

Abstract
Reading frame maintenance is a critical property of ribosomes. However, a number of genetic elements have been described that can induce ribosomes to shift on mRNAs, the most well understood of which are a class that directs ribosomal slippage by one base in 5′ (-1) direction. This is referred to as programmed -1 ribosomal frameshifting (-1 PRF). Recently, a new -1 PRF promoting element was serendipitously discovered in a study examining the effects of stretches of adenosines in the coding sequences of mRNAs. Here, we discuss this finding, recent studies describing how -1 PRF is used to control gene expression in eukaryotes, and how -1 PRF is itself regulated. The implications of dysregulation of -1 PRF on human health are examined, as are possible new areas in which novel -1 PRF promoting elements might be discovered.
Keywords: Ribosome, translation, frameshifting, polyA track, pseudoknot, NMD, telomere, ribosomopathy, cancer, SCA26, miRNA
Introduction
We are living in a golden age of scientific discovery, from the detection of exoplanets and the near certainty of extraterrestrial life, to the visualization of atomic scale molecular machines in action. While new findings tend to be consistent with generally accepted theories, they also reveal interesting exceptions to the general rules. In so doing, they both further illuminate these rules, and help to reveal the deeper mysteries of the natural world. In this essay we discuss how the discovery of cis-acting mRNA elements that subvert normal mRNA decoding is adding a new dimension to our understanding of how cellular gene expression is regulated.
A quick tutorial on protein synthesis
mRNAs are decoded by ribosomes as groups of three contiguous nucleotides (codons) on messenger RNAs (mRNAs). Each codon specifies either an amino acid or, in three cases, instructions to stop protein synthesis. Ribosomes are programmed to identify the right place to start (initiation, usually at an AUG codon encoding methionine in eukaryotes and archaea and formyl-methionine in prokaryotes), and then proceed to decode the genetic information in the mRNA by marching (elongation) down the mRNA in the 5′ to 3′ direction 1 codon at a time until they reach one of the stop codons (termination). Central to this is that ribosomes must maintain the reading frame as defined by the initiation codon, in order to properly decode the information contained in the mRNA. Translational reading frame maintenance is an under-appreciated area of research as compared to a large body of literature on e.g. mechanisms underlying tRNA charging by aminoacyl-tRNA synthetases, or the accurate decoding of codons [1]. From this, it follows that bad things must happen when ribosomes fail to maintain reading frame. And indeed, this is true…with some exceptions.
PolyA tracts and mRNA decay
To explore these exceptions, we begin with a recent paper by Arthur and co-workers [2] describing why consecutive runs of A residues rarely occur in the protein coding regions of mRNAs, and their surprising elucidation of how such polyA tracts are used to regulate gene expression. Prior studies in nucleated (eukaryotic) cells had shown that if ribosomes bypass a normal stop codon they becomes stalled on the mRNA’s polyA tail. This is presumably due to the fact that the AAA codon encodes the basic amino acid lysine, and the demonstrated ability of poly-basic polypeptides such as poly-lysine promote ribosome stalling [3, 4], presumably through their interactions with the negatively charged ribosomal exit tunnel [5]. The stalled ribosomes are recognized by a complex of proteins that remove the ribosome for recycling [6]. During this process, the complex also recruits an endonuclease which cleaves the mRNA and initiates its degradation. This is called “Non-Stop” Decay (NSD) because the failure of ribosomes to stop where they are supposed to results in destruction of the mRNA (reviewed in [7]). While on the subject of mRNA decay, two additional mechanisms merit discussion. A second is initiated when ribosome stalling occurs in the protein coding sequence, typically due to strong mRNA structures that are difficult to unwind. Here, the same (or similar) machinery is recruited to save the ribosome and kill the message: this is called “No-Go” Decay (NGD) [8, 9]. Thirdly, if a ribosome encounters a stop codon in the wrong context, e.g. far away from a polyA tail (called a Premature Termination Codon, or PTC), mRNA degradation proceeds through an independent process called the Nonsense-Mediated mRNA Decay pathway (NMD) (recently reviewed in [10]). Note that although the ribosome is at fault in these cases, it is the messenger that pays the price.
