

Abstract
Obsessive-compulsive disorder (OCD) is a debilitating neuropsychiatric condition estimated to afflict 1-3% of the world population. The estimated financial impact in the treatment and management of OCD is in the billions of dollars annually in the US alone. At present there is a marked lack of evidence on the specific causes of OCD. Current hypotheses largely focus on the serotonin (5-HT) system on the basis of the effectiveness of selective serotonin reuptake inhibitors (SSRIs) in alleviating symptoms of patients with OCD, yet a considerable fraction of patients are non-responsive or minimally responsive to these agents. Despite this fact, SSRIs have remained the primary pharmacological treatment avenue for OCD. In recent years, multiple lines of evidence have implicated glutamatergic synaptic dysfunction within the cortico-striatal-thalamo-cortical (CSTC) brain circuit in the etiology of OCD and related disorders, thereby prompting intensified effort in the development and evaluation of agents that modulate glutamatergic neurotransmission for the treatment of OCD. With this in mind, here we review the following topics with respect to synaptic dysfunction and the neural circuitry underlying OCD: (1) evidence supporting the critical involvement of the CSTC circuit, (2) genetic studies supporting the involvement of glutamatergic dysfunction, (3) insights from genetic animal models of OCD, and (4) preliminary findings with glutamatergic neurotransmission-modulating agents in the treatment of OCD. Given the putative mechanistic overlap between OCD and the broader OC-spectrum of disorders, unraveling the synaptic basis of OCD has potential to translate into more effective treatments for an array of poorly understood human disorders.
Keywords: OCD, Glutamate, Glutamatergic, Direct pathway, Indirect pathway, Striatonigral, Striatopallidal, CSTC, Fronto-subcortical, Synapse, Neurotransmission, Genetics, SAPAP3, Riluzole, Tourette's syndrome, Trichotillomania, Neuropsychiatric disorder, SSRI, N-acetylcysteine, Memantine, Orbitofrontal cortex, SLC1A1, GRIN2B, NMDA receptor, D-cycloserine, Corticostriatal, Cortico-striatal
What is OCD?
OCD Symptoms, Worldwide Prevalence, and Economic Impact
Obsessive-compulsive disorder is a debilitating neuropsychiatric condition characterized by persistent intrusive thoughts (obsessions) and the expression of ritualistic repetitive behaviors (compulsions) which are generally enacted in an effort to somehow alleviate intense anxiety caused by specific obsessions. The majority of OCD sufferers are aware or become aware that their obsessions are irrational and their repetitive actions excessive, which only further exacerbates feelings of distress and anxiety due to an inability to prevent such symptoms. As a significant proportion of time each day (>1 hour by definition) is generally absorbed by obsessions and compulsions, these symptoms can render sufferers incapacitated in their daily functioning [1, 2]. Commonly reported obsessions and compulsions are shown in Table 1. Although OCD can develop at any age, it most often develops in late adolescence to young adulthood. Left untreated, symptoms can persist throughout the lifetime, although the specificity and severity of symptoms may change with time.
Table 1. Common Symptoms in OCD [1, 2].
| Obsessions |
|---|
| excessive doubt about task completion |
| fear of contamination or un-cleanliness |
| need for symmetry or exactness |
| fear of causing harm to others |
| excessive concern over right and wrong |
| intrusive inappropriate sexual thoughts |
| Compulsions |
| checking |
| washing/cleaning |
| counting |
| ordering/arranging |
| repeating |
| hoarding |
| praying |
OCD is estimated to afflict roughly 1-3% of the world population [3-5] thus making it one of the most prevalent psychiatric disorders in our society. At last glance, the estimated financial impact in the treatment and management of OCD is several billions of dollars annually in the United States alone [6], and this cost is expected to rise with improvements in diagnosis, reporting, and availability of effective treatments.
Although the exact causes of OCD are unknown, many lines of evidence suggest that OCD has a genetic basis. Specifically, the concordance rate for OCD is greater among pairs of monozygotic twins (80-87%) than among pairs of dizygotic twins (47-50%, [7, 8]). Family studies of OCD also indicate that the risk to first degree relatives is 3-12 times greater than that for the general population [9]. Despite intensive efforts, no common alleles have been directly linked to the pathogenesis of OCD, which is likely to reflect a complex model of genetic susceptibility.
The OC-Spectrum of Disorders
Based on commonalities in symptoms, comorbidity, neurocircuitry, and response to drug treatments, OCD has been suggested to share etiology with a growing list of other neuropsychiatric disorders. These include Body dysmorphic disorder (BDD), Tourette syndrome (TS), trichotillomania (TTM; compulsive hair pulling), compulsive skin-picking (CSP; also called dermatillomania) [10, 11]. Several other disorders have been suggested for inclusion (e.g. additional impulse control disorders and various eating disorders) although no consensus has emerged as to the appropriate boundary of the OC spectrum—which is likely attributed to a poor understanding of many of these human disorders. Taken together, OC-spectrum disorders may conceivably affect as much as 10% of the general population [12].
Current Treatments for OCD
Despite a lack of understanding about the precise neurobiological mechanisms underlying OCD, treatments have been developed that are at least partially effective in alleviating symptoms. These treatments can be subdivided into either pharmacological or behavioral therapies.
Pharmacological Therapies
The current first-line drugs in the treatment of OCD are selective serotonin reuptake inhibitors (SSRIs). The SSRIs fluoxetine, fluvoxamine, sertraline, and paroxetine have been approved by the Food and Drug Administration (FDA) for the treatment of OCD, although paroxetine is not approved for use in pediatric cases. SSRIs act by blocking the presynaptic serotonin transporter (5-HTT) that normally mediates the reuptake of synaptically released serotonin. All of the currently available SSRIs that have been investigated seem to exhibit similar efficacy in treating OCD symptoms. On average, SSRI responders see a modest but significant reduction of symptoms, whereas 40-60% of patients are unresponsive to SSRI monotherapy [13, 14].
Notably, higher doses of SSRIs over longer periods of time (2-3 months) are necessary to achieve optimal anti-obsessional benefits in comparison to their implementation in the treatment of major depression [14]. This delay in therapeutic benefit in the treatment of OCD is suggested to reflect the time course of serotonin receptor desensitization resulting from chronically elevated synaptic serotonin levels [15]. Long-term modifications at serotonin receptors are also indicated by the fact that acute interventions known to decrease serotonin transmission fail to disrupt the ongoing benefits of SSRI treatment in OCD patients [16].
The tricyclic antidepressant clomipramine is often still considered a first-line drug in treating OCD symptoms, and there is some evidence to suggest superior efficacy relative to SSRIs; however, clomipramine is less well tolerated than the SSRIs and causes a broad range of side effects—likely due to anticholinergic effects [17]. Thus, clomipramine may be best utilized for patients that are refractory to SSRI treatment.
Augmenting agents are employed when there is a partial response to SSRI monotherapy. Since this is the most likely outcome for the majority of cases, the use of augmenting agents is common, and this strategy is generally favored over switching to a different SSRI monotherapy since a partial response has been established. Examples of augmenting agents that are commonly employed include the antipsychotics risperidone and haloperidol. A detailed guideline (algorithm) for OCD pharmacotherapy can be found elsewhere [18].
