium salt] (ADPβS), N-ethylmaleimide (NEM) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Sigma-Aldrich (St. Louis, MO) and MRS2365 from Tocris Bioscience (Bristol, UK). 1400W, PTIO, L-NAME and Propylamine Propylamine NONOate (PAPA NONOate) were purchased from Cayman Chemical (Ann Arbor, MI). The Cx43 hemichannel mimetic peptide 43Gap26 and panx1 mimetic peptide 10panx were purchased from Anaspec (Fremont, CA).
Solutions
Live tissue was maintained in Dulbecco's Modified Eagle Medium: Nutrient Mixture F12 (DMEM/F12; Life Technologies) containing 3 μM nicardipine and 1 μM scopolamine during microdissection and incubations. LMMPs were perfused with modified Krebs buffer containing (in mM): 121 NaCl, 5.9 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 10 HEPES, 21.2 NaHCO3, 1 pyruvic acid and 8 glucose (pH adjusted to 7.4 with NaOH) with 3 μM nicardipine and 1 μM scopolamine.
Data and statistical analysis
Average fluorescence intensity was quantified by measuring average fluorescence using the in-program measure function in ImageJ software, version 1.49 (NIH). Neuron packing density was determined by counting the number of HuC/D-immunoreactive neurons per ganglionic area in ten ganglia per LMMP preparation using the cell counter plug-in tool in ImageJ. All results are presented as mean ± standard error of the mean (SEM) and statistical differences determined using an ANOVA or t-test as appropriate with a P value of <0.05 considered significant (GraphPad Prism). For Ca2+ and NO imaging, traces represent the average change in fluorescence (ΔF/F) over time for all glial cells within a single ganglion.
All authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
Purinergic activation of enteric glial cells drives glial Ca2+ responses and enteric neuron death
Enteric neuron death during intestinal inflammation requires the activation of neuronal P2X7Rs and neuronal ATP release through panx1 channels9. Neuronal ATP released via panx1 is rapidly hydrolysed to ADP by eNTPDase2 and ADP recruits Ca2+ responses in the surrounding enteric glial cells following stimulation of P2Y1Rs (Fig. 1A)9. In agreement with our previous work in mice, we find that activation of P2Y1Rs with the agonist ADP stimulates Ca2+ responses in enteric glial cells within the myenteric plexus of the human jejunum (Fig. 1B). Likewise, activation of neuronal P2X7Rs with the agonist BzATP in the human myenteric plexus recruits Ca2+ responses in the surrounding enteric glial cells (Fig. 1B). Previous reports suggest that P2Y1Rs are expressed by enteric neurons in the guinea pig ENS18 and by enteric glia in the mouse ENS9,19. Our data support the conclusion that P2Y1Rs are primarily localized to enteric glia in the mouse myenteric plexus because P2Y1R agonists including ADP (100 μM), the non-hydrolysable ADP analog ADPβS (100 μM) or MRS2365 (1 μM) primarily drive cellular activity in enteric glial cells and not neurons (Fig. 1C–D, data not shown for ADP and MRS2365). Likewise, immunohistochemistry with specific antibodies against an extracellular loop of the P2Y1R shows that P2Y1Rs are primarily localized to enteric glial cell processes in the myenteric plexus (Fig. 1E).
Figure 1.

Activation of enteric glial P2Y1Rs drives neuron death through a Cx43-dependent mechanism. (A) Schematic depicting how activation of neuronal P2X7Rs elicits Ca2+ responses in enteric glia. (B) Representative traces of in situ Ca2+ imaging of human myenteric glia. As in mice, human glia respond to the P2Y1R agonist, ADP and to the neuronal release of ATP stimulated by the P2X7R agonist, BzATP. Gray traces show responses of individual glial cells within a ganglion and the averaged response of all glia within the ganglion is overlaid in green. Traces are representative of recordings from at least 4 myenteric ganglia from the human jejunum. (C–D) In situ Ca2+ imaging of the mouse myenteric plexus demonstrates that P2Y1R agonists primarily elicit Ca2+ responses in enteric glia. (C) Representative images of Fluo-4 fluorescence in a myenteric ganglion from the mouse colon at rest (baseline) and at peak stimulation (at time = 60seconds) with the non-hydrolysable P2Y1R agonist, ADPβS. Arrows point to representative enteric glia. (D) Averaged Ca2+ responses of glia (green line) and neurons (magenta dashed line) within a myenteric ganglion from the mouse colon in response to ADPβS. Traces are representative of responses in over 5 myenteric ganglia. (E) Representative mouse myenteric ganglion showing immunoreactivity for P2Y1Rs (magenta). Enteric glia are labeled with the glial cell marker glial fibrillary acidic protein (GFAP; green) and arrowheads highlight areas of co-localization (scale bar, 30 μM). The boxed region in the overlay image at top right is expanded in the bottom right panel to highlight a glial cell with dense P2Y1R expression. (F) Mean packing density of HuC/D-immunoreactive neurons in myenteric ganglia after in situ activation of P2Y1Rs with the agonists ADP, ADPβS and MRS2365 or activation of P2X7Rs with BzATP in the presence or absence of the Cx43 inhibitor 43Gap26 (n = 3-5 animals, **P≤0.01, ***P≤0.005 ANOVA). (G) Mean packing density of live enteric neurons in myenteric ganglia following in situ neuroinflammation with BzATP, ADP or buffer control, quantified by Calcein-AM fluorescence. Representative images show viable cells, labeled with fluorescent Calcien-AM (green), in mouse myenteric ganglia from control and BzATP-treated tissues. Dead (non-fluorescent) neurons are denoted by asterisks (scale bar, 30 μM), (n = 3-4 animals).