Arthur and colleagues mined sequence data to find that runs of polybasic amino acids are universally underrepresented in the protein coding regions of mRNAs. This engendered the hypothesis that these might be used as regulatory elements by acting as “translational attenuators” akin to the NSD process. Surprisingly however, only runs of polyA, but not repeated AAG codons (which also encodes poly-lysine) or repeated CGA or AGG (encoding poly-arginine), conferred strong translational attenuation effects. Thus, something other than basic amino acid mediated ribosome stalling had to be operating.
Slip sliding away
Thirty years ago, the HIV/AIDS epidemic was dominating the headlines and the virus was just beginning to be characterized. Elucidation of retroviral genomic sequences by many different groups revealed a novel feature: overlapping and mutually out of frame open reading frames. This soon led to the discovery of special “slippery” sequences able to program elongating ribosomes to slip from one reading frame to another in a process that is called programmed -1 ribosomal frameshifting (-1 PRF) [11]. In the intervening years, we and others have characterized the nature of these -1 PRF signals, demonstrating the importance of -1 PRF on virus propagation by ensuring synthesis of the correct stoichiometries of viral proteins [12–14]. More recently, it has been recognized that ribosomal frameshifting and other forms of translational recoding are widely used in all three domains of life [15–18]. Here, we focus on -1 PRF in eukaryotes, where computational searches for “classic” -1 PRF signals suggested that up to 10% of genes may be regulated by this mechanism [19]. Counterintuitively, unlike viruses in which -1 PRF is used to synthesize two (or more) proteins from a single mRNA (Figure 1A), more than 99% of -1 PRF events were predicted to direct elongating ribosomes to PTCs (Figure 1B). Follow up studies revealed that these ‘genomic’ -1 PRF signals function as mRNA destabilizing elements through NMD from yeast to humans [20–22]. Thus, while -1 PRF generally serves to expand the genomic coding content of viruses, in eukaryotes it appears to be primarily employed as a post-transcriptional regulatory mechanism. However, while classic -1 PRF signals require slippery stretches of nucleotides, slippage is greatly stimulated by strong downstream RNA structural elements that induce elongating ribosomes to pause at the slippery sequences. Interestingly, a strong RNA secondary structural element can also promote significant rates of -1 PRF even in absence of an upstream canonical slippery site, albeit to a lesser extent [22]. Whether the stimulatory elements actively help ribosomes to slip, or passively enhance kinetic partitioning between reading frames remains to be determined. We and others have also recently shown that -1 PRF can be regulated in a sequence-specific manner through interaction of -1 PRF signals with trans-acting nucleic acids, e.g. naturally occurring miRNAs and synthetic oligonucleotides [21, 23–25]. However, while polyA is one of the allowable “slippery” sequences within the coding region of an mRNA, the surprising finding was that it can direct efficient -1 PRF in the absence of any other stimulating element. Indeed, Arthur and colleagues demonstrated that as few as 9 A’s in a row were able to promote a significant fraction of ribosomes to shift reading frame. In the context of naturally occurring polyA sequences, these were shown to direct ribosomes to PTCs, destabilizing mRNAs through NMD, thus limiting protein expression. The ability of these short polyA sequences to promote frameshifting at rates of ~10% in the absence of a downstream stimulatory structural element is rather surprising. While not discussed by the authors, we suggest that the presence of poly-lysine in the ribosome exit tunnel may cause ribosomes to pause over the slippery polyA sequence, thus enhancing their ability to kinetically partition into the -1 frame. If so, this suggests that at least two different -1 PRF mechanisms convergently evolved as mRNA destabilizing elements to control gene expression in eukaryotic cells. Additionally, Gene Ontogeny analysis of polyA track containing messages identified by Arthur et al. reveals that approximately 12% are located in mRNAs encoding trans-acting regulatory factors involved in stress response and apoptosis. While this suggests a regulatory role for these sequences, no suggestion for how such regulation may be effected has been presented. In addition, the fact that polyA sequences do not promote slippage into the +1 frame is also interesting: while not discussed by the authors, perhaps this observation is instructive about the fundamental nature of how ribosomes naturally maintain reading frame.