Behavioral Therapies
A specific form of cognitive-behavioral therapy called exposure and response prevention (ERP) is an established and effective treatment for some OCD symptoms [19, 20]. ERP involves having the OCD patient repeatedly expose themselves to anxiety-inducing stimuli linked to frequent obsessions (e.g. contact with contaminated objects), followed by a prevention of the rituals that typically ensue (e.g. hand washing). In this way the patient undergoes habituation to the aversive stimuli with repeated exposures. The response prevention aspect is critical in allowing the patient to learn that the decreasing anxiety experienced with repeated exposure is not linked to the completion of compulsion-induced rituals [21]. ERP has been shown to be equally or perhaps more effective in treating OCD symptoms than pharmacological treatments alone [22, 23], and the rate of relapse following cessation of treatment is significantly lower for ERP compared to pharmacological treatments alone [24]. Thus, cognitive-behavioral therapy may be implemented as a first-line treatment and is a particularly valuable treatment option for SSRI refractory patients and patients that refuse pharmacological treatment altogether. Other structured psychotherapies such as cognitive therapy without ERP are also a valuable treatment option, particularly in cases where specific OCD symptoms are not amenable to ERP therapy [25].
Combined Therapy
There is some evidence that combined pharmacological and behavioral treatments (SSRIs or clomipramine combined with cognitive-behavioral therapy) are more effective than SSRI or clomipramine monotherapy, although it is unclear at this time if combined regimes offer significant benefits over cognitive-behavioral therapy alone [22, 26]. Even the most rigorous treatment regimes available today offer incomplete reprieve for most. Advances in our understanding of the basic neurobiological mechanisms underlying OCD are necessary to guide the development of improved treatment options.
Evidence Supporting the Corticostriatal-Thalamo-Cortical Circuit as a Primary Site of Synaptic Dysfunction in OCD
Anatomy and Function of the CSTC Circuit
In 1983, Penney and Young displayed remarkable insight in their description of the CSTC circuit and the role of this circuit in the control of normal movement, as well as in the context of movement disorders [27]. In particular, their model mechanistically explains the central role of the basal ganglia within this CSTC circuit for the selection of which behaviors will be carried out and the exclusion of unwanted behaviors.
Subsequent work has firmly established that the CSTC circuit is composed of numerous parallel open loops that functionally link the frontocortical and subcortical areas [28-30]. It is believed that different sets of frontocortical-subcortical loops participate in specialized motor and cognitive functions as determined by the specific frontocortical area involved [31]. Cortical neurons send dense projections into the striatum which form glutamatergic cortico-striatal synapses onto medium spiny neurons (MSNs). In turn, GABAergic MSNs connect to the major output structures of the basal ganglia, the globus pallidus pars internalis (GPi) and substantia nigra pars reticulata (SNr), by way of two anatomically distinct pathways that are thought to have opposing roles. Dopamine receptor type 1 (D1R) expressing MSNs in the striatum compose the “direct pathway” (striatonigral), whereas dopamine receptor type 2 (D2R) expressing MSNs in the striatum compose the “indirect pathway” (stiatopallidal) [32]. The direct pathway axons terminate in GPi/SNr, whereas the indirect pathway involves a more complex relay successively involving the globus pallidus pars externalis (GPe) and subthalamic nucleus (STN). In a simplified model, the output structures of the basal ganglia provide inhibitory tone to the thalamus, and this tone is modulated by the balance of activity through the direct and indirect pathways (the so-called “accelerator-brake” model) [33]. In this way, a shift of balance favoring activity in the direct pathway will “disinhibit” the thalamus and thereby promote the selection of behavioral sequences. Conversely, a shift of balance favoring activity in the indirect pathway has a net effect of reinforcing or strengthening the inhibitory tone to the thalamus and thereby inhibits the selection of behavioral sequences. The thalamus sends excitatory inputs back to the cortex, thereby completing the CSTC loop (Fig. 1). Although highly simplified, this model of the CSTC circuit provides a useful framework for understanding CSTC circuit dysfunction in OCD.
Fig. (1). The Cortico-striatal-thalamo-cortical loop is a major site of synaptic dysfunction in OCD.
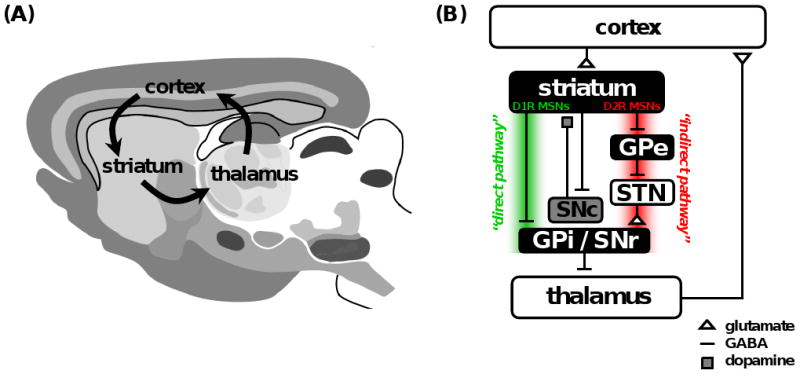
(A) Diagram of a mouse brain (sagittal view) illustrating the major brain regions that compose the CSTC loop. Hyperactivity in specific cortical-subcortical loops involving areas such as the orbitofrontal cortex is hypothesized to lead to the expression of OCD symptoms. (B) Simplified circuit diagram of the CSTC loop in mammals. MSN, medium spiny neuron; GPe, globus pallidus pars externalis; GPi, globus pallidus pars internalis; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulate; STN, subthalamic nucleus; D1R, D1-type dopamine receptor; D2R, D2-type dopamine receptor. Panel (A) adapted from [32].
Imaging Studies Implicating CSTC Circuit Dysfunction in OCD
The use of non-invasive neuroimaging methods to probe for abnormalities in the brains of human patients with OCD has led to extensively replicated findings supporting the involvement of the CSTC circuit in OCD (thoroughly reviewed in [34]). Numerous early studies using Positron Emission Tomography (PET) or single photon emission CT (SPECT) have consistently reported increased glucose metabolism and blood flow (pretreatment baseline or during symptom provocation) in one or more of the following regions: orbitofrontal cortex, anterior cingulate cortex, caudate, and thalamus [35-40]. These regions are the principle components of the CSTC circuit (Fig. 1). As a complement to this approach, additional PET and SPECT studies were performed on OCD patients before and after treatment. These studies reported decreased glucose metabolism and blood flow within the same CSTC regions, mainly using SSRI or clomipramine treatment [41-45], but also with behavioral therapy [43, 46] and neurosurgical lesion [47]. Qualitatively similar results in OCD patients (i.e. alterations within the CSTC circuit) have been reported for both the pre- and post treatment assessments using magnetic resonance spectroscopy (MRS) to measure regional concentrations of amino acid neurotransmitters in general [48-50]. The observed changes with MRS are often cited as reflecting alterations in glutamate, although this is not the only possibility (see [51] for further discussion on the limitations of MRS in this context).
According to a prominent model put forth by Baxter et al. [34, 52] which integrates findings from key imaging studies and extends the proposals of others [53, 54], OCD symptoms may be caused by hyperactivity in the orbitofrontal-subcortical loops as a consequence of imbalance in the basal ganglia pathways. Persistent activation of the direct pathway has been suggested, which is proposed to lead to inappropriate, potentially repetitive release of cognitive and motor sequences. The release of such cognitive and motor sequences or “action chunks” become pathological because they are no longer under the control of intent despite being consciously performed [55]. See also [56] for a detailed review regarding the orbitofrontal-subcortical model of OCD in the context of neuroimaging studies.