We tested how direct activation of glial cells with P2Y1R agonists affects neuron survival in isolated preparations of the mouse ENS. Activation of glial P2Y1Rs with ADP, ADPβS or MRS2365 decreased the ganglionic density of HuC/D-immunoreactive neurons by 24 ± 4%, 23 ± 4% and 19 ± 4%, respectively (Fig. 1F). Importantly, P2Y1R activation drove enteric neuron death to an equal extent as activation of neuronal P2X7Rs with BzATP (300 μM) (23 ± 2%, Fig. 1F). To confirm that the loss of HuC/D-immunoreactivity truly reflects a loss of neurons and not merely a loss of HuC/D-immunoreactivity in neurons, we assessed neuronal viability using the fluorescent dye Calcien-AM which labels live, membrane-intact cells14. Treatment with BzATP (300 μM) and ADP (100 μM) decreased neuronal density in myenteric ganglia by ~50% compared to buffer control (53 ± 4% and 52 ± 3% respectively, Fig. 1G), consistent with results determined by HuC/D-immunoreactivity. These results show that the activation of glial P2Y1Rs is sufficient to induce enteric neuron death driven through P2X7R activation.
In situ glial-driven neuron death requires Cx43 hemichannels
The activation of glial P2Y1Rs drives Ca2+ responses and triggers mechanisms that lead to Cx43 hemichannel opening in mice4. Astroglial Cx43 hemichannel opening in the context of neuroinflammation releases mediators that contribute to the development of neuropathic pain and neurodegeneration in the central nervous system (CNS)20,21. Thus, we tested if glial-driven enteric neuron death requires Cx43 hemichannel opening by activating glial P2Y1Rs in the presence of the specific Cx43 hemichannel mimetic peptide, 43Gap26 (100 μM) which inhibits Cx43 hemichannel opening22. The neurotoxic effects of both P2Y1R agonists (ADP and ADPβS) and P2X7R agonist (BzATP) were lost in the presence of 43Gap26, which protected against decreased ganglionic neuron density (92 ± 2%, 89 ± 3% and 108 ± 8% for ADP, ADPβS and BzATP with 43Gap26 respectively, compared to buffer control, Fig. 1F). These results strongly suggest that glial cells are responsible for driving neuron death because Cx43 hemichannels are confined to enteric glial cells within the ENS4.
Glial Cx43 hemichannels are required for in vivo neuron death during inflammation
To more accurately test the role of glial Cx43 hemichannels in inflammatory neuropathy, we combined in vivo DNBS-colitis with a targeted gene deletion approach to conditionally ablate Cx43 in GFAP-expressing glial cells following the administration of tamoxifen4 (inducible and conditional Cx43 knockout; Cx43i-cKO; Fig. 2). In agreement with our previous work, the density of HuC/D-immunoreactive myenteric neurons was reduced by 24% (2057 ± 68 vs. 1565 ± 139 neurons mm−2) at the peak of DNBS colitis in control mice (littermates treated with tamoxifen but lacking cre recombinase in glia, Cre−; Fig. 2A). However, mice with a conditional ablation of Cx43 hemichannels in glia (Cre+) were resistant to the neurotoxic effects of in vivo inflammation (2057 ± 68 neurons mm−2 in Cre−/Saline controls vs. 1850 ± 117 neurons mm−2 in Cre+/DNBS, Fig. 2A). This effect is not likely due to an alteration in the inflammation driven by DNBS because Cx43i-cKO (Cre+) mice exhibited the same pattern of weight loss and macroscopic damage scores as littermate controls (Fig. 2B–C). Further, the ablation of Cx43 in glia did not affect normal neuron packing density in healthy animals (2057 ± 68 neurons mm−2 in Cre−/Saline animals vs. 2149 ± 101 neurons mm−2 in Cre+/Saline; Fig. 2A). Collectively, these data show that the expression of glial Cx43 hemichannels is a requirement for inflammatory neuropathy in the ENS.
Figure 2.
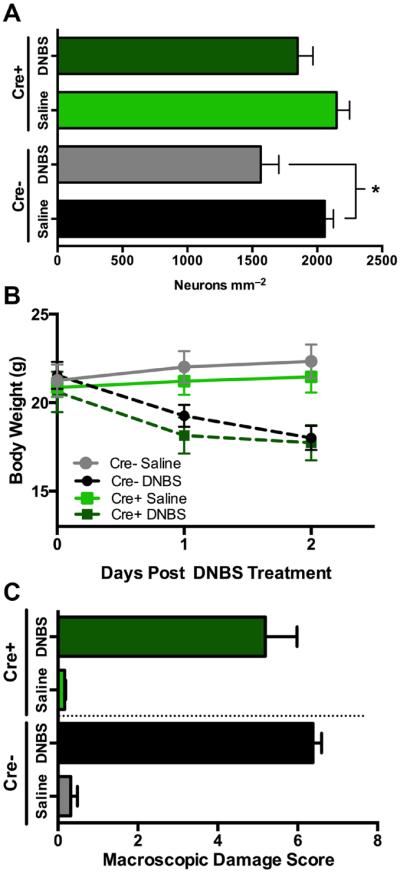
Genetic ablation of glial Cx43 limits neuropathy without modifying overt inflammation. (A) Mean packing density of myenteric neurons following DNBS-colitis in transgenic mice with an inducible ablation of Cx43 in glial cells (Cx43i-cKO, Cre+) and their littermate controls (Cre−). Weight loss pattern and macroscopic damage for experimental groups shown in (B) and (C), respectively (n = 5–7 animals, *P≤0.05, ANOVA).
Stimulation of enteric glial P2Y1Rs elicits Cx43-dependent ATP release
One possible mechanistic explanation for glial-driven neuron death is that glial Cx43 hemichannel opening modulates P2X7R activation threshold by augmenting levels of extracellular ATP. In support, astroglial Cx43 hemichannels are highly permeable to ATP23,24 and neurotoxic activation of P2X7Rs requires a conformational change that only high concentrations of ATP are capable of inducing by occupying all four ATP binding sites25. We tested if purinergic activation of enteric glia drives Cx43-dependent ATP release by stimulating glial P2Y1Rs while monitoring extracellular ATP release with ATP-sensitive microelectrodes15. In these experiments, we either directly stimulated glial P2Y1Rs with the non-hydrolysable agonist ADPβS or indirectly generated endogenous ADP by activating neuronal P2X7R-dependent ATP release with the agonist BzATP. We found that stimulating glial P2Y1Rs with ADPβS elicits robust ATP release from enteric glia (Fig. 3A–B). P2Y1R-driven glial ATP release was completely dependent upon Cx43 because ATP release was absent in the presence of the Cx43 mimetic peptide, 43Gap26 and in tissue from Cx43i-cKO (Cre+) mice (Fig. 3A–B). Likewise, stimulation of P2X7R-dependent neuron-glia communication generated high levels of extracellular ATP (Fig. 3C–D). Interestingly, panx1-dependent ATP release from enteric neurons accounted for only approximately 1/3 of total ATP release while Cx43-dependent release from glia accounted for the majority of ATP release. These results indicate that enteric glia have the potential to release large quantities of ATP through Cx43 hemichannels following stimulation of P2Y1Rs.
