

Abstract
Attempts to reproduce eight, putative, enantioselective dibromination and chlorohydroxylation reactions from oftcited literature studies are described. The reactions were performed with full fidelity to the original report wherever possible. Analysis of the enantiomeric composition was performed by chiral stationary phase HPLC or SFC (CSP-HPLC or CSP-SFC), as opposed to the original report, which used chiral shift reagent NMR spectroscopy. After careful study, the reported levels of enantioselectivity were found to be incorrect. Possible explanations for the false positive results are discussed.
Graphical abstract

The halofunctionalization of alkenes with electrophilic halogenating agents has long been a staple of stereoselective synthesis, prized for its predictable constitutional selectivity and relative stereochemical course.1 However, catalytic dihalogenation methods, in which the absolute configuration of the products is controlled from an achiral starting material, have proved elusive until recently.2 Enantioselective bromination is of particular interest due to the ability of bromide to act as a leaving group in stereospecific substitution reactions. Additionally, over 3500 organobromine compounds are known in nature, the majority isolated from marine organisms.3
Despite the extensive history of dihalogenation and other halofunctionalization reactions of alkenes, methods to form stereodefined dihalogenated products from olefins are rare. In the reactions of olefins with Br2, vicinal dibromides are formed via bromide ion attack on either an alkene-Br2 π-complex or a bromiranium ion, resulting in anti-stereospecific addition of Br2 across the double bond.4 However, strategies for enantioselectivity may be thwarted in a number of ways: (1) A racemic background reaction may occur from the formation of molecular bromine (or its equivalent) when both electrophilic and nucleophilic bromine sources are present; (2) facial selectivity must also be controlled to produce an enantioenriched alkene-Br2 π-complex or bromiranium ion intermediate; (3) bromiranium ions are configurationally unstable in the presence of excess olefin, via an alkene-to-alkene transfer pathway;5 and (4) the regioselectivity of bromide addition must also be controlled, since attack on either carbon atom of a non-C2-symmetric bromiranium ion intermediate yields enantiomeric products.
In 2011, Nicolaou et al. reported a dichlorination reaction of allylic alcohols using (DHQ)2PHAL as the catalyst and 4-Ph(C6H4)ICl2 as the chlorinating agent.6 Dichloride products were formed in a wide range of selectivities (50:50 to 90.5:9.5 er). More recently, Burns et al. developed an enantioselective dibromination of allylic alcohols using dibromomalonate as an electrophilic bromine source and BrTi(Oi-Pr)3 as a nucleophilic bromine source, with a TADDOL-derived catalyst.7 The dibromide products were formed with good enantioselectivities (85.5:14.5–92.5:7.5 er). This work has been extended to enantioselective bromochlorination with constitutional site selectivities of 6:1–>20:1 and enantioselectivities of 89:11–98.5:1.5 er.8
The Wacker oxidation has also been modified for halofunctionalization of alkenes. The use of high chloride ion concentrations under Wacker-type conditions results in the conversion of ethylene to ethylene chlorohydrin, instead of the expected acetaldehyde product.9 An enantioselective variant of this reaction was subsequently reported by Henry and coworkers, using chiral, non-racemic palladium(II) bisphosphine catalysts 3a to achieve the enantioselective generation of chlorohydrins 2 from terminal alkenes 1.10 The possibility of a facile background reaction stemming from dihalogen formation was reduced by the use of chloride as the sole halogen source. A limited number of chlorohydrins were produced, with constitutional site selectivities of 5.5:1–>95:1, and reported enantioselectivities of 64:36–91:9 er (Scheme 1a). In all cases, yields were reported based solely on O2 uptake.
Scheme 1.