Last-Resort Treatment Options for OCD Disrupt CSTC Circuits
This hypothesis of frontocortical hyperactivity in OCD also fits well with the reported beneficial effects of certain invasive neurosurgical interventions for treatment refractory OCD. Anterior cingulotomy, anterior capsulotomy, and subcaudate tractotomy and capsulotomy are all designed to disrupt connections between frontocortical and subcortical areas within the CSTC circuit, and a significant percentage of patients improve following these surgeries [57, 58]. High-frequency deep brain stimulation (DBS) has also been explored for treatment resistant OCD as an alternative to permanent ablation/lesion options. In cases where DBS was successfully employed, the lessening of OCD symptoms was accompanied by a reduction in the activity of orbitofrontal cortical regions as measured by PET [59, 60]—thus mirroring the earlier findings using more conventional treatment options. The site of DBS in these studies was the anterior internal capsule, which may lead to disruption of the connections between thalamus and orbitofrontal cortex [60]. Additional studies have shown effectiveness of DBS in the nucleus accumbens (NAc) region in OCD [61, 62], and an investigation into the detailed mechanism of NAc DBS in rats found that activity in orbitofrontal cortex was reduced through a complex form of recurrent inhibition involving antidromic activation of cortico-striatal axon collaterals [63]
OCD Precipitated by Basal Ganglia Disruptions
Symptoms of OCD have been reported in several cases involving brain lesions. Laplane [64] reported OCD-like symptoms in several patients that had bilateral lesions of the basal ganglia. Another study reported 5 cases of OCD occurring late in life, all involving lesions of the basal ganglia [65]. Thus, these lesion studies are consistent with the proposed critical involvement of the CSTC circuit, although such lesions must impact the CSTC circuit in a fundamentally different way than the aforementioned neurosurgical lesions shown to alleviate rather than exacerbate OCD symptoms.
In addition to OCD symptoms brought on by brain lesions, acquired forms of OCD can also result from infection. Rheumatic fever is triggered by group A beta-hemolytic streptococcal infections (GAS) in children. A fraction of children with rheumatic fever develop a movement disorder known as Sydenham's chorea (SC). Available evidence suggests that the pathophysiology of SC involves an autoimmune mechanism whereby antibodies triggered by the GAS infection cross-react with neurons in the basal ganglia causing inflammation and compromised basal ganglia function [66]. It has long been recognized that OCD symptoms frequently accompany SC, and several clinical studies have substantiated this association [67-69]. Cases of OCD and TS are also observed in children following GAS infection, but in the absence of clinically defined SC, and this group has been referred to as pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) [70].
Involvement of Other Brain Circuits in OCD
Defects in other brain regions are also likely to play a role in OCD and should be mentioned. For example, there is evidence for involvement of the limbic system with respect to the anxiety component of OCD. Altered amygdala volume and dysfunctional amygdala activation have been reported in OCD patients [71-73], and the amygdala has a well established role in the expression of fear and anxiety [74, 75]. In addition, although the CSTC circuit is proposed to mediate some aspects of disgust relevant to fear of contamination in OCD, there is further evidence that discrete circuits in the insular cortex are a primary loci of disgust (reviewed in [76]).
Genetic Studies Supporting Glutamatergic Dysfunction in OCD
Although numerous lines of evidence have implicated dysfunction of the CSTC circuit in OCD, the precise nature of the defect(s)—including information regarding which neurotransmitter systems are primarily affected—has not been detailed. No causal relationship has been established in the etiology of OCD. Most hypotheses on the causes of OCD have focused on the serotonin system on the basis of positive benefits of SSRIs in treating OCD symptoms [77]. However, the limited effectiveness of SSRIs suggests the critical involvement of other neurotransmitter systems.
Evidence for the involvement of the glutamatergic system in OCD is steadily mounting. Frontocortical hyper-glutamatergic function has been proposed to account for the CSTC abnormalities observed in imaging studies of OCD [34], and in support of this model, a recent small study demonstrated elevated CSF glutamate levels in adult OCD patients compared to normal controls [78]. The model also implies that genes involved in glutamatergic neurotransmission are good candidates for genetic association studies in OCD—and indeed, several studies (as detailed below) have reported associations in just the last few years. It is important to keep in mind that numerous genetic studies support the involvement of the serotonergic and dopaminergic neurotransmitter systems in OCD (though many of the findings are controversial), and an extensive review of these studies can be found elsewhere [77].
GRIN2B
In 2004, Arnold et al. [79] provided the first report of a positive association between a glutamatergic system gene and OCD. This study demonstrated preliminary association of one variant in the GRIN2B gene encoding the N-methyl-D-aspartate (NMDA) receptor subunit 2B. This gene is highly expressed in the CSTC circuit, and NMDA-type glutamate receptors have a well established role in basic synaptic transmission, synaptic plasticity mechanisms underlying learning and memory, and neuropsychiatric disorders (see [80, 81] for detailed reviews). Of particular interest, the subunit composition of NMDA receptors is dynamically regulated during development and is important in determining the functional properties of the receptor [81]. Interestingly, Arnold et al. hypothesized the involvement of NMDA receptor gene variations in OCD based on neuroimaging findings of deficient striatal activation during learning tasks in OCD patients [82], thereby demonstrating the potential utility of integrating neuroimaging and genetic studies. The findings of this study have yet to be replicated and thus should be regarded as preliminary.
SLC1A1/EAAC1
In 2006, Arnold et al. [83] and Dickel et al. [84] simultaneously published independent studies demonstrating that variants in the solute carrier family 1, member 1 gene SLC1A1 are associated with OCD. SLC1A1 encodes the neuronal glutamate transporter excitatory amino acid carrier 1 (EAAC1). These studies were in follow-up to the first genome-wide linkage study for OCD that provided evidence suggestive of an OCD susceptibility gene in the chromosomal region 9p24 [85], a result that was later validated in a focused replication study [86]. The region 9p24 contains numerous predicted and known genes; however, of the known genes in this region at the time, SLC1A1 was the only one with expression in the brain according to Arnold et al. [83]. Of particular interest, SLC1A1 is highly expressed in brain regions comprising CSTC circuits [87, 88]. A third independent investigation [89] also confirmed an association between OCD and SLC1A1 for both single marker and haplotype single nucleotide polymorphisms (SNPs) in the 3′ region of the gene, consistent with the earlier studies [83, 84]. Although the findings of the three studies are not identical, it has been suggested that the associated SNPs and haplotypes identified thus far may be in linkage disequilibrium with a yet unsampled, potentially causal SNP in the 3′ region of SLC1A1 [89]. In all three studies, subgroup analysis by gender revealed specific associations of this gene with OCD for males but not females. Thus, considerable consistency has been established in the reported associations between SLC1A1 and OCD.
The work on SLC1A1 also yields important lessons for evaluating the results of other candidate gene studies. An initial attempt failed to support association of SLC1A1 with OCD (but did confirm SLC1A1 as a positional candidate by mapping this gene within the 9p24 region) [90]. This earlier negative result has been attributed to small sample size and incomplete coverage of the SLC1A1 gene region, and indeed association of SLC1A1 with OCD became apparent in a more thorough study by the same group in 2006 [84]. In light of this, a negative result in attempted gene association studies only provides strong evidence for the lack of an association when the sample size is appropriately large and the selected markers provide excellent coverage of the gene region under investigation.
GRIK2 & GRIK3
Delorme et al. [91] investigated potential association between OCD and the ionotropic kainate receptors GRIK2 and GRIK3. This study found no evidence for association for three SNPs in GRIK2 and one SNP in GRIK3, however, a separate transmission disequilibrium test revealed a significant under-transmission of the I867 allele of SNP rs2238076 in OCD. Notably, there is increasing evidence for GRIK2 variations associated with autism [92-94], a disorder with manifestations that include compulsive-repetitive behavior similar to OCD.
SAPAP3
SAP90/PSD95-associated protein 3 (SAPAP3) is a post-synaptic scaffolding protein at excitatory synapses that is highly expressed in the striatum [95]. Recent evidence from the SAPAP3 null mouse demonstrates the importance of this protein in regulating cortico-striatal synaptic function [96]. Thus, SAPAP3 is an important component within the CSTC circuit. This is further established by the finding that the SAPAP3 null mice exhibit OCD-like behaviors (see also the following section on genetic mouse models for further details) [96]. These findings prompted two recent genetic studies of SAPAP3 and OCD.