Figure 3.

Stimulation of enteric glial P2Y1Rs elicits Cx43-dependent ATP release. (A,C) Representative traces and (B,D) quantified measurements of ATP release from the mouse myenteric plexus obtained with ATP-selective electrodes. Glial P2Y1Rs were directly stimulated with the P2Y1R agonist, ADPβS (A,B; 100 μM), or by eliciting neuron-to-glia communication with the neuronal P2X7R agonist, BzATP (C,D; 300 μM), in the presence or absence of the Cx43 mimetic peptide, 43gap26 (100 μM), the pannexin-1 mimetic peptide, 10Panx (100 μM), or in tissue from Cx43i-cKO mice following the selective ablation of glial Cx43 (Cre+ KO or Cre− littermate controls). n = 3 animals.
Oxidative stress coincides with neuron death during in vivo inflammation and is required for P2X7R-driven neuron death in situ
Next, we asked whether the neurotoxic opening of glial Cx43 channels requires potentiation of channel function by other factors associated with inflammation. We initially focused on oxidative stressors because Cx43 hemichannel opening is potentiated by oxidative stress26 and oxidative stress is considered a key mechanism in the pathogenesis of gut inflammation1,27,28. In support, we observed high levels of oxidative stress in myenteric ganglia during DNBS-colitis (Fig. 4A,C). Measures of oxidative stress included neuronal thiol oxidation ratios (ratio of oxidized/reduced neuronal glutathione) (Fig. 4A) and ganglionic concentrations of superoxide measured by fluorescence of the superoxide-specific fluorescent indicator dihydroethidium (DHE) (Fig. 4C). We confirmed that our measures truly reflected oxidative stress by administration of the antioxidant N-acetyl cysteine (NAC; 5 g/L) in vivo prior to and during the induction of DNBS-colitis (Fig. 4A). Treatment with NAC prevented increased oxidative stress during DNBS-colitis (Fig. 4A) without altering the acute inflammatory insult, as there was no change in macroscopic damage score with NAC treatment (Fig. 4B). Importantly, we investigated the mechanistic significance of oxidative stress and found that treatment with NAC prevented P2X7R-driven neuron death in situ (Fig. 4D–E). Because our results above show that glial pathways mediate P2X7R-driven neuron death, these results indicate that oxidative stress contributes to the activation of neurotoxic glial mechanisms.
Figure 4.

Oxidative stress coincides with neuron death during in vivo inflammation and is required for P2X7R-driven neuron death in situ. (A) Thiol oxidation measurements (ratio of fluorescently labeled oxidized/reduced glutathione) in myenteric neurons from healthy (saline) or inflamed (DNBS-colitis) animals drinking normal water (H2O) or water containing the antioxidant N-Acetyl Cysteine (NAC; 5 g/L) (n = 5 animals, **** P < 0.001, ANOVA). (B) Macroscopic damage score for animals treated in (A). (C) Ganglionic fluorescence of the superoxide indicator dihydroethidium (DHE, 2 μM) in the myenteric plexus of healthy (saline) or inflamed (DNBS-colitis) mice (n = 5–6 animals, * P < 0.05, unpaired t-test). (D) Representative myenteric ganglia from in situ preparations treated with BzATP in the presence or absence of the antioxidant NAC. Neurons are labeled with the neuronal marker HuC/D, with dead neurons denoted by asterisks (scale bar = 30 μM). (E) Mean packing density of HuC/D-immunoreactive neurons in myenteric ganglia following in situ activation of P2X7Rs with BzATP (300 μM) in the presence of NAC (n = 4 animals, * P < 0.05, ANOVA).
Enteric glia contribute to oxidative stress via local nitric oxide (NO) production during inflammation
Glial cells can directly contribute to the local generation of oxidative stressors by up-regulating the activity of iNOS29,30; an enzyme that produces large amounts of NO during inflammation31. We assessed the glial contribution to local NO levels within enteric ganglia from healthy and inflamed animals by measuring cellular NO concentrations in both neurons and glial cells with the NO-sensitive fluorescent dye DAF-FM (Fig. 5A). DAF-FM fluorescence reliably reflected intracellular NO concentrations as treatment with the NO donor PAPA NONOate elevated fluorescence while treatment with the pan-NOS inhibitor L-NAME decreased fluorescence (Fig. 5A). NO content was equally distributed between neurons and glia in non-inflamed animals and this distribution was not altered up to 6hours following DNBS-colitis (Fig. 5B–C). However, we observed an elevation of glial NO content during the peak of the inflammatory response 48hours after initiation of DNBS-colitis (Fig. 5B–C). Further, increases in immunoreactivity for nitrated proteins, another measure of NO concentrations, correlated with changes in glial NO content post DNBS-colitis treatment (Fig. 5D). Nitrated protein immunoreactivity primarily co-localized with GFAP immunoreactivity within myenteric ganglia (Fig. 5E), suggesting that glial cells are the main sites of nitration and other NO-mediated modifications in the myenteric plexus during inflammation. Importantly, the kinetics of glial NO production and protein nitration coincide with the appearance of neuron death during colitis in mice9 and guinea pigs32.
Figure 5.