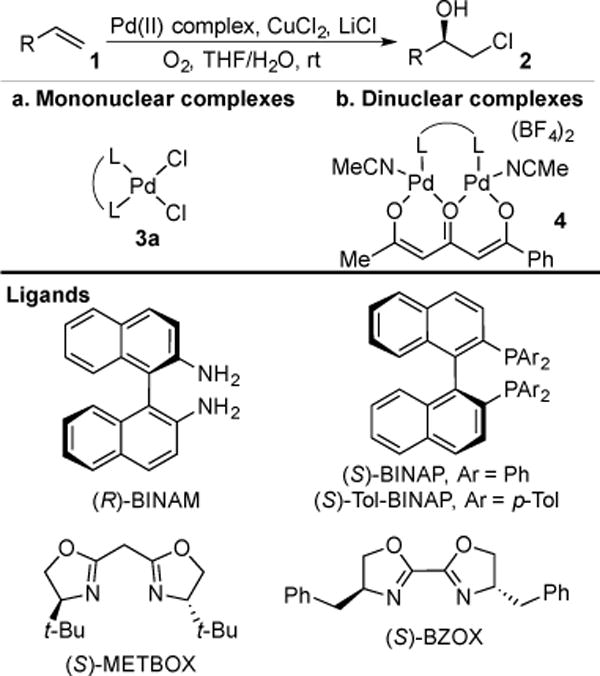
Henry’s Wacker-Type Chlorohydrin Synthesis
Enantioselectivity was improved in later work by Henry and coworkers, by the use of dinuclear palladium(II) complexes 4 with bridging triketone ligands, although no direct comparisons to the mononuclear catalysts were made.11 Terminal olefins 1 were oxidized to chlorohydrins 2 with constitutional site selectivities of 2.3:1–>95:1 and enantioselectivities of 57.5:42.5–97:3 er (Scheme 1b).
Henry and coworkers later reported an extension of this work to the enantioselective dibromination of olefins, using similar Wacker-type conditions with 2.5–17.7 mol % (per Pd atom) palladium(II) catalyst 3b or 4 (Scheme 2).12 The reactions were performed in aqueous THF solutions of varying composition, containing copper(II) bromide (~2 M) and lithium bromide (0.13–0.29 M). The concentration of alkene substrate 5 varied between 0.06 and 0.47 M. Since these reaction conditions (i.e. concentrations of LiBr, CuBr2, catalyst and alkene, and solvent THF/H2O ratio) varied so considerably among substrates, direct comparisons are of limited value. The higher intrinsic nucleophilicity of bromide ion compared to H2O, coupled with the high bromide ion concentrations employed led to the formation of vicinal dibromides 6 rather than bromohydrins as the major products. Although the yields of dibromides reported were highly variable (31–95%),13 good to excellent enantioselectivities were claimed (90:10–98.5:1.5 er), with the exception of methyl (E)-cinnamate (57:43 er). Enantiomeric ratios were determined by 1H NMR spectroscopic analysis using Eu(hfc)3 (europium tris[3-(heptafluoropropyl-hydroxymethylene)-d-camphorate), although neither spectra nor details of concentrations were given. Optical rotation data were provided for only two of the nine, dibromide products.
Scheme 2.