Figure 1. Frameshifting on viral compared to cellular mRNAs.

A. In viruses, PRF events result in synthesis of C-terminally extended fusion proteins. In many virus families (e.g. Retroviridae, Totiviridae), rates of -1 PRF determine the stoichiometric ratios of capsid (pink Gag) to replicase (pink+blue Gag-pol) proteins. Correct ratios are critical for viral particle assembly. B. Canonical ‘genomic’ -1 PRF signals or poly(A) tracks (blue triangle) can direct an elongating ribosome to a -1 frame premature termination codon (PTC). The recognition of the PTC by the ribosome results in activation of nonsense mediated mRNA decay (NMD) pathway and subsequent degradation of the transcript through a process of decapping and deadenylation followed by exonucleolytic degradation by Xrn1p (5′ → 3′) and the Ski complex (3′ → 5′).
Regulation of Gene expression by -1 PRF
In our studies, we have validated -1 PRF signals in mRNAs encoding proteins involved in numerous cellular processes, including telomere maintenance [22] and the immune response [21]. Critically, the availability of a set of sequences that direct -1 PRF at rates ranging from 1% – 70% enabled the relationship between rates of -1 PRF and mRNA abundance to be determined:
where x = −1 PRF efficiency and mRNA abundance is a function (f) of x (Figure 2). Since this is an inverse exponential relationship, small changes in frameshifting can have large effects on gene expression. This suggests that -1 PRF is a translational attenuation mechanism that functions to balance gene dosage. Intriguingly, most -1 PRF signals promote 1% – 10% frameshifting: this lies in the linear range of the plot where changes in -1 PRF efficiencies are predicted to promote the largest changes in mRNA abundances.
Figure 2. The relationship between frameshift efficiency and mRNA abundance.
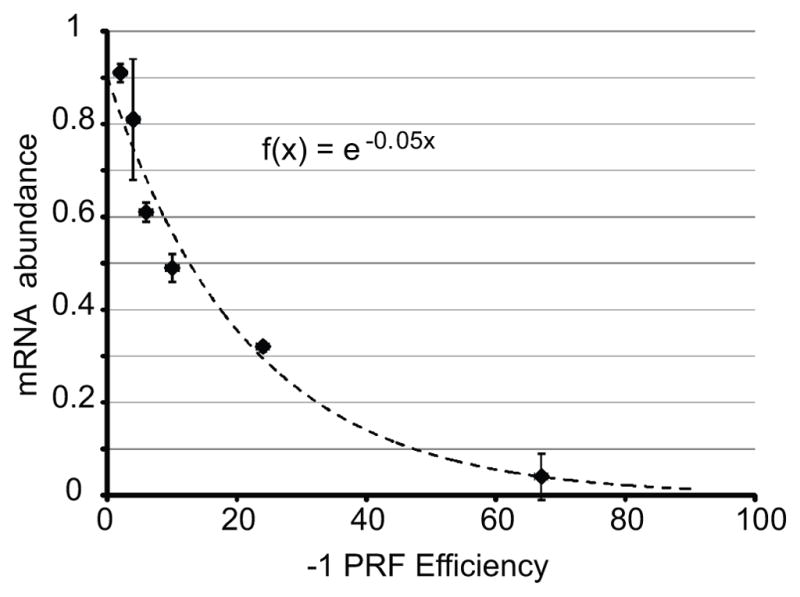
The mathematical relationship between 1 PRF efficiency and mRNA abundance was determined using frameshift signals derived from the yeast EST1, EST2, STN1 and CDC13 mRNAs. These were cloned into a reporter gene and cellular mRNA steady-state abundances were plotted relative to the abundance of the reporter without frameshift signals. Plotting of these data fit to the logarithmic function f(x) = e−0.05x (adapted from [22])
What might be the consequences of perturbing this delicate balance? In elegant studies using B. subtilis, Losick and coworkers have shown that switching between two morphological cell types is controlled not by the absolute numbers of regulatory molecules, but by their relative ratios [26]. This represents an important paradigmatic change: rather the Galilean quantitative numerical emphasis, it returns our understanding of biological regulation to the classical ratiometric Euclidian view. Curiously, included in the first report of yeast mutants in which -1 PRF was globally elevated was the observation that many of the mutants had temperature-dependent cell cycle defects [27]. Twenty years later, the basis for this was elucidated: in yeast, telomere maintenance is controlled by -1 PRF [22]. Global increases