Züchner et al. [97] performed SAPAP3 gene resequencing analysis in OCD and TTM case samples. This study identified seven rare non-synonymous heterozygous SAPAP3 variants. The combined load of these rare SAPAP3 mutations was 4.2% of the OCD/TTM case samples compared to only 1.1% in control samples, providing tentative support for a role of SAPAP3 in OCD and TTM. The majority of the variants represented missense mutations, some of which are predicted to be possibly detrimental to protein function based on computer algorithms. These findings await further validation, including analysis of the functional relevance of these rare SAPAP3 mutations.
A relatively large family-based gene association study of SAPAP3 in OCD and grooming disorders (GD) has been conducted by Bienvenu et al. [98]. This study explored 6 SNPs spanning region 1p35 of the SAPAP3 gene. The preliminary evidence suggests that multiple variations in the SAPAP3 gene are associated with GD. No clear association between SAPAP3 variants and OCD was reported, although it is noteworthy that GDs without OCD were uncommon in this study. With this in mind the authors suggest the possibility that SAPAP3 variants may be involved in a subtype of OCD involving pathological grooming behaviors.
Summary of Genetic Studies in OCD
Studies reporting genetic linkage or association which are subsequently replicated merit continued focus. As detailed above, the most reliable evidence for gene association in OCD exists for the glutamate transporter gene SLC1A1, which together with recent reports of association for other genes directly involved in glutamatergic signaling at synapses has provoked a shift in attention to glutamatergic neurotransmission as a target for OCD pharmacotherapy. It is critical to point out that evidence for genetic association does not imply causality, and that OCD is a complex disorder that is likely to have a polygenic basis. Furthermore, genetic association studies leave open the mechanism by which specific genetic variations may disrupt CSTC circuit function in OCD. Progress in this area will most likely be facilitated by detailed investigations in animal models of OCD.
Genetic Animal Models of OCD
Animal models serve as invaluable platforms for the development, screening, and refinement of pharmacological agents for the treatment of human disorders. They are also amenable to invasive research techniques not suitable for studies involving human patients. At present there are several behavioral, pharmacological, and genetic animal models of OCD that vary greatly in the extent to which they model the human condition (reviewed extensively in [99, 100]). Arguably, genetic animal models hold the most promise for rapid progress in our understanding of the basic neurobiology of OCD. This is true because the specific genetic disruption is often known, and in many cases there exists prior knowledge (such as mRNA or protein expression pattern) such that the investigators possess a priori mechanistic insight that can help explain many observed phenotypes. With this in mind, we selectively overview the existing genetic animal models (summarized in Table 2), including two very recently characterized genetic mouse models of OCD. The validity of each model is discussed along with how the model has (or has not) contributed to our understanding of synaptic dysfunction in OCD.
Table 2. Evaluation of Genetic Animal Models of OCD.
| Genetic Model | Face Validity* | CSTC Circuit Expressionˆ | CSTC Synaptic Dysfunction | Response to SSRIs |
|---|---|---|---|---|
| D1CT Tg | +++ | ++ | ++ | nd |
| Hoxb8 mutant | +++ | +++ | nd | nd |
| 5-HT2C null | + | + | nd | nd |
| ArKO | ++ | ? | nd | nd |
| DAT KD | + | +++ | +++ | nd |
| SAPAP3 null | +++ | +++ | +++ | ++ |
D1CT Tg, transgenic mouse line in which neuropotentiating cholera toxin is expressed under the D1-type dopamine receptor promoter elements; Hoxb8 mutant, homozygous loss-of-function for mammalian transcription factor Hoxb8; 5-HT2C null, null mutant mouse for the 5-HT2C subtype of the serotonin receptor; ArKO (for aromatase knock-out), estrogen deficient mouse line resulting from elimination of aromatase expression; DAT KD, dopamine transporter knock-down mice with DAT levels reduced to 10% of wild-type values; SAPAP3 null, null mutant mouse for the postsynaptic scaffolding protein SAPAP3.
Note that face validity incorporates both behavioral similarity AND specificity.
CSTC circuit expression refers to the extent to which the gene or Tg is expressed in these brain regions.
nd - not demonstrated.
D1CT-7 Transgenic Mice
The first genetic mouse model of OCD was reported in 1999 by Campbell et al. [101]. Transgenic mice were generated in which the expression of an intracellular form of the neuropotentiating cholera toxin (CT) is controlled by the D1-type dopamine receptor promoter elements (termed D1CT mice). D1CT mouse lines have chronically elevated neuronal activity restricted to D1 positive subsets of neurons in the brain.
In one particular line of D1CT mice (D1CT-7) transgene expression was found to be predominantly restricted to D1 positive neurons within the intercalated nucleus of the amygdala and within cortical areas that project to orbitofrontal cortex and striatum. Chronic potentiation of these D1 positive neuronal subsets was suggested to stimulate corticostriatal glutamatergic transmission, although this has not been directly validated. D1CT-7 mice exhibited repetitive biting of littermates that was unrelated to aggression, repetitive leaping, and extended bouts of repetition during normal behaviors [101]. These mice also exhibited increased anxiety levels according to several behavioral measures [102]. Thus, the D1CT mice present behavioral features with similarity to OCD. The contention that hyper-glutamatergic transmission in cortical regions projecting to striatum drives the observed OCD-like behaviors provides a potential overlap with the putative neural underpinnings of human OCD. Direct measurements of cortico-striatal transmission supporting this contention would solidify the status of D1CT mice as a useful tool in OCD research.
Interestingly, a subsequent study provided evidence for TS-like behaviors in D1CT-7 mice, arguing that these mice may more accurately serve as a model for OCD with comorbid TS [103]. TS-like behaviors were ameliorated by small doses of the noradrenergic agent clonidine, which can have beneficial effects in TS patients [104]. Clonidine alone is not considered an effective treatment for OCD symptoms, and importantly, the response to SSRIs has not been evaluated thus far in D1CT mice.
Hoxb8 Mutant Mice
Hoxb8 is a member of the large Hox (homeobox-containing) family of mammalian transcription factors best known for their role in early development [105]. Intriguingly, Hoxb8 is broadly expressed in adult brain, with expression in the olfactory bulb, basal ganglia, hippocampus, cortex, cerebellum, and brain stem [106]. To address the role of Hoxb8 in the adult brain, Greer and Capecchi [106] created mice homozygous for a loss-of-function allele of Hoxb8. These mice showed excessive grooming that led to hair removal and skin lesions both of self and wild-type cage mates. The excessive grooming was manifest in the absence of any skin abnormalities and developed with 100% penetrance. The authors proposed that the behaviors of Hoxb8 mutant mice mirror those observed for OCD and TTM, an OC-spectrum disorder.
The extensive expression of Hoxb8 in virtually all regions of the CSTC circuit that is disrupted in OCD indicates strong potential for the involvement of Hoxb8 gene disruption in forms of human OCD and/or TTM. Such broad expression of Hoxb8 throughout the adult brain may also account for possible atypical features that could be unrelated to OCD, such as the observed excessive grooming of wild-type cage mates, which in principle could represent abnormal aggression. The Hoxb8 mutant mice have not been tested for response to SSRIs, and the neural basis for their OCD-like behaviors remains entirely unexplored.