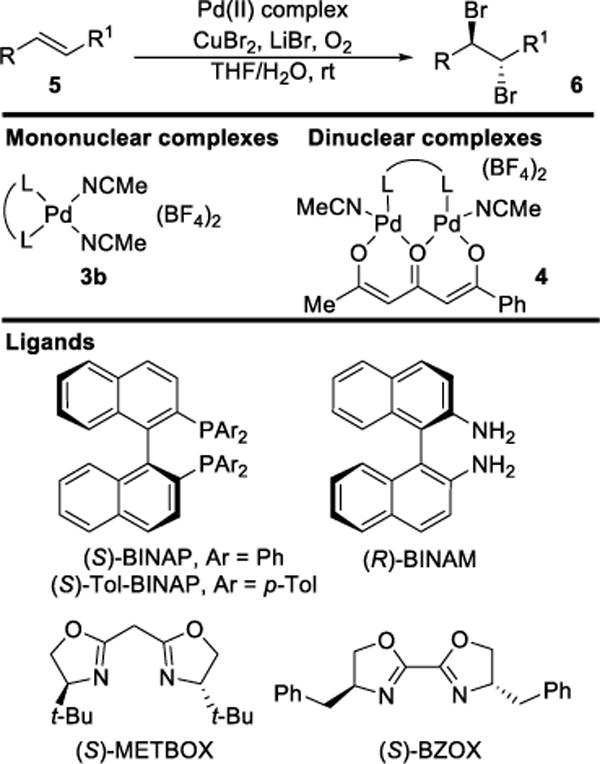
Henry’s Asymmetric Dibromination of Alkenes
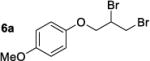
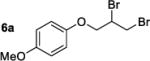
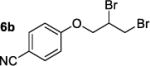
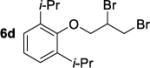
We have attempted to replicate four reactions in the original report from Henry and coworkers (Table 1, Entries 1–7). The allylic ether dibromination procedures were repeated as rigorously as the described procedures allow, albeit on a smaller scale (0.25 mmol versus 2.8–3.7 mmol allylic ether in the original work). To vouchsafe that scale was not a critical factor, one trial, Table 1, Entry 3, was performed on the original 3.0 mmol scale and was allowed to run for the original reaction time of 6 days. Although dibromides 6 revert back to the corresponding allylic ethers 5a–d over several weeks in light, running reactions and isolating products in the dark affected neither the yields nor enantioenrichment of products. Workup and chromatographic isolation of products was performed immediately and identically to the original protocol so as to obviate any possibility of epimerization over time.
Table 1.
Comparison of Results for the Dibromination and Chlorohydroxylation of Several Substrates
| Run | Ligand | Complex Type | Complex Nuclearity | Time, d | Product | Yield, % | er | Lit. yield, % | Lit. er |
|---|---|---|---|---|---|---|---|---|---|
| 1 | (R)-BINAPa,b | 4 | 2c | 1.17 |
|
86d | 50:50 | 95 | 98:2 |
| 2 | (R)-BINAPa,e | 4 | 2c | 1.17 |
|
86d | 50:50 | 95 | 98:2 |
| 3 | (R)-BINAPa | 3 | 1c | 6 |
|
78f | 50:50 | 95 | 98:2 |
| 4 | (R)-BINAPa | 3 | 1c | 0.25 |
|
65d | 50:50 | – | – |
| 5 | (R)-Tol-BINAPa | 4 | 2 | 1 |
|
85d | 50:50 | 95 | 98.5:1.5 |
| 6 | (R)-BINAPa,e | 4 | 2 | 1 |
|
69d | 50:50 | 95 | 97.5:2.5 |
| 7 | (R)-BINAPa | 3 | 1 | 1 |
|
90d | 50:50 | 95 | 97:3 |
| 8 | (R)-Tol-BINAP | 3 | 1 | 14 |
|
14 | 50:50 | NR | 90:10 |
| 9 | (R)-BINAPa,b | 4 | 2 | 5 |
|
40 | 50:50 | 92 | 96.5:3.5 |
| 10 | (R)-Tol-BINAP | 3 | 1 | 10 |
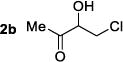
|
33g | 50:50 | NR | 91:9 |
| 11 | (R)-BINAPa,b | 4 | 2 | 10 |
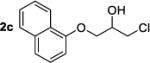
|
29 | 50:50 | 95h | 90:10 |
NR = not reported.
The (S)-enantiomer of the ligands was used in the original work.
The dinuclear catalyst was prepared according to Henry’s procedure.
The nuclearity of the catalyst used is ambiguous in the original text.
The yield is the average of two runs, each within 2% of the average.
The dinuclear catalyst was prepared according to a modification of Henry’s procedure.
3.0 mmol scale.
1.0 mmol scale.
The product was mischaracterized in the original work.