in -1 PRF imbalances the stoichiometries of key proteins that are either intrinsic to telomerase itself (Est1p and Est2p), or involved in telomerase recruitment to chromosome ends (Stn1p, Cdc13p). We hypothesize changes in the relative ratios of these proteins inhibits telomerase activity and/or recruitment to chromosome ends, triggering “checkpoint arrest” as cells work to repair this particular DNA synthesis defect before committing to cell division. This accounts for the observed cell cycle arrest phenotypes upon global dysregulation of -1 PRF. Notably, this phenomenon was also observed upon inhibition of NMD, consistent with the epistatic relationship of NMD to -1 PRF. Importantly, this places pressure on cells to select for mutations that bypass this quality control checkpoint, most commonly by “repairing” telomeres using the double stranded break repair apparatus. Unfortunately, this approach requires templating by homologous sequences, i.e. the ends of other chromosomes, which can lead to chromosome fusion, breakage during mitosis, and eventually aneuploidy. Unpublished finding from our laboratory have recently validated -1 PRF signals in some human mRNAs encoding telomere maintenance proteins, suggesting the evolutionary conservation of -1 PRF in chromosome end maintenance.
Bad Ribosomes
As noted above, we estimate that ~10% of nuclear encoded mRNAs harbor classic -1 PRF signals, and another ~2% harbor slippery polyA sequences; a respectable number of genes. Thus, it follows that defects in -1 PRF due to defective ribosomes may be significant drivers of disease. The importance of ribosomes and human health has emerged over the past decade through the emergence of a class of diseases collectively called Ribosomopathies [28]. Originally associated with genetically inherited anemias, it is becoming clear that mutations that affect ribosome biogenesis and/or ribosome function have dire consequences. Interestingly, while patients with classic ribosomopathies such as Shwachman-Bodian-Diamond syndrome and X-linked dyskeratosis congenita (X-DC) initially present with hypo-proliferative cellular disorders, e.g. anemias, should they survive this phase of the disease, they are at much higher risk of developing cancers of the same tissue bed, i.e. cellular hyper-proliferation. This seeming paradox is known in the field of hematology as Dameshek’s Riddle [29]. Using X-DC as a model, we showed that rRNA hypopseudouridylation decreases the affinity of ribosomes for at least two classes of RNA ligands, internal ribosomal entry signals (IRES) and tRNAs [30]. While this leads to decreased expression of IRES containing mRNAs (emerging as an important driver of cancer [31]), this biochemical defect also renders tRNAs more likely to slip at -1 PRF signals. The effects of X-DC associated mutations on -1 PRF and gene expression is currently a topic of intense research. Unpublished findings from our laboratory also suggests that mutations associated with other classic ribosomopathies affect various aspects of translational fidelity, including -1 PRF, and hence gene expression.