5-HT2C Null Mice
The 5-HT2C subtype of the serotonin receptor family is a G-protein coupled receptor (GPCR) that is broadly expressed in the brain and is proposed to mediate many of the central nervous system actions of serotonin, including those underlying the therapeutic benefit of SSRI treatment in OCD [15]. The initial characterization of mice lacking the 5-HT2C receptor indicated that these mice were hyperphagic, leading to obesity, and were prone to spontaneous death from severe seizures [107]. In a much later study, Chou-Green et al. [108] characterized compulsive aspects of 5-HT2C null mouse behavior that were more representative of OCD-like behaviors. These mice exhibit increased chewing of non-nutritive clay, ‘neat’ patterns of chewing on plastic screens, and perseverative head dipping behaviors. The chewing abnormalities have been likened to the human OCD symptom of checking, ordering, smoothing, or washing, whereas perseverative head dipping has been likened to the human symptom of compulsive checking. Despite these similarities and the known involvement of serotonin signaling in the CSTC circuit, the presence of such dominant atypical features (namely obesity and seizure) in the 5-HT2C null mice poses a formidable challenge in applying this model in a context that will be informative specifically to OCD.
DAT Knock-Down Mice
The dopamine transporter (DAT) gene encodes the membrane transporter protein that is responsible for the clearance and recycling of synaptically released dopamine. Based on evidence implicating abnormal dopaminergic neurotransmission and involvement of the DAT protein in schizophrenia and attention deficit hyperactivity disorder (ADHD), Zhuang et al. [109] set out to create a model of these conditions by knock-down (KD) of DAT levels to only 10% of wild-type values. The resulting hyperdopaminergic mutant mice did not exhibit confounding growth defects as did the preexisting DAT null mice, but did recapitulate hyperactivity in response to novelty similar to what was previously observed in DAT null mice. Amphetamine administration resulted in reduced hyperactivity, consistent with the beneficial effects of amphetamine and methylphenidate in the treatment of ADHD, and lending support for the DAT KD mouse as a model for ADHD.
A later study employing hyperdopaminergic DAT KD mice characterized overly-rigid grooming behavior that was referred to as sequential super-stereotypy [110]. Because overly rigid sequential patterns of action, language, and thought are characteristic of the symptoms of OCD and TS, by analogy, the DAT KD mice can be said to model these conditions in a very straightforward sense. However, despite the extensive characterization of sequential super-stereotypy in DAT KD mice, additional studies with DAT KD mice have provided evidence to suggest that behavioral abnormalities in these mice model aspects of mania, bipolar disorder [111], and drug addiction [112] in addition to ADHD, OCD, and TS. Thus, a consideration of the specificity of behavioral features (i.e. lack of atypical features) in addition to the similarity of symptoms with respect to OCD in humans is valuable in assessing face validity. This consideration is particularly relevant in evaluating the 5-HT2C null and DAT KD mice as models for OCD.
The importance of the DAT protein in dopaminergic neurotransmission in the basal ganglia suggests extensive involvement of CSTC circuit abnormalities in sequential super-stereotypy in the DAT KD mouse model. This has been directly demonstrated in recordings from brain slice of DAT KD mice [113]. DAT KO mice were found to have altered cortico-striatal excitatory transmission as well as diminished modulation of synaptic responses by cocaine and amphetamine, which may result from reduced function of D2 receptors in response to chronically elevated dopamine levels in the striatum [113]. The question still remains as to whether these synaptic abnormalities are germane to OCD-like behaviors versus one or more of the various other disorders the DAT KO mice are suggested to model. The effects of SSRI treatment on the reported synaptic alterations and the sequential super-stereotypy have not been investigated.
ArKO Mice
The enzyme aromatase converts androgen to estrogen. Hill et al. [114] eliminated aromatase expression by targeted disruption of the Cyp19 gene, thereby producing estrogen deficient mice (designated ArKO mice). The absence of estrogen in adult male (but not female) mice led to compulsive behaviors including excessive grooming, barbering, and wheel running. ArKO male mice therefore exhibit behaviors that share similarities with symptoms of OCD patients.
The OCD-like behaviors of male ArKO mice are suggested to be caused by dysregulation of estrogen-induced catechol-O-methyltransferase (COMT) expression in the hypothalamus. This disruption is likely to influence dopamine signaling since COMT is a major enzyme regulating dopamine degradation [115]. In contrast, estrogen deficiency had no impact on COMT expression in the frontal cortex of male ArKO mice. This profile of brain region specific regulation of COMT by estrogen in ArKO mice is not in accord with CSTC circuit dysfunction in OCD. Given that some evidence exists to support the involvement of the hypothalamus in grooming and running wheel behaviors [116, 117], it seems plausible that the compulsive behaviors exhibited by male ArKO mice have an etiology that is distinct from human OCD involving CSTC circuit dysfunction. In light of the evidence that low COMT activity is a risk factor in human OCD, specifically in males [17, 118-120], and given that only two brain regions were analyzed in the initial study, perhaps a more detailed evaluation of COMT expression in the brain is warranted in male ArKO mice. Such extension could clarify the neurocircuitry involved and thus resolve what appears to be a mechanistic disconnect.
Estrogen replacement therapy for three weeks attenuated OCD-like compulsive behaviors in male ArKO mice, indicating that developmental defects resulting from the loss of estrogen are not likely to be involved in the observed phenotype. However, this finding does not rule out the involvement of potentially confounding effects related to estrogen depletion in non-neuronal tissues. Overall, the relevance of this model to OCD is promising but at present is not well established. Future assessment of the potential benefits of SSRIs and further clarification of the neurocircuitry involved will be required to extend the validity of the ArKO mouse model of OCD beyond simply face validity.
SAPAP3 Null Mouse Model of OCD
SAP90/PSD95-associated protein 3 (SAPAP3) belongs to a family of four homologous genes encoding SAPAP proteins that are widely yet differentially expressed in the nervous system [95]. The SAPAP family of proteins is found at excitatory synapses and was originally identified as a family of proteins that interacts with postsynaptic density-95 (PSD-95) and Shank family members [121-123]. By virtue of their interactions with each other and with various signaling molecules, these three groups of major scaffolding proteins at the postsynaptic density (PSD) of excitatory synapses are proposed to be central players in the dynamic regulation of glutamatergic synaptic signaling [124].
SAPAP3 is the only family member that is strongly expressed in the striatum [95], thus offering a unique opportunity to explore the specific function of SAPAP3 at glutamatergic synapses in this brain region. Genetic deletion of the SAPAP3 gene in mice caused behavioral abnormalities consisting of increased anxiety and compulsive self-grooming to the point of facial hair loss and skin lesions [96]. These features share similarity with various aspects of symptoms exhibited by human patients with OCD, as demonstrated for other genetic animal models of OCD. Consistent with the predicted function of the SAPAP3 protein, SAPAP3 null mice were also found to have defects in glutamatergic transmission at cortico-striatal synapses (a feature that is only known to be shared by the DAT KD mice thus far, but has been suggested as a central defect for D1CT mice), and remarkably, both synaptic and behavioral defects were rescued by virus-mediated reintroduction of SAPAP3 specifically into the striatum [96]. Further work is necessary to elucidate the exact nature of the glutamatergic synaptic defects in these mice.
Given the high expression and postsynaptic localization of SAPAP3 at glutamatergic synapses in the striatum, these findings fit well within the existing framework of glutamatergic dysfunction in the CSTC circuit in OCD. Finally, the SAPAP3 null mouse model is the only genetic mouse model of OCD for which predictive validity has been established in that chronic administration of fluoxetine successfully alleviated measures of anxiety and compulsive grooming. Thus, the SAPAP3 null mouse model provides significant advancement over the existing genetic animal models of OCD—particularly from the perspective of CSTC circuit dysfunction. With this in mind, the SAPAP3 null mouse model should serve as a useful tool in the screening of novel drugs for the treatment of OCD.