The catalyst used in Table 1, Entry 1 was produced by replication of Henry’s described method with as complete fidelity as possible. This procedure afforded a mixture of compounds, as determined by 31P NMR spectroscopy. The dinuclear palladium complexes used in Table 1, Entries 2, 5 and 6 were synthesized by a modification of Henry’s method, in which NaH rather than Et3N was used as a Brønsted base.11b This procedure afforded compounds that appeared to be pure by 31P NMR spectroscopy, and circumvented a laborious separation of triethylammonium tetrafluoroborate from the complex. The pure complexes were stored in a moisture- and oxygen-free environment, and were found to decompose over time in solution, and under vacuum, with decomposition observable by NMR spectroscopy after just a few minutes under vacuum. The complexes were therefore characterized and used immediately after purification. The decomposition product (readily visible as a multiplet exhibiting P-P coupling by 31P NMR spectroscopy) could be removed by extensive washing of the solid with anhydrous, degassed toluene under an argon atmosphere. Nonetheless, the method of preparation of the dinuclear complex had no impact on the reaction outcome (Table 1, entries 1–2). For operational simplicity, the mononuclear Pd(II) bisphosphine complexes 3 used in entries 3–4 were generated in situ from Pd(MeCN)4(BF4)2 and the requisite chiral bisphosphine.14 The mononuclear catalyst used in Table 1, Entry 7 was prepared under Ar and found to be stable in the absence of air and moisture.15 The reactions were run under positive pressure from an O2 manifold.
Moderate to high yields of dibrominated products 6 were obtained, with small amounts of side products observable in the 1H NMR spectra. Both the crude product mixtures and the chromatographically pure dibromides were analyzed by CSP-HPLC or CSP-SFC to assess enantioenrichment. In every case examined, the vicinal dibromides were racemic (Table 1). It is notable that in our hands, the complete conversion of starting material was achieved in significantly shorter times than those quoted in the original work: 24 or 28 h versus 4–7 days. It is possible that the original reactions were monitored by O2 uptake whereas our experiments were monitored by TLC. A repeat of the dibromination of 5a was arbitrarily stopped after 6 h (Table 1, Entry 4) to determine whether an initial enantioselective process may occur, only to be compromised by epimerization under the reaction conditions. In this case as well the product was racemic.
In the light of these results, four chlorohydroxylation reactions from Henry and coworkers’ earlier reports were also investigated (Table 1, Entries 8–11).10 These experiments were performed on a 0.5–1.0 mmol-scale (original scale: 3.7–6.5 mmol where reported). However, analysis by CSP-SFC and chiral stationary phase gas chromatography (CSP-GC) again revealed that all chlorohydrin products were racemic.
In their original report, Henry et al. propose a mechanism that bypasses a bromiranium ion intermediate (Scheme 3), instead suggesting that the palladium complex (shown as mononuclear with a chiral ligand abbreviated as L) coordinates olefin 5. Free bromide then attacks the activated olefin I from the opposite face, yielding an alkylpalladium(II) complex II. Cu(II) may stereoretentively oxidize the C–Pd(II) bond to form the alkyl bromide without overall oxidation state change at Pd.16 It is unknown whether this bromide is derived from the coordination sphere of Pd or that of Cu in complex II.
Scheme 3.