An understanding of how somatically acquired mutations that affect translational reading frame maintenance may also underlie other cancers is beginning to take shape. Exome sequencing initially revealed a conserved mutation in the gene encoding ribosomal protein L10 (also known as uL16 [32]) in ~10% of T-cell acute lymphoblastic leukemias [33], and followup studies revealed that this mutation promotes globally increased rates of -1 PRF [34, 35]. Current efforts are aimed at characterizing how this affects gene expression and drives carcinogenesis. Additionally, while ribosomopathies are currently defined as diseases caused by defective ribosomes, it is not inconceivable that mutations that affect other components of the translational apparatus may also be pathogenic. Case in point: spinocerebellar ataxia 26 (SCA26) is an autosomal dominant disease caused by a mutation in EEF2, encoding the translational translocase eukaryotic elongation factor 2 (eEF2), which promotes increased rates of -1 PRF both in yeast [36], and in patient-derived cells (unpublished). Altered rates of ribosomal slippage may not only affect gene expression by altering mRNA abundances. The increased expression of C-terminally truncated dead end polypeptides may also burden the cellular protein degradation apparatus. Indeed, yeast cells harboring the EEF2-SCA26 mutation show a more robust unfolded protein response (UPR) induction in response to antioxidant and heat shock challenges [36]. This may be of importance with regard to neurological disease in particular as Purkinje neurons are particularly vulnerable to a wide variety of molecular and cellular insults [37]. The links between -1 PRF and proteostaic insult represent a critical, underexplored area of investigation. Additionally, unlike the stringent quality control mechanisms governing ribosome biogenesis [38], no such control governing trans-acting factors like eEF2 are known. Thus, diseases associated with mutations in this class of proteins should be rare and may be expected to present as hypoproliferative only.
Sequence-specific regulation of frameshifting
If -1 PRF is widely used to control gene expression, then it stands to reason that it should be subject to regulation. However, given that global changes in -1 PRF appear to be deleterious, regulation of -1 PRF would have to be sequence-specific. microRNAs (miRNAs) and other non-coding RNAs naturally participate in sequence-specific interactions with mRNAs, and thus present the logical places to look for trans-acting regulators of individual -1 PRF signals. With this in mind, computational methods were used to identify two miRNAs that interact with and stimulate -1 PRF promoted by a sequence element in the mRNA encoding CCR5, a cytokine receptor that is used as a co-receptor for HIV-1 [21]. A series of genetic and biochemical experiments revealed that one of these, miR-1224, directly interacts with the CCR5 -1 PRF stimulating mRNA pseudoknot. Presumably, the interaction stabilizes the pseudoknot, rendering it more difficult for ribosomes to resolve. This would increase ribosome pause times at the slippery site, stimulating kinetic partitioning into the -1 frame. Theoretically, miRNAs may also have -1 PRF inhibitory activities by being able to destabilize -1 PRF promoting mRNA downstream elements. In support of this, siRNA knockdown of the cellular miRNA processing apparatus stimulated -1 PRF promoted by some frameshift signals, and inhibited -1 PRF promoted by others [21]. Non-coding RNA stimulation of -1 PRF at polyA sequences may also be possible. In support of this, hybridization of antisense linked nucleic acids (LNAs) immediately 3′ of heptameric slippery sequences was sufficient to promote efficient -1 frameshifting in the absence of any other stimulatory element [39]. Natural attenuation of -1 PRF by stem-loop structures immediately 5′ of coronavirus slippery sequences has also been reported [40], a phenomenon that can be replicated by hybridization of oligonucleotides complementary to sequences lying just upstream of slippery sites [41]. These studies establish the role of ncRNAs in regulating translational fidelity and suggest that regulation of -1 PRF may not be limited to miRNAs. Missing however, is any hypothesis or model explaining how polyA-directed -1 PRF may be specifically regulated. The demonstration of protein-induced transactivation of frameshifting in porcine reproductive and respiratory syndrome virus [42] may provide a clue in this respect; perhaps polyA-mediated -1 PRF may be regulated by polyA-binding proteins.