As a final consideration regarding genetic animal models, it will also be critical to tests drugs that have been shown not to be effective in treating OCD, as suggested by Joel [99], as it is well established that SSRIs are only effective in about half of OCD patients. Furthermore, without establishing a drug response profile involving multiple drugs, caution is necessary when interpreting “negative results” of drug screening using animal models of OCD. Investigators using animal models in this manner—particularly in the testing of SSRIs—should also consider implementing criteria to distinguish treatment responders from non-responders, which may lead to a system for specifically exploring treatment resistant OCD in animal models.
Glutamatergic Neurotransmission-Based Pharmacotherapy for OCD and Oc-Spectrum Disorders: Implications for New Drug Development
The recent findings concerning the neural basis of OCD arising from diverse studies validate an intensified focus on glutamatergic synaptic dysfunction within the CSTC circuit. Thus, it is not surprising that numerous glutamatergic neurotransmission-modulating agents are currently being tested in the treatment of OCD. Many of these drugs have previously been approved for treatment of other disorders, which should speed progress in the search for novel OCD treatments since information on tolerability and pharmacokinetics are expected to be readily be available. Table 3 summarizes candidate glutamatergic neurotransmission modulating agents for the treatment of OCD as discussed in detail below.
Table 3. Summary of Candidate Glutamatergic Neurotransmission Modulating Agents for the Treatment of OCD.
| Drug Name | FDA Approval | Mechanism(s) of Action | OCD Studies | Effectiveness |
|---|---|---|---|---|
| riluzole | ALS | reduction of synaptic glutamate release via multiple presynaptic mechanisms; stimulation of glutamate uptake by astocytes | Pittenger et al. 2008 [135] Coric et al. 2005 [130] Grant et al. 2007 [132] Coric et al. 2003 [129] Coric et al. 2007 [134] |
slightly more than half of the 32 total OCD patients treated open-label with riluzole have shown significant improvements |
| topiramate | epilepsy | block of voltage-dependent Na+ and K+ channels; reduction of AMPA/kainate receptor channel conductance; positive modulatory actions on GABAA receptors | Van Ameringen et al. 2006 [137 ] Hollander & Dell'Osso, 2006 [138 ] * Ozkara et al. 2005 [140 ] |
12/17 cases showed improvements; topiramate induced OCD symptoms in one isolated case report |
| lamotrigine | epilepsy, bipolar disorder | inhibition of voltage-dependent Na+ channels to reduce synaptic glutamate release from neurons | Kumar & Khanna, 2000 [139 ] * Alkin et al. 2007 [141 ] * Kuloglu et al. 2008 [142 ] * Kemp et al. 2007 [143 ] |
1/8 OCD patients showed benefit; lamotrigine induced OCD symptoms in 7 cases from 3 other studies |
| N-acetylcysteine | acetaminophen toxicity | stimulation of the glial cystine/glutamate exchanger resulting in activation of mGluR2 receptors which dampens presynaptic glutamate release from neurons during bouts of excessive neuronal activity | Lafleur et al. 2006 [146 ] | a single case report indicates potential benefit as augmenting agent |
| LY354740 | N/A | agonist of type II metabotropic glutamate receptors; dampens presynaptic glutamate release from neurons during bouts of excessive neuronal activity | No studies in OCD patients | N/A |
| memantine | Alzheimer's disease | NMDA-type glutamate receptor open-channel blocker (moderate antagonist relative to dizocilpine) | Pasquini & Biondi, 2006 [150 ] Poyurovsky et al. 2005 [151 ] |
2/3 OCD patients treated with memantine showed improvements |
| amantadine | influenza A virus infection, Parkinson's disease | NMDA-type glutamate receptor open-channel blocker (moderate antagonist relative to dizocilpine) | No studies in OCD patients | N/A |
| dizocilpine (MK-801) | N/A | NMDA-type glutamate receptor open-channel blocker (high-affinity antagonist relative to memantine/amantadine) | No studies in OCD patients | exacerbates OCD symptoms in the D1CT mouse model [154 ] |
| D-cycloserine (DCS) | antibiotic treatment for tuberculosis | partial agonist of NMDA-type glutamate receptors | Wilhelm et al. 2008 [160 ] Kushner et al. 2007 [161 ] ˆ Storch et al. 2007 [162 ] |
optimal DCS administration may improve treatment response when combined with behavioral therapy |
OCD symptoms induced by drug treatments in patients that were not previously diagnosed with OCD, therefore these are not technically OCD studies per se.
This study found no evidence of improvements with DCS treatment, however it was suggested that these findings were due to sub-optimal timing of DCS administration.
N/A, not applicable.
Riluzole – a Glutamatergic Neurotransmission Modulating Agent
Riluzole is one such drug that has received considerable attention in the OCD field in recent years. This drug was previously approved by the FDA for the treatment of amyotrophic lateral sclerosis (ALS) and has a reasonably good record of safety in that context [125]. Riluzole is suspected to reduce glutamate release in the brain via multiple presynaptic mechanisms [126, 127], although it may also act by stimulating glutamate uptake by astrocytes [128]. In the first open-label trial of riluzole augmentation for treatment resistant OCD by Coric et al. [129, 130], 7 out of 13 patients showed improvements on riluzole, with 5 qualifying as treatment responders by additional criteria. Members of this same research group have recently followed-up with another open-label trial of riluzole employing similar design and with an equal number of patients. Again, 7 out of 13 patients showed significant improvements during the course of treatment with riluzole [131]. An open-label trial of riluzole in children with treatment resistant OCD produced qualitatively similar results, with 4 out of 6 patients classified as riluzole treatment responders [132]. Interestingly, a recent case study reported that riluzole was effective in treating the combined symptoms of OCD and pathological/compulsive skin picking (CSP) [133], and another case study found similar benefits in a patient with TTM [134], suggesting that this agent may be effective in treating other OC-spectrum disorders. Taken together, the results of these open-label trials with riluzole have been remarkably consistent, with roughly half of the patients treated thus far experiencing significant treatment response. For a very recent comprehensive review on riluzole's mechanism of action and use in psychiatric conditions see [135].
Anticonvulsant/Antiepileptic Drugs
Topiramate is classified as an anticonvulsant/antiepileptic drug (AED) that is approved by the FDA for treating epilepsy. The structure of topiramate differs from other known AEDs, which may indicate a unique mechanism of action. Proposed mechanisms of action include block of voltage-dependent Na+ and K+ channels, a positive modulatory effect on GABAA receptors, and a negative modulatory effect on AMPA/kainite-type glutamate receptor phosphorylation and ion channel conductance, and these actions may involve neurons and astrocytes [136]. One or more of these mechanisms may account for the anti-glutamatergic properties of topiramate. In an open-label retrospective study of topiramate augmentation in treatment resistant OCD, 11 of the 16 patients analyzed showed improvement in OCD symptoms [137]. An additional single case report for topiramate plus the SSRI paroxetine in treatment resistant OCD also supports the potential beneficial effects of this drug [138]. Lamotrigine is also an AED that has been approved by the FDA for the treatment of epilepsy and bipolar disorder. Evidence to merit the use of lamotrigine as an augmenting agent in OCD treatment is less convincing than for topiramate. In a case study of 8 OCD patients that previously failed to respond to SSRIs alone, augmentation with lamotrigine only benefited one patient [139]. It should also be noted there have been case reports for both topiramate and lamotrigine potentially causing OCD-like symptoms [140-143].
mGluR2 Targeting Agents
One of the more promising avenues to explore will be drugs that modulate metabotropic glutamate receptors (mGluRs). In particular, mGluR2 (which is strongly expressed in the forebrain) has extra- or peri-synaptic localization on presynaptic membranes of excitatory axons and function as an autoreceptor [144]. Thus, activation of mGluR2 on excitatory neurons negatively modulates neurotransmitter release. Importantly, excessive neuronal activity leading to glutamate spillover seems to be required for activation of mGluR2, suggesting that its function may be to protect against pathological bouts of glutamatergic hyperexcitability [144]. Two agents aimed at modulating mGluR2 function by distinct routes have recently been proposed for the treatment of OCD.