Enteric glia contribute to oxidative stress by producing nitric oxide (NO) during inflammation. (A–C) In situ NO imaging with the NO sensitive dye, DAF-FM. (A) Representative traces of mean glial NO responses increasing following treatment with the NO donor PAPA NONOate (solid green line, 100 μM) and decreasing in the presence pan-nitric oxide synthase (NOS) inhibitor L-NAME (dashed black line, 100 μM). (B) Representative images of DAF-FM fluorescence in myenteric ganglia from healthy (saline) or inflamed (DNBS-colitis) mice. Arrows point to representative glial cells and representative neurons (or the lack thereof) are denoted by asterisks (scale bar, 30 μM). (C) Quantification of DAF-FM fluorescence in observable myenteric neurons and glia in the healthy (saline) or inflamed (DNBS) colon at 6hours (left) and 48hours (right) after the initiation of DNBS-colitis (n = 5–10 animals, *P < 0.05, t-test compared to glia-saline). (D) Percentage of nitrotyrosine immunoreactive (nT +ve) ganglia in the myenteric plexus of saline DNBS-treated animals at 6hours (left) and 48hours (right) after the initiation of DNBS-colitis (n = 3-5 animals, * P < 0.05, unpaired t-test). (E) Representative myenteric ganglion showing immunoreactivity for nitrated proteins (nTYR; magenta). Enteric glia are labeled with the glial cell marker GFAP (green) and yellow arrowheads highlight areas of co-localization (scale bar, 20 μM). The boxed region in the overlay image is shown at a higher magnification to highlight immunoreactivity of nitrated proteins on glial processes.
NO potentiates glial ATP release and promotes enteric neuron death through a Cx43-dependent mechanism
Given that nitrosylation of Cx43 hemichannels is associated with increased Cx43 channel opening in astrocytes26, we hypothesized that NO would potentiate the Cx43-dependent release of ATP driven by glial P2Y1R activation. In support, we found that the Cx43-dependent release of ATP from glia stimulated by the P2Y1R agonist ADPβS was potentiated in the presence of the NO donor PAPA NONOate (Fig. 6A). This suggests that NO production during intestinal inflammation can contribute to the activation of the neurotoxic Cx43 pathway in enteric glia.
Figure 6.

Nitric oxide (NO) potentiates ATP release from glia and promotes neuron death in situ through a mechanism that involves glial Cx43 hemichannels. (A) ATP release from myenteric ganglia following stimulation of glial P2Y1Rs with ADPβS (100 μM) alone or in the presence of the NO donor PAPA NONOate (n = 4, * P < 0.05, unpaired t-test). (B) Mean packing density of HuC/D-immunoreactive myenteric neurons following direct glial P2Y1R stimulation with ADP and inhibition of iNOS (1400W; 10 μM) or P2X7Rs (A740003; 10 μM). Inhibition of iNOS or P2X7Rs protects against P2Y1R-driven neuron death (*P≤0.01, ANOVA as compared to ADP, n = 3-4 animals). (C) Mean packing density of myenteric neurons following application of BzATP or NO-modifying drugs in wild type (bottom bars) or iNOS-knockout mice (iNOS−/−, top two green bars). Inhibition (1400W; 10 μM) or ablation (iNOS−/−) of iNOS protects against P2X7R-driven neuron death. The NO donor, PAPA NONOate (100 μM), drives neuron death to an equal extent as BzATP but the combination is not additive. Like BzATP, PAPA NONOate–driven neuron death requires iNOS (blocked by 1400W and in iNOS−/− mice) and Cx43 hemichannel opening (blocked by 43Gap26).
*P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001, ANOVA as compared to BzATP, n = 3–11 animals.
In situ, we observed an equal extent of neuron death in whole-mounts of myenteric plexus incubated with the NO donor PAPA NONOate as in preparations exposed to the neuronal P2X7R agonist BzATP (24 ± 5% vs. 21 ± 4%, Fig. 6C), further supporting a pathogenic role for NO in the context of neuron death. Importantly, the neurotoxic effects of PAPA NONOate and BzATP were not additive. This finding suggests that NO and P2X7R agonists drive neuron death through a common mechanism. Indeed, BzATP and PAPA NONOate-driven neuron death were both entirely dependent upon Cx43 hemichannel opening because the neurotoxic effects of these compounds were abolished in the presence of 43Gap26. Likewise, neurons were protected against BzATP-induced death in tissue from iNOS null (iNOS−/−) mice and by blocking NO production from iNOS with the inhibitor, 1400W (Fig. 6C). Collectively, these data show that the production of NO by iNOS during inflammation is essential for the activation of neurotoxic pathways mediated through glial Cx43 hemichannels and strongly suggest that NO contributes to neuropathy by potentiating glial ATP release via Cx43 hemichannels.
Mechanisms of neuron death downstream of glial P2Y1R activation involve glial iNOS and neuronal P2X7Rs
To determine the sequence of signaling events downstream of glial P2Y1R activation that lead to neuron death, we activated glial P2Y1Rs in situ with the specific agonist ADP (100 μM) in the presence of drugs to inhibit either iNOS or P2X7Rs. The inhibition of glial iNOS with 1400W33 completely abolished the neurotoxic effect of glial P2Y1R activation (118 ± 13% vs. 72 ± 7%; Fig. 6B). Likewise, the inhibition of neuronal P2X7Rs with A74000334 protected against neuron death driven by glial P2Y1R activation (90 ± 5%, vs. 72 ± 7%; Fig. 6B). Taken together, these results suggest a cyclical signaling mechanism where neuronal P2X7R-panx1-mediated ATP release activates glial P2Y1Rs and intracellular signaling mechanisms that lead to glial NO production and the potentiation of ATP release from glial Cx43 hemichannels which causes neuron death through actions on neuronal P2X7Rs.