Conclusion and prospective
Arthur and co-workers serendipitously discovered that there is more than one way to program -1 ribosomal frameshifting. What other ribosomal frameshift promoting elements may be out there, and how might they be identified? An approach for discovering new -1 PRF signals that has worked particularly well with viruses and bacteria has been to first detect evolutionarily conserved reading frames and then, using molecular genetics tools, identify and characterize the translational recoding elements [16]. Mining of ribosome profiling data to pinpoint frameshifted ribosomes, and from there identifying the elements that made them shift reading frame presents a new and promising approach [21, 43]. The recent discovery of modified bases in mRNAs and their regulation [44, 45], raises the question of how these may affect the ability of ribosomes to maintain reading frame. Additionally, although we have known about mRNA editing for quite some time (reviewed in [46]), its role in creating or ablating -1 PRF signals remains completely unexplored. Furthermore, identifying the trans-acting factors (miRNAs, other ncRNAs and even proteins) that regulate specific -1 PRF signals, characterizing how their expression is regulated, and how dysregulation may be linked to disease is another open research area. Lastly, as outlined in Figure 3, -1 PRF and NMD present as therapeutic targets, not only with regard to viral diseases, e.g. HIV/AIDS, but also as a potential modality to fine tune and correct errors in gene expression, either using small molecules that target specific -1 PRF signals to correct the expression of specific genes (e.g. using synthetic RNA-like molecules) or using therapeutics that correct for changes in gene expression due to global defects in -1 PRF.
Figure 3. From genes to disease and points of therapeutic intervention.

Mutations in genes that participate in translation that alter global rates of -1 PRF elicit downstream post-transcriptional surveillance pathways, e.g. NMD that alter the transcriptome. This leads to altered gene expression (proteomic changes) and progression to disease states. Therapeutic approaches may include use of synthetic polynucleotide analogs (miRNAs and related derivitives) targeting specific -1 PRF signals designed to fine tune frameshifting rates. Given the epistatic relationship of NMD to -1 PRF, targeting this pathway using small molecule inhibitors presents another therapeutic modality.
Acknowledgments
We would like to thank our colleagues, in particular Susan Baserga, Kim de Keersmaecker, Arlen Johnson, Davide Ruggero, John Woolford, and many others in the translational control field for participating in stimulating and creative discussions about the topics covered in this essay in an ongoing discussion over the past two decades. The discussion of Dr. Losick’s findings in the context of Galilean versus Euclidian mathematical schemes was inspired by lectures by Dr. Losick, and Dr. Mark A. Peterson (Dept. of Mathematics, Mount Holyoke College). We also wish to acknowledge members of the Dinman lab, past and present. This work was supported by grant to JDD from the National Institutes of Health (R01 HL119439, R01 GM117177).