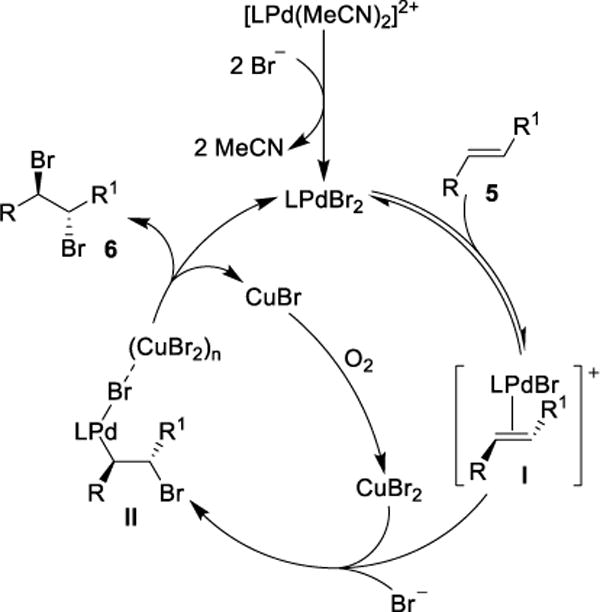
Catalytic Cycle for Dibromide Formation
Henry et al. claim the rate does not decrease during the course of the reaction, i.e. overall zeroth order.12 Qaseer later reported a similar system using Wacker-type conditions and a racemic dinuclear Pd(II) catalyst 4 to generate vicinal dibromides, and reported a zeroth order rate of O2 uptake.17 However, measurement of O2 uptake is insufficient to substantiate claims related to the rate of product formation. For example, the yield for the dibromination of methyl cinnamate (catalyzed by a mononuclear palladium(II) bisoxazoline complex) is reported as 80% (Table S1, Entry 6) based on O2 uptake. However, the amount of recovered starting material is 30% by mass.12 This inconsistency exemplifies that O2 uptake is not a reliable method for monitoring product formation.
The ability to reproduce the formation of the dibromides and chlorohydrins from multiple substrates following the original procedure, but to obtain uniformly racemic products, presents a quandary. The possibility that important details are missing for preparing the catalysts or executing the reactions cannot be excluded. Certainly many possibilities exist for generating Br2 under the reaction conditions and of course, CuBr2 itself is also capable of effecting the dibromination of alkenes at room temperature.18 However, in our opinion, the more likely explanation for the disparity is the authors’ use of chiral shift reagent NMR analysis to determine enantiomeric composition of the dibromide products. Unfortunately, the spectral data are not provided. Our own attempt to observe signal separation in a racemic sample of 1-(2,3-dibromopropoxy)-4-methoxybenzene 5a using Eu(hfc)3 was inconclusive. After portion-wise addition of four equivalents of the chiral shift reagent, some degree of signal separation was observed, showing roughly equal quantities of each enantiomer. However, the level of signal broadening precluded any quantitative determination of enantiomeric ratios.
In conclusion, four dibromination and four chlorohydroxylation reactions of allylic ethers catalyzed by chiral mono- or dinuclear palladium(II) complexes reported by Henry and coworkers were repeated. Although the reaction yields were reproduced, the dibromide and chlorohydrin products were generated in racemic form.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (R01 GM085235) for generous financial support.
Footnotes
Supporting Information Available. Full experimental procedures, analyses, characterization data, and NMR data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chung W-J, Vanderwal CD. Acc Chem Res. 2014;47:718–728. doi: 10.1021/ar400246w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tan CK, Yeung Y-Y. Chem Commun. 2013;49:7985–7996. doi: 10.1039/c3cc43950j. [DOI] [PubMed] [Google Scholar]; (c) Ranganathan S, Muraleedharan KM, Vaish NK, Jayaraman N. Tetrahedron. 2004;60:5273–5308. [Google Scholar]