NO positively modulates glial Cx43 hemichannels and negatively modulates neuronal panx1 hemichannels
NO could directly modulate a number of receptors and channels involved in the P2X7R-panx1 neuron death pathway including glial Cx43 hemichannels and/or neuronal panx1 hemichannels and P2X7Rs35. To determine whether the neurotoxic effect of NO we observe is due to the potentiation of neuronal (P2X7R-panx1) or glial (Cx43) signaling mechanisms, we measured glial activity induced indirectly by the activation of neuronal P2X7R-panx1 or directly via the activation of glial P2Y1Rs in the presence of the NO donor PAPA NONOate (100 μM). We previously demonstrated that enteric glia can be used as endogenous “sniffer cells” to measure the activity of the neuronal P2X7R-panx1 pathway9 and that glial network activity in response to the P2Y1R agonist ADP reflects glial Cx43 activity4. Our results show that glial Ca2+ responses downstream of neuronal P2X7R-panx1 activation are significantly blunted in the presence of PAPA NONOate (72% decrease in peak ΔF/F vs. control, Fig. 7A–B). Glial Ca2+ responses downstream of neuronal P2X7R activation are entirely dependent on the neuronal release of ATP through panx19 and blunted glial responses suggest that NO negatively regulates P2X7R-panx1-dependent neuron-to-glia communication. This result is in agreement with other studies showing that NO inhibits panx1 channel activity35,36. An alternate explanation for this result is that NO decreased the ability of glia to respond to neuronal activation. We tested this possibility by directly activating glial cells with ADP. Instead of decreasing glial responsiveness, we find that NO significantly potentiates glial Ca2+ responses to ADP (35% increase in peak ΔF/F vs. control, Fig. 7C–D). This outcome suggests that glial Cx43 hemichannel opening is facilitated by NO because Ca2+ responses through the enteric glial network are mediated by Cx434. Our other data support this conclusion by showing that NO potentiates glial Cx43-dependent ATP release (Fig. 6A). Together, these results strongly support the conclusion that the sensitization of glial release mechanisms, rather than neuronal signaling components, is the primary cause of neuron death.
Figure 7.

NO positively modulates glial Cx43 hemichannels and negatively modulates neuronal panx1 hemichannels. (A–B) Representative Ca2+ imaging traces (A) and averaged peak ΔF/F responses (B) show that NO (NO donor PAPA NONOate) blunts glial Ca2+ responses downstream of neuronal P2X7R-panx1 stimulation with BzATP (300 μM). (C–D) Representative Ca2+ imaging traces (C) and averaged peak ΔF/F responses (D) of glial Ca2+ responses during direct glial P2Y1R activation with ADP (100 μM) in the presence or absence of the NO donor PAPA NONOate. Note that NO potentiates glial network responses driven by the direct agonist ADP. *P<0.05, ****P<0.001, T-test compared to control, n= 51–139 individual cells in 3–7 ganglia.
DISCUSSION
Our observations provide the first evidence that enteric glial cells play an active role in the death of enteric neurons during gut inflammation. Specifically, our data show that mediators of inflammation such as NO potentiate the gating of glial Cx43 hemichannels and subsequently, neuron death. Based on these data, we propose a model where neuronal P2X7-panx1-mediated purine release during inflammation or neuron stress, drives glial activation leading to pathogenic Cx43-dependent ATP release by glial cells and enteric neuron death by activation of neuronal P2X7Rs (Fig. 8).
Figure 8.

Proposed model of the mechanisms involved in glial cell-driven enteric neuron death during acute inflammation. The activation of neuronal P2X7Rs leads to ATP release through panx1 channels that recruits activity in the surrounding glia by activating glial P2Y1Rs. Intracellular signaling pathways downstream of P2Y1R activation drive glial iNOS production and glial NO potentiates Cx43 hemichannel-dependent ATP release. Glial NO may also feed back on neurons to block panx1 channel activity. Large quantities of ATP are released through glial Cx43 and act on neuronal P2X7Rs to drive neuron death.
Enteric glial cells are generally thought to support the function and survival of enteric neurons and models of glial ablation support this conclusion as glial ablation produces a rapid loss of enteric neurons5,6. Thus, a loss of the supportive roles of glial cells is postulated as a potential mechanism for the development of ENS dysfunction in inflammatory bowel disease2,5. In contrast, newer data show that the chronic activation of astroglia in the context of neuroinflammation leads to the release of neurotoxic mediators from glial cells7. Our results support a similar scenario in the ENS during inflammation where a gain, rather than a loss, of glial cell function leads to the death of enteric neurons. These findings highlight the dichotomous roles of enteric glial cells during health and disease and suggest that either a loss of protective glial function or gain of pathological glial functions can drive ENS dysfunction but presumably through very different mechanisms.
Physiologically, enteric glial cells possess neuroprotective properties and are integral in maintaining enteric neuron populations. Enteric glia secrete neuroprotective compounds such as reduced glutathione and the prostaglandin derivative 15-deoxy-PGJ23,37. Further, intraganglionic enteric glia express a number of receptors and enzymes that can respond to and degrade neuroactive compounds, thus preventing aberrant neuronal activation38,39. Finally, glial cells can directly participate in neuron signaling by responding to and releasing neurotransmitters as previously shown for Cx43 hemichannels in hippocampal astrocytes in the CNS40. In support of a functional role for Cx43 hemichannels in the enteric nervous system, we observed that ablation of Cx43 in enteric glial cells blunts glial network activity and disrupts the neuronal regulation of gastrointestinal transit under physiological conditions neuronal regulations of gastrointestinal transit under physiological conditions4.
Here, we find that glial Cx43 hemichannel opening is required for neuron death during inflammation and that the selective ablation of glial Cx43 is neuroprotective in gut pathology. We postulate that these observations reflect the necessity for Cx43 hemichannels in both physiological and pathophysiological functions of enteric glia, where the pathological potential of glial Cx43 is evident when potentiated by inflammatory mediators such as NO. Indeed, here, NO potentiated Cx43-mediated ATP release from enteric glia, driving neuron death. Although inhibition of Cx43 was neuroprotective, the therapeutic potential of Cx43 inhibition may be limited given the essential role of glial Cx43 in normal gut function. However, selective inhibition of pathological channel opening by NO has the potential to disrupt pathology without affecting physiological functions. Thus, drugs such as GW274150 that display excellent iNOS selectivity and little toxicity41 may represent important new therapeutics in the treatment of functional gastrointestinal disorders.