References
- 1.Cochella L, Green R. Fidelity in protein synthesis. Curr Biol. 2005;15:R536–40. doi: 10.1016/j.cub.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 2.Arthur LL, Pavlovic-Djuranovic S, Koutmou KS, Green R, Szczesny P, Djuranovic S. Translational control by lysine-encoding A-rich sequences. Sci Adv. 2015;1:e1500154. doi: 10.1126/sciadv.1500154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu J, Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito-Harashima S, Kuroha K, Tatematsu T, Inada T. Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 2007;21:519–24. doi: 10.1101/gad.1490207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimitrova LN, Kuroha K, Tatematsu T, Inada T. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J Biol Chem. 2009;284:10343–52. doi: 10.1074/jbc.M808840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, et al. Rli1/ABCE1 Recycles Terminating Ribosomes and Controls Translation Reinitiation in 3′UTRs In Vivo. Cell. 2015;162:872–84. doi: 10.1016/j.cell.2015.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vasudevan S, Peltz SW, Wilusz CJ. Non-stop decay--a new mRNA surveillance pathway. Bioessays. 2002;24:785–8. doi: 10.1002/bies.10153. [DOI] [PubMed] [Google Scholar]
- 8.Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–4. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Passos DO, Doma MK, Shoemaker CJ, Muhlrad D, et al. Analysis of Dom34 and its function in no-go decay. Mol Biol Cell. 2009;20:3025–32. doi: 10.1091/mbc.E09-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Celik A, Kervestin S, Jacobson A. NMD: At the crossroads between translation termination and ribosome recycling. Biochimie. 2015;114:2–9. doi: 10.1016/j.biochi.2014.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacks T, Varmus HE. Expression of the Rous Sarcoma Virus pol gene by ribosomal frameshifting. Science (80-) 1985;230:1237–42. doi: 10.1126/science.2416054. [DOI] [PubMed] [Google Scholar]
- 12.Firth AE, Brierley I. Non-canonical translation in RNA viruses. J Gen Virol. 2012;93:1385–409. doi: 10.1099/vir.0.042499-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller WA, Jackson J, Feng Y. Cis- and trans-regulation of luteovirus gene expression by the 3′ end of the viral genome. Virus Res. 2015;206:37–45. doi: 10.1016/j.virusres.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plant EP, Rakauskaite R, Taylor DR, Dinman JD. Achieving a golden mean: mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins. J Virol. 2010;84:4330–40. doi: 10.1128/JVI.02480-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cobucci-Ponzano B, Rossi M, Moracci M. Translational recoding in archaea. Extremophiles. 2012;16:793–803. doi: 10.1007/s00792-012-0482-8. [DOI] [PubMed] [Google Scholar]
- 16.Baranov PV, Atkins JF, Yordanova MM. Augmented genetic decoding: global, local and temporal alterations of decoding processes and codon meaning. Nat Rev Genet. 2015;16:517–29. doi: 10.1038/nrg3963. [DOI] [PubMed] [Google Scholar]
- 17.Caliskan N, Katunin VI, Belardinelli R, Peske F, et al. Programmed -1 Frameshifting by Kinetic Partitioning during Impeded Translocation. Cell. 2014;157:1619–31. doi: 10.1016/j.cell.2014.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dinman JD. Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip Rev RNA. 2012;3:661–73. doi: 10.1002/wrna.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belew AT, Hepler NL, Jacobs JL, Dinman JD. PRFdb: a database of computationally predicted eukaryotic programmed -1 ribosomal frameshift signals. BMC Genomics. 2008;9:339. doi: 10.1186/1471-2164-9-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Belew AT, Advani VM, Dinman JD. Endogenous ribosomal frameshift signals operate as mRNA destabilizing elements through at least two molecular pathways in yeast. Nucleic Acids Res. 2010;39:2799–808. doi: 10.1093/nar/gkq1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, et al. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature. 2014;512:265–9. doi: 10.1038/nature13429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Advani VM, Belew AT, Dinman JD. Yeast telomere maintenance is globally controlled by programmed ribosomal frameshifting and the nonsense-mediated mRNA decay pathway. Translation. 2013;1:38–47. doi: 10.4161/trla.24418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olsthoorn RC, Laurs M, Sohet F, Hilbers CW, et al. Novel application of sRNA: stimulation of ribosomal frameshifting. RNA. 2004;10:1702–3. doi: 10.1261/rna.7139704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson CM, Anderson CB, Howard MT. Antisense-induced ribosomal frameshifting. Nucleic Acids Res. 2006;34:4302–10. doi: 10.1093/nar/gkl531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howard MT, Gesteland RF, Atkins JF. Efficient stimulation of site-specific ribosome frameshifting by antisense oligonucleotides. RNA. 2004;10:1653–61. doi: 10.1261/rna.7810204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norman TM, Lord ND, Paulsson J, Losick R. Stochastic Switching of Cell Fate in Microbes. Annu Rev Microbiol. 2015;69:381–403. doi: 10.1146/annurev-micro-091213-112852. [DOI] [PubMed] [Google Scholar]