N-acetylcysteine (NAC) is an amino acid derivative that stimulates the glial cystine/glutamate exchanger and leads to elevated extracellular glutamate levels. This rise in glutamate levels in the extrasynaptic space activates inhibitory mGluR2 receptors that dampen synaptic release of glutamate from neurons [145]. This mechanism of action prompted a case study of NAC in combination with the SSRI fluvoxamine in treatment-refractory OCD that yielded evidence for significant improvement in OCD symptoms [146]. Larger, well controlled studies are needed to further explore the potential of NAC in treating OCD symptoms. Since elevating extracellular/extrasynaptic glutamate by stimulating cystine exchange could also activate NMDA receptors similar to spillover effects of synaptically released glutamate [147], this aspect should not be overlooked in searching for the appropriate therapeutic dose of NAC.
LY354740 is an mGluR2/3 (type II) agonist. This drug showed potential in treating generalized anxiety disorders in humans, which might have relevance to OCD, but evidence to support this claim are restricted almost entirely to results from animal models of anxiety [148]. In contrast, the mGluR 2/3 antagonists MGS0039 and LY341495 have been shown to inhibit marble burying in mice (considered by some to represent OCD-like behaviors) to a similar extent as SSRIs [149]. The agonist LY354740 attenuated this effect but did not affect marble burying behavior on its own. Clearly, further preclinical work is needed to clarify the therapeutic potential of mGluR2/3 agonists and antagonists in the context of OCD. Testing these drugs in a better established animal model for OCD-like behaviors will help to dissociate effects on compulsive behaviors versus generalized anxiety.
NMDA-Type Glutamate Receptor Targeting Agents
Memantine is an NMDA receptor open channel blocker (antagonist) that has received FDA approval for the treatment of Alzheimer's disease (AD). Attractive features of memantine action include its low affinity and voltage-dependence of NMDA receptor blockade, which may permit low level NMDA receptor activation during normal bouts of synaptic activity. Evidence from a small number of case studies supported a positive effect of memantine in some patients with treatment refractory OCD [150, 151]; however, other attempts to treat a limited number of OCD patients with memantine have proved unsuccessful [51].
Amantadine is structurally and functionally related to memantine and has recently been shown to inhibit marble burying in mice [152]. The overall effectiveness of amantadine in this model was more favorable than memantine and riluzole. This result led the authors to propose the use of amantadine in the treatment of OCD. Amantadine has not been tested in OCD but has been approved by the FDA as an antiviral medication and is used in the treatment of Parkinson's disease (PD). Curiously, an in vitro study comparing amantadine and memantine action in neurons from different brain regions suggested that at therapeutically relevant doses amantadine is more active in the striatum, whereas memantine is more active in other brain structures [153]. The basis for this functional dichotomy is unknown, but nonetheless may hint at greater potential for amantadine over memantine in treating OCD.
Dizocilpine (also called MK-801) is another NMDA receptor open pore blocker. This drug seems to exacerbate rather than ameliorate OCD symptoms based on tests in the D1CT mice [154]. This result may reflect elevations in forebrain glutamate by dizocilpine similar to a mechanism described for phencyclidine [155], but which seems paradoxical for a glutamate receptor antagonist. This effect is most likely mediated by block of NMDA receptors located on GABAergic interneurons that normally suppress forebrain glutamate release from excitatory neurons [154]. No such effect has been described for memantine or amantadine, although the possibility should be explored. Dizocilpine action at NMDA receptors differs significantly from memantine and amantadine (e.g. trapping versus partial-trapping channel block [156]) making it a more potent NMDA receptor antagonist that drastically interferes with brain function.
D-cycloserine (DCS) is a partial agonist of the NMDA receptor [157, 158]. Since NMDA receptor activation is critical for learning and memory, DCS is regarded as a cognitive enhancer. DCS facilitates amygdala-dependent fear extinction (a process requiring NMDA receptor dependent long-term potentiation) [159], suggesting that it may be helpful in conjunction with extinction-based behavioral therapies for human OCD patients. Three trials examining DCS augmentation of behavioral therapy in OCD have been completed. Two studies demonstrated more rapid or more pronounced treatment response with DCS [160], at least for initial sessions [161], while the third study did not find evidence of improvements with DCS [162]. The negative result of Storch et al. [162] may have resulted from high DCS doses or sub-optimal timing of DCS administration with respect to the behavioral therapy [163].
While not all glutamatergic neurotransmission-modulating agents have proven beneficial in early trials for the treatment of OCD, the majority of these agents do seem to influence symptom severity (i.e. ameliorate or exacerbate), indicating a critical involvement of the glutamatergic neurotransmitter system in OCD. However, at this early stage the evidence surrounding even the most promising glutamatergic neurotransmission-modulating agent riluzole consists of just a few dozen OCD cases. No strong conclusions should be drawn about the usefulness of riluzole (or any other such drug) for the treatment of OCD prior to the completion of properly controlled, large-scale clinical trials.
Summary
In recent years, diverse lines of evidence have converged to implicate glutamatergic synaptic dysfunction within the CSTC brain circuit in the etiology of OCD. Genetic studies on the glutamatergic system, though relatively few thus far, have consistently supported this model and may ultimately establish the causal link(s) in OCD. These findings have led to new targets for the treatment of this often difficult to treat disorder, and the initial results with several agents that modulate the glutamatergic neurotransmitter system have been promising—suggesting perhaps that more efficient treatment options for OCD may soon be attainable. Most of the agents in testing are believed to have the net effect of reducing glutamate output or signaling, which may serve to normalize pathologically elevated glutamatergic transmission in the CSTC circuit in OCD. Alternatively, DCS acts as an enhancer of glutamatergic signaling through NMDA receptors, and is likely to exert its effects through actions on the fear extinction circuit of the amygdala that controls anxiety. These interpretations are tentative at this early stage, but may provide useful insights for the development of novel glutamatergic neurotransmission-modulating agents. The implementation of animal models of OCD for preclinical drug screening can significantly speed progress in this area, and although the lack of demonstrated predictive validity has been a major criticism of OCD models in recent years, at least one genetic animal model has emerged that has both predictive validity and has provided clear evidence for the involvement of CSTC circuit dysfunction. Utilizing such tools to explore and define the synaptic basis of OCD should greatly advance our knowledge concerning a broad spectrum of poorly understood human disorders.
Acknowledgments
We are grateful to Molly Heyer for critically reading the manuscript. The work performed in the laboratory of the authors is supported by a McKnight Neuroscience of Brain Disorders Award, a Hartwell Foundation Individual Bio-medical Research Award, and grants from the National Institutes of Health.