Cx43 hemichannels are subject to post-translational modifications including S-nitrosylation26 by increased oxidant concentrations. Here, we show an increase in tyrosine nitration, another NO-mediated post-translational modification, that correlates with increased glial NO following in vivo inflammation. Nitrotyrosine immunoreactivity is primarily localized to enteric glial cells, suggesting that glial proteins are susceptible to NO-mediated post-translation modifications. This supports our hypothesis that glial Cx43 is a candidate of post-translational nitrosylation as a result of increased NO production during in vivo inflammation. We observed glial NO production during in vivo inflammation using a fluorescent NO marker during the initiation (6hours) and peak (48hours) of inflammation. Although we did not observe a significant increase in glial NO content at 6hours after initiating inflammation, this could be due to the high variability between the inflammatory stages of individual ganglia at this early stage. However, our nitration data show a similar timeline of nitration in glia so it is likely that glial NO production occurs in response to active inflammation and is not necessarily responsible for the initiation of inflammation.
One alternate interpretation of our findings is that NO production by glia and oxidative stress in neurons directly affects the P2X7Rs and panx1 hemichannels on enteric neurons; leading to neuron death independent of glial mechanisms9. Indeed, NO modulates the activity of hemichannels including those composed of panx135,42,43 and Cx4326,44. However, we do not believe that our data support this interpretation because of several reasons. Firstly, we show that NO significantly blunts glial activity initiated by the neuronal release of ATP through P2X7R-panx1. In contrast, NO potentiated Cx43-dependent intercellular communication between enteric glia in response to direct P2Y1R stimulation. Likewise, the neurotoxic effect of the NO donor was completely abolished by the Cx43 mimetic peptide or the genetic ablation of glial iNOS. Further, our data show that glial cell release of ATP through Cx43 is the primary contributor to extracellular ATP levels following stimulation of either neuronal P2X7Rs or glial P2Y1Rs while neuronal panx1 opening contributes only minor amounts of ATP. Finally, the selective ablation of glial Cx43 alone was able to protect enteric neurons during inflammation in vivo. We interpret these findings as indicating that the potentiation of neurotoxic mechanisms is primarily occurring in glial cells although we recognize that a combination of effects on neuronal and glial pathways could be required for neuron death.
Our in vivo model of Cx43 ablation utilizes cre/lox technology and requires expression of cre recombinase, driven by the GFAP promoter, in response to tamoxifen administration. Although enteric glial cells are a heterogeneous population, the majority of glia in the myenteric plexus are GFAP positive45 making this a suitable promoter for our study. Importantly, our model is dependent upon transcription of cre from the GFAP promoter and thus variability in GFAP protein expression with inflammation and time would not likely have a major effect on cre activity and Cx43 excision45. In our experimental paradigm, inflammation was induced after the ablation of Cx43 was induced. However, enteric glia are subject to gliogenesis in response to injury and inflammation46. Thus, any newly generated glia following inflammation would not have been exposed to tamoxifen and would express functional Cx43 hemichannels. Notably, the timeline for gliogenesis46 is substantially longer than that of our current inflammatory model32 so we do not believe that glial turnover was a major confounding factor in these experiments.
GFAP is also expressed by astrocytes in the CNS47,48 and tamoxifen treatment in our mouse model induces the ablation of Cx43 hemichannels in enteric glia and CNS astrocytes. Disruption of astrocytic Cx43 gap junction and hemichannel function results in CNS dysfunction and this may have exerted confounding effects in our disease model. However, we do not observe unusual weight loss or gut inflammation in these animals in the absence of inflammatory stimuli and we observed comparable weight loss and inflammation to wild-type animals during inflammation (Fig. 2). Thus, dysfunction in the brain-gut axis does not seem to be a significant contributor to disease outcome in our in vivo model.
Purinergic activation of enteric glial cells is a central component of our proposed signaling mechanism (Fig. 8). We show that glial purinergic responses are mediated through P2Y1Rs that are primarily localized to enteric glial cells in the mouse myenteric plexus. This supports previous work demonstrating glial P2Y1R expression and activity in cultured glial cells19 and in whole mount preparations in mouse and guinea pig9,49,50. Previous work suggests P2Y1R expression on enteric neurons18,51. However, we demonstrate that activation with the P2Y1R agonist ADP elicits Ca2+ responses in glial cells, a phenomenon observed in the CNS notwithstanding P2Y1R expression on both neurons and astrocytes52. Given the present data, it is tempting to speculate that the P2Y1R-mediated slow excitatory postsynaptic potentials recorded in enteric neurons51 are actually downstream of glial activation and this will be an interesting hypothesis to test in future experiments.
In conclusion, our findings have uncovered a novel role for enteric glia in the pathogenesis of enteric neuropathies. Enteric neuropathy is increasingly recognized as a key pathological finding in functional gastrointestinal disorders including irritable bowel syndrome53 and slow transit constipation54. Inflammation is thought to produce persistent gut dysfunction through effects on the ENS including enteric neuron death and the functional remodeling of enteric circuitry. Indeed, our prior results show that an acute inflammatory event triggers enteric neuron death and leads to gut motor dysfunction that persists despite resolution of active inflammation9. Our current observations suggest that the activation of enteric glial cells is necessary for at least a portion of these permanent effects on the ENS. Thus, new therapies that modulate the pathophysiological functions of enteric glial cells could lead to the development of more effective treatments for functional bowel disorders.
SYNOPSIS.
Death of enteric neurons contributes to motility dysfunction in gastrointestinal disorders. Our work provides the first evidence of glial activation as a driver of enteric neurodegeneration.
Acknowledgments
Grant Support: This project was supported by grants from the Pharmaceutical Researchers and Manufacturers Association of America Foundation (PhRMA Foundation, to B. Gulbransen), the Crohn's and Colitis Foundation of America (CCFA, to B. Gulbransen), Biotechnology and Biological Sciences Research Council (BBSRC BB/I025409/1, to B. Patel) and the National Institutes of Health (NIH; HD065879 and R01DK103723, to B. Gulbransen).
Abbreviations
- Ca2+
calcium
- Cx43
connexin-43
- ENS
enteric nervous system
- panx1
pannexin-1
- P2X7R
P2X7 receptor
- P2Y1R
P2Y1 receptor
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- ATP
adenosine triphosphate
- ADP
adenosine diphosphate
- LMMP
longitudinal muscle myenteric plexus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: No conflicts of interest exist.