- 2.(a) Hennecke U, Wilking M. Synlett. 2014;25:1633–1637. [Google Scholar]; (b) Cheng YA, Yu WZ, Yeung Y-Y. Org Biomol Chem. 2014;12:2333–2343. doi: 10.1039/c3ob42335b. [DOI] [PubMed] [Google Scholar]; (c) Chen J, Zhou L. Synthesis. 2014;46:586–595. [Google Scholar]; (d) Denmark SE, Kuester WE, Burk MT. Angew Chem Int Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hennecke U. Chem Asian J. 2012;7:456–465. doi: 10.1002/asia.201100856. [DOI] [PubMed] [Google Scholar]; (f) Snyder SA, Brucks AP. In: Asymmetric Synthesis II: More Methods and Applications. Christmann M, Brässe S, editors. Wiley; Weinheim: 2013. pp. 147–156. [Google Scholar]; (g) Castellanos A, Fletcher SP. Chem Eur J. 2011;17:5766–5776. doi: 10.1002/chem.201100105. [DOI] [PubMed] [Google Scholar]; (h) Snyder SA, Treitler DS, Brucks AP. Aldrichimica Acta. 2011;44:35–48. [Google Scholar]
- 3.(a) Dictionary of Natural Products. dnp.chemnetbase.com (accessed Aug 1, 2015); (b) Gribble G. Chemosphere. 2003;52:289–297. doi: 10.1016/S0045-6535(03)00207-8. [DOI] [PubMed] [Google Scholar]; (c) Gribble GW. Progress in the Chemistry of Organic Natural Products. Vol. 68. Springer; Vienna: 1996. pp. 1–498. [PubMed] [Google Scholar]
- 4.(a) Lenoir D, Chiappe C. Chem Eur J. 2003;9:1036–1044. doi: 10.1002/chem.200390097. [DOI] [PubMed] [Google Scholar]; (b) Brown RS. Acc Chem Res. 1997;30:131–137. [Google Scholar]; (c) Ruasse MF. Adv Phys Org Chem. 1993;28:207–291. [Google Scholar]; (d) Schmid GH. In: The Chemistry of Double-Bonded Functional Groups. Patai S, editor. Wiley; New York: 1989. pp. 679–731. [Google Scholar]
- 5.Denmark SE, Burk MT, Hoover AJ. J Am Chem Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]
- 6.Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. J Am Chem Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu DX, Shibuya GM, Burns NZ. J Am Chem Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]
- 8.Hu DX, Seidl FJ, Bucher C, Burns NZ. J Am Chem Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]
- 9.Stangl H, Jira R. Tetrahedron Lett. 1970;11:3589–3592. [Google Scholar]
- 10.Hamed O, Henry PM. Organometallics. 1998;17:5184–5189. [Google Scholar]
- 11.(a) El-Qisairi A, Qaseer HA, Henry PM. J Organomet Chem. 2002;656:168–176. [Google Scholar]; (b) El-Qisairi A, Henry PM. J Organomet Chem. 2000;603:50–60. [Google Scholar]; (c) El-Qisairi A, Hamed O, Henry PM. J Org Chem. 1998;63:2790–2791. doi: 10.1021/jo005627e. [DOI] [PubMed] [Google Scholar]
- 12.El-Qisairi AK, Qaseer HA, Katsigras G, Lorenzi P, Trivedi U, Tracz S, Hartman A, Miller JA, Henry PM. Org Lett. 2003;5:439–441. doi: 10.1021/ol0273093. [DOI] [PubMed] [Google Scholar]
- 13.The true yield of methyl crotonate dibromination is indecipherable. Henry et al. report a yield of 80% based on oxygen uptake, yet 30% starting material was also recovered.
- 14.Hatano M, Mikami K. J Am Chem Soc. 2003;125:4704–4705. doi: 10.1021/ja0292748. [DOI] [PubMed] [Google Scholar]
- 15.Nesper R, Pregosin PS, Püntener K, Wörle M. Helv Chim Acta. 1993;76:2239–2249. [Google Scholar]
- 16.(a) Hamed O, Henry PM. Organometallics. 1998;17:5184–5189. [Google Scholar]; (b) Henry PM. J Org Chem. 1974;39:3871–3874. [Google Scholar]; It is possible that a transient Pd(IV) species may be formed during this step. Pd(IV) is competent for dichlorination of alkenes, see:; (c) McCall AS, Wang H, Desper JM, Kraft S. J Am Chem Soc. 2011;133:1832–1848. doi: 10.1021/ja107342b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qaseer HA. Pol J Chem. 2007;81:31–38. [Google Scholar]
- 18.(a) Rodebaugh R, Debenham JS, Fraser-Reid B, Snyder JP. J Org Chem. 1999;64:1758–1761. doi: 10.1021/jo9718509. [DOI] [PubMed] [Google Scholar]; (b) Baird WC, Surridge JH, Buza M. J Org Chem. 1971;36:3324–3330. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.