- 27.Dinman JDJD, Wickner RBRB. Translational maintenance of frame: mutants of Saccharomyces cerevisiae with altered -1 ribosomal frameshifting efficiencies. Genetics. 1994;136:75–86. doi: 10.1093/genetics/136.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCann KL, Baserga SJ. Genetics. Mysterious ribosomopathies. Science. 2013;341:849–50. doi: 10.1126/science.1244156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dameshek W. Riddle: what do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and “hypoplastic” leukemia have in common? Blood. 1967;30:251–4. [PubMed] [Google Scholar]
- 30.Jack K, Bellodi C, Landry DM, Niederer RO, et al. rRNA Pseudouridylation Defects Affect Ribosomal Ligand Binding and Translational Fidelity from Yeast to Human Cells. Mol Cell. 2011;44:660–6. doi: 10.1016/j.molcel.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr Opin Genet Dev. 2011;21:474–83. doi: 10.1016/j.gde.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ban N, Beckmann R, Cate JH, Dinman JD, et al. A new system for naming ribosomal proteins. Curr Opin Struct Biol. 2014;24:165–9. doi: 10.1016/j.sbi.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Keersmaecker K, Atak ZK, Li N, Vicente C, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013;45:186–90. doi: 10.1038/ng.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sulima SO, Gülay SP, Anjos M, Patchett S, et al. Eukaryotic rpL10 drives ribosomal rotation. Nucleic Acids Res. 2014;42:2049–63. doi: 10.1093/nar/gkt1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sulima SO, Patchett S, Advani VM, De Keersmaecker K, et al. Bypass of the pre-60S ribosomal quality control as a pathway to oncogenesis. Proc Natl Acad Sci. 2014;111:5640–5. doi: 10.1073/pnas.1400247111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hekman KE, Yu GY, Brown CD, Zhu H, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet. 2012;21:5472–83. doi: 10.1093/hmg/dds392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orr HT. Cell biology of spinocerebellar ataxia. J Cell Biol. 2012;197:167–77. doi: 10.1083/jcb.201105092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graille M, Séraphin B. Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat Rev Mol Cell Biol. 2012;13:727–35. doi: 10.1038/nrm3457. [DOI] [PubMed] [Google Scholar]
- 39.Yu CH, Noteborn MH, Olsthoorn RC. Stimulation of ribosomal frameshifting by antisense LNA. Nucleic Acids Res. 2010;38:8277–83. doi: 10.1093/nar/gkq650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cho C-P, Lin S-C, Chou M-Y, Hsu H-T, et al. Regulation of programmed ribosomal frameshifting by co-translational refolding RNA hairpins. PLoS One. 2013;8:e62283. doi: 10.1371/journal.pone.0062283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu H-T, Lin Y-H, Chang K-Y. Synergetic regulation of translational reading-frame switch by ligand-responsive RNAs in mammalian cells. Nucleic Acids Res. 2014;42:14070–82. doi: 10.1093/nar/gku1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Treffers EE, Napthine S, Tas A, et al. Transactivation of programmed ribosomal frameshifting by a viral protein. Proc Natl Acad Sci U S A. 2014:1–10. doi: 10.1073/pnas.1321930111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michel AM, Choudhury KR, Firth AE, Ingolia NT, et al. Observation of dually decoded regions of the human genome using ribosome profiling data. Genome Res. 2012;22:2219–29. doi: 10.1101/gr.133249.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yue Y, Liu J, He C. RNA N 6 -methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29:1343–55. doi: 10.1101/gad.262766.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–6. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenthal JJC. The emerging role of RNA editing in plasticity. J Exp Biol. 2015;218:1812–21. doi: 10.1242/jeb.119065. [DOI] [PMC free article] [PubMed] [Google Scholar]