Author Contributions: BDG was responsible for the overall project conception, design and supervision. Experiments performed by IAB, JLM, BDG and BAP. Manuscript was written by IAB and BDG with all authors contributing to editing.
REFERENCES
- 1.De Giorgio R, Guerrini S, Barbara G, et al. Inflammatory neuropathies of the enteric nervous system. Gastroenterology. 2004;126(7):1872–1883. doi: 10.1053/j.gastro.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 2.Neunlist M, Rolli-Derkinderen M, Latorre R, et al. Enteric glial cells: recent developments and future directions. Gastroenterology. 2014;147(6):1230–1237. doi: 10.1053/j.gastro.2014.09.040. [DOI] [PubMed] [Google Scholar]
- 3.Abdo H, Derkinderen P, Gomes P, et al. Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J. 2010;24(4):1082–1094. doi: 10.1096/fj.09-139519. [DOI] [PubMed] [Google Scholar]
- 4.McClain JL, Grubišić V, Fried D, et al. Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology. 2014;146(2):497–507. e1. doi: 10.1053/j.gastro.2013.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush TG, Savidge TC, Freeman TC, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93(2):189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- 6.Cornet A, Savidge TC, Cabarrocas J, et al. Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn's disease? Proc Natl Acad Sci USA. 2001;98(23):13306–13311. doi: 10.1073/pnas.231474098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayo L, Trauger SA, Blain M, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med. 2014;20(10):1147–1156. doi: 10.1038/nm.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bi F, Huang C, Tong J, et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci USA. 2013;110(10):4069–4074. doi: 10.1073/pnas.1218497110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gulbransen BD, Bashashati M, Hirota SA, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18(4):600–604. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke H, Krügel U, Schmidt R, Grosche J, Reichenbach A, Illes P. P2 receptor-types involved in astrogliosis in vivo. Br J Pharmacol. 2001;134(6):1180–1189. doi: 10.1038/sj.bjp.0704353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacMicking JD, Nathan C, Hom G, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81(4):641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 12.Mullett SJ, Di Maio R, Greenamyre JT, Hinkle DA. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J Mol Neurosci. 2013;49(3):507–511. doi: 10.1007/s12031-012-9904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic. Biol. Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- 14.Palma PFR, Baggio GL, Spada C, Silva RDA, Ferreira SIACP, Treitinger A. Evaluation of annexin V and Calcein-AM as markers of mononuclear cell apoptosis during human immunodeficiency virus infection. Braz J Infect Dis. 2008;12(2):108–114. doi: 10.1590/s1413-86702008000200003. [DOI] [PubMed] [Google Scholar]
- 15.Patel BA, Rogers M, Wieder T, O'Hare D, Boutelle MG. ATP microelectrode biosensor for stable long-term in vitro monitoring from gastrointestinal tissue. Biosens Bioelectron. 2011;26(6):2890–2896. doi: 10.1016/j.bios.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 16.Patel BA. Mucosal adenosine triphosphate mediates serotonin release from ileal but not colonic guinea pig enterochromaffin cells. Neurogastroenterol Motil. 2014;26(2):237–246. doi: 10.1111/nmo.12254. doi:10.1111/nmo.12254. [DOI] [PubMed] [Google Scholar]
- 17.Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm Bowel Dis. 2009;15(11):1678–1685. doi: 10.1002/ibd.20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wood JD, Liu S, Drossman DA, Ringel Y, Whitehead WE. Anti-enteric neuronal antibodies and the irritable bowel syndrome. J Neurogastroenterol Motil. 2012;18(1):78–85. doi: 10.5056/jnm.2012.18.1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomes P, Chevalier J, Boesmans W, et al. ATP-dependent paracrine communication between enteric neurons and glia in a primary cell culture derived from embryonic mice. Neurogastroenterol Motil. 2009;21(8):870–e62. doi: 10.1111/j.1365-2982.2009.01302.x. [DOI] [PubMed] [Google Scholar]
- 20.Chen MJ, Kress B, Han X, et al. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia. 2012;60(11):1660–1670. doi: 10.1002/glia.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Froger N, Orellana JA, Calvo C-F, et al. Inhibition of cytokine-induced connexin43 hemichannel activity in astrocytes is neuroprotective. Mol Cell Neurosci. 2010;45(1):37–46. doi: 10.1016/j.mcn.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Boitano S, Evans WH. Connexin mimetic peptides reversibly inhibit Ca(2+) signaling through gap junctions in airway cells. Am J Physiol Lung Cell Mol Physiol. 2000;279(4):L623–L630. doi: 10.1152/ajplung.2000.279.4.L623. [DOI] [PubMed] [Google Scholar]
- 23.Kang J, Kang N, Lovatt D, et al. Connexin 43 hemichannels are permeable to ATP. J Neurosci. 2008;28(18):4702–4711. doi: 10.1523/JNEUROSCI.5048-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19(2):520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan Z, Khadra A, Li S, Tomic M, Sherman A, Stojilkovic SS. Experimental Characterization and Mathematical Modeling of P2X7 Receptor Channel Gating. J Neurosci. 2010;30(42):14213–14224. doi: 10.1523/JNEUROSCI.2390-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Retamal MA, Cortés CJ, Reuss L, Bennett MVL, Sáez JC. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: induction by oxidant stress and reversal by reducing agents. Proc Natl Acad Sci USA. 2006;103(12):4475–4480. doi: 10.1073/pnas.0511118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med (Maywood) 2012;237(5):474–480. doi: 10.1258/ebm.2011.011358. [DOI] [PubMed] [Google Scholar]
- 28.Roberts JA, Lukewich MK, Sharkey KA, Furness JB, Mawe GM, Lomax AE. The roles of purinergic signaling during gastrointestinal inflammation. Curr Opin Pharmacol. 2012;12(6):659–666. doi: 10.1016/j.coph.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green CL, Ho W, Sharkey KA, McKay DM. Dextran sodium sulfate-induced colitis reveals nicotinic modulation of ion transport via iNOS-derived NO. Am J Physiol Gastrointest Liver Physiol. 2004;287(3):G706–G714. doi: 10.1152/ajpgi.00076.2004. [DOI] [PubMed] [Google Scholar]
- 30.Turco F, Sarnelli G, Cirillo C, et al. Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut. 2014;63(1):105–115. doi: 10.1136/gutjnl-2012-302090. [DOI] [PubMed] [Google Scholar]
- 31.Zhang N, Weber A, Li B, et al. An inducible nitric oxide synthase-luciferase reporter system for in vivo testing of anti-inflammatory compounds in transgenic mice. J Immunol. 2003;170(12):6307–6319. doi: 10.4049/jimmunol.170.12.6307. [DOI] [PubMed] [Google Scholar]
- 32.Linden DR, Couvrette JM, Ciolino A, et al. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol Motil. 2005;17(5):751–760. doi: 10.1111/j.1365-2982.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- 33.Garvey EP, Oplinger JA, Furfine ES, et al. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. 1997;272(8):4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 34.Honore P, Donnelly-Roberts D, Namovic MT, et al. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J Pharmacol Exp Ther. 2006;319(3):1376–1385. doi: 10.1124/jpet.106.111559. [DOI] [PubMed] [Google Scholar]
- 35.Lohman AW, Weaver JL, Billaud M, et al. S-nitrosylation inhibits pannexin 1 channel function. J Biol Chem. 2012;287(47):39602–39612. doi: 10.1074/jbc.M112.397976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poornima V, Vallabhaneni S, Mukhopadhyay M, Bera AK. Nitric oxide inhibits the pannexin 1 channel through a cGMP-PKG dependent pathway. Nitric Oxide. 2015;47:77–84. doi: 10.1016/j.niox.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Abdo H, Mahé MM, Derkinderen P, Bach-Ngohou K, Neunlist M, Lardeux B. The omega-6 fatty acid derivative 15-deoxy-Δ12,14-prostaglandin J2 is involved in neuroprotection by enteric glial cells against oxidative stress. J Physiol. 2012;590(Pt 11):2739–2750. doi: 10.1113/jphysiol.2011.222935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braun N, Sévigny J, Robson SC, Hammer K, Hanani M, Zimmermann H. Association of the ecto-ATPase NTPDase2 with glial cells of the peripheral nervous system. Glia. 2004;45(2):124–132. doi: 10.1002/glia.10309. [DOI] [PubMed] [Google Scholar]
- 39.Lavoie EG, Gulbransen BD, Martín-Satué M, Aliagas E, Sharkey KA, Sévigny J. Ectonucleotidases in the digestive system: focus on NTPDase3 localization. Am J Physiol Gastrointest Liver Physiol. 2011;300(4):G608–G620. doi: 10.1152/ajpgi.00207.2010. [DOI] [PubMed] [Google Scholar]
- 40.Chever O, Lee CY, Rouach N. Astroglial Connexin43 Hemichannels Tune Basal Excitatory Synaptic Transmission. J Neurosci. 2014;34(34):11228–11232. doi: 10.1523/JNEUROSCI.0015-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. 2012;2012:318087. doi: 10.1155/2012/318087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brunse S, Locovei S, Schmidt M, et al. The potassium channel subunit Kvbeta3 interacts with pannexin 1 and attenuates its sensitivity to changes in redox potentials. FEBS J. 2009;276(21):6258–6270. doi: 10.1111/j.1742-4658.2009.07334.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhang L, Deng T, Sun T, Liu K, Yang Y, Zheng X. Role for nitric oxide in permeability of hippocampal neuronal hemichannels during oxygen glucose deprivation. J Neurosci Res. 2008;86(10):2281–2291. doi: 10.1002/jnr.21675. [DOI] [PubMed] [Google Scholar]
- 44.Retamal MA, Froger N, Palacios-Prado N, et al. Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci. 2007;27(50):13781–13792. doi: 10.1523/JNEUROSCI.2042-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boesmans W, Lasrado R, Vanden Berghe P, Pachnis V. Heterogeneity and phenotypic plasticity of glial cells in the mammalian enteric nervous system. Glia. 2015;63(2):229–241. doi: 10.1002/glia.22746. [DOI] [PubMed] [Google Scholar]
- 46.Joseph NM, He S, Quintana E, Kim Y-G, Núñez G, Morrison SJ. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest. 2011;121(9):3398–3411. doi: 10.1172/JCI58186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25(9–10):1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 48.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gulbransen BD, Sharkey KA. Purinergic neuron-to-glia signaling in the enteric nervous system. Gastroenterology. 2009;136(4):1349–1358. doi: 10.1053/j.gastro.2008.12.058. [DOI] [PubMed] [Google Scholar]
- 50.Gulbransen BD, Bains JS, Sharkey KA. Enteric glia are targets of the sympathetic innervation of the myenteric plexus in the guinea pig distal colon. J Neurosci. 2010;30(19):6801–6809. doi: 10.1523/JNEUROSCI.0603-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu HZ, Gao N, zhu MX, et al. Slow excitatory synaptic transmission mediated by P2Y1 receptors in the guinea-pig enteric nervous system. J Physiol. 2003;550(Pt 2):493–504. doi: 10.1113/jphysiol.2003.041731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fam SR, Gallagher CJ, Salter MW. P2Y(1) purinoceptor-mediated Ca(2+) signaling and Ca(2+) wave propagation in dorsal spinal cord astrocytes. J Neurosci. 2000;20(8):2800–2808. doi: 10.1523/JNEUROSCI.20-08-02800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindberg G, Törnblom H, Iwarzon M, Nyberg B, Martin JE, Veress B. Full-thickness biopsy findings in chronic intestinal pseudo-obstruction and enteric dysmotility. Gut. 2009;58(8):1084–1090. doi: 10.1136/gut.2008.148296. [DOI] [PubMed] [Google Scholar]
- 54.Bassotti G, Villanacci V, Cathomas G, et al. Enteric neuropathology of the terminal ileum in patients with intractable slow-transit constipation. Hum Pathol. 2006;37(10):1252–1258. doi: 10.1016/j.humpath.2006.04.027. [DOI] [PubMed] [Google Scholar]