References
- 1.Diagnostics and Statistical Manual of Mental Health Disorders. 4th. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 2.Math SB, Janardhan RYC. Issues in the pharmacological treatment of obsessive-compulsive disorder. Int J Clin Pract. 2007;61(7):1188–97. doi: 10.1111/j.1742-1241.2007.01356.x. [DOI] [PubMed] [Google Scholar]
- 3.Karno M, Golding JM, Sorenson SB, Burnam MA. The epidemiology of obsessive-compulsive disorder in five US communities. Arch Gen Psychiatry. 1988;45(12):1094–9. doi: 10.1001/archpsyc.1988.01800360042006. [DOI] [PubMed] [Google Scholar]
- 4.Weissman MM, Bland RC, Canino GJ, et al. The cross national epidemiology of obsessive compulsive disorder. The Cross National Collaborative Group. J Clin Psychiatry. 1994;55(Suppl):5–10. [PubMed] [Google Scholar]
- 5.Rasmussen SA, Eisen JL. The epidemiology and differential diagnosis of obsessive compulsive disorder. J Clin Psychiatry. 1994;55(Suppl):5–10. discussion 1-4. [PubMed] [Google Scholar]
- 6.DuPont RL, Rice DP, Shiraki S, Rowland CR. Economic costs of obsessive-compulsive disorder. Med Interface. 1995;8(4):102–9. [PubMed] [Google Scholar]
- 7.Inouye E. Similar and dissimilar manifestations of obsessive-compulsive neuroses in monozygotic twins. Am J Psychiatry. 1965;121:1171–5. doi: 10.1176/ajp.121.12.1171. [DOI] [PubMed] [Google Scholar]
- 8.van Grootheest DS, Cath DC, Beekman AT, Boomsma DI. Twin studies on obsessive-compulsive disorder: a review. Twin Res Hum Genet. 2005;8(5):450–8. doi: 10.1375/183242705774310060. [DOI] [PubMed] [Google Scholar]
- 9.Grados MA, Walkup J, Walford S. Genetics of obsessive-compulsive disorders: new findings and challenges. Brain Dev. 2003;25(Suppl 1):S55–61. doi: 10.1016/s0387-7604(03)90010-6. [DOI] [PubMed] [Google Scholar]
- 10.Phillips KA. The obsessive-compulsive spectrums. Psychiatr Clin North Am. 2002;25(4):791–809. doi: 10.1016/s0193-953x(02)00024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stein DJ. Neurobiology of the obsessive-compulsive spectrum disorders. Biol Psychiatry. 2000;47(4):296–304. doi: 10.1016/s0006-3223(99)00271-1. [DOI] [PubMed] [Google Scholar]
- 12.Dell'Osso B, Altamura AC, Mundo E, Marazziti D, Hollander E. Diagnosis and treatment of obsessive-compulsive disorder and related disorders. Int J Clin Pract. 2007;61(1):98–104. doi: 10.1111/j.1742-1241.2006.01167.x. [DOI] [PubMed] [Google Scholar]
- 13.Kaplan A, Hollander E. A review of pharmacologic treatments for obsessive-compulsive disorder. Psychiatr Serv. 2003;54(8):1111–8. doi: 10.1176/appi.ps.54.8.1111. [DOI] [PubMed] [Google Scholar]
- 14.Jenike MA. Clinical practice. Obsessive-compulsive disorder. N Engl J Med. 2004;350(3):259–65. doi: 10.1056/NEJMcp031002. [DOI] [PubMed] [Google Scholar]
- 15.Goddard AW, Shekhar A, Whiteman AF, McDougle CJ. Serotoninergic mechanisms in the treatment of obsessive-compulsive disorder. Drug Discov Today. 2008;13(78):325–32. doi: 10.1016/j.drudis.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Barr LC, Goodman WK, McDougle CJ, et al. Tryptophan depletion in patients with obsessive-compulsive disorder who respond to serotonin reuptake inhibitors. Arch Gen Psychiatry. 1994;51(4):309–17. doi: 10.1001/archpsyc.1994.03950040053007. [DOI] [PubMed] [Google Scholar]
- 17.Denys D. Pharmacotherapy of obsessive-compulsive disorder and obsessive-compulsive spectrum disorders. Psychiatr Clin North Am. 2006;29(2):553–84. xi. doi: 10.1016/j.psc.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Koran LM, Hanna GL, Hollander E, Nestadt G, Simpson HB. Practice guideline for the treatment of patients with obsessive-compulsive disorder. Am J Psychiatry. 2007;164(7 Suppl):5–53. [PubMed] [Google Scholar]
- 19.Meyer V. Modification of expectations in cases with obsessional rituals. Behav Res Ther. 1966;4(4):273–80. doi: 10.1016/0005-7967(66)90023-4. [DOI] [PubMed] [Google Scholar]
- 20.Franklin ME, Abramowitz JS, Kozak MJ, Levitt JT, Foa EB. Effectiveness of exposure and ritual prevention for obsessive-compulsive disorder: randomized compared with nonrandomized samples. J Consult Clin Psychol. 2000;68(4):594–602. [PubMed] [Google Scholar]
- 21.Abramowitz JS. The psychological treatment of obsessive-compulsive disorder. Can J Psychiatry. 2006;51(7):407–16. doi: 10.1177/070674370605100702. [DOI] [PubMed] [Google Scholar]
- 22.Foa EB, Liebowitz MR, Kozak MJ, et al. Randomized, placebo-controlled trial of exposure and ritual prevention, clomipramine, and their combination in the treatment of obsessive-compulsive disorder. Am J Psychiatry. 2005;162(1):151–61. doi: 10.1176/appi.ajp.162.1.151. [DOI] [PubMed] [Google Scholar]
- 23.Kobak KA, Greist JH, Jefferson JW, Katzelnick DJ, Henk HJ. Behavioral versus pharmacological treatments of obsessive compulsive disorder: a meta-analysis. Psychopharmacology (Berl) 1998;136(3):205–16. doi: 10.1007/s002130050558. [DOI] [PubMed] [Google Scholar]
- 24.Simpson HB, Liebowitz MR, Foa EB, et al. Post-treatment effects of exposure therapy and clomipramine in obsessive-compulsive disorder. Depress Anxiety. 2004;19(4):225–33. doi: 10.1002/da.20003. [DOI] [PubMed] [Google Scholar]
- 25.Steketee G, Frost R. Compulsive hoarding: current status of the research. Clin Psychol Rev. 2003;23(7):905–27. doi: 10.1016/j.cpr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 26.POTS-Team. Cognitive-behavior therapy, sertraline, and their combination for children and adolescents with obsessive-compulsive disorder: the Pediatric OCD Treatment Study (POTS) randomized controlled trial. JAMA. 2004;292(16):1969–76. doi: 10.1001/jama.292.16.1969. [DOI] [PubMed] [Google Scholar]
- 27.Penney JB, Jr, Young AB. Speculations on the functional anatomy of basal ganglia disorders. Annu Rev Neurosci. 1983;6:73–94. doi: 10.1146/annurev.ne.06.030183.000445. [DOI] [PubMed] [Google Scholar]
- 28.Graybiel AM. Neurochemically specified subsystems in the basal ganglia. Ciba Found Symp. 1984;107:114–49. doi: 10.1002/9780470720882.ch7. [DOI] [PubMed] [Google Scholar]
- 29.Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci. 1986;9:357–81. doi: 10.1146/annurev.ne.09.030186.002041. [DOI] [PubMed] [Google Scholar]
- 30.Parent A, Hazrati LN. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Brain Res Rev. 1995;20(1):128–54. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- 31.Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53(2):647–54. doi: 10.1016/s0022-3999(02)00428-2. [DOI] [PubMed] [Google Scholar]
- 32.Gerfen CR. Basal ganglia. In: Paxinos G, editor. The Rat Nervous System. 3rd. San Diego: Elsevier Academic Press; 2004. pp. 455–508. [Google Scholar]
- 33.Graybiel AM. The basal ganglia. Curr Biol. 2000;10(14):R509–11. doi: 10.1016/s0960-9822(00)00593-5. [DOI] [PubMed] [Google Scholar]
- 34.Saxena S, Rauch SL. Functional neuroimaging and the neuroanatomy of obsessive-compulsive disorder. Psychiatr Clin North Am. 2000;23(3):563–86. doi: 10.1016/s0193-953x(05)70181-7. [DOI] [PubMed] [Google Scholar]
- 35.Baxter LR, Jr, Phelps ME, Mazziotta JC, Guze BH, Schwartz JM, Selin CE. Local cerebral glucose metabolic rates in obsessive-compulsive disorder. A comparison with rates in unipolar depression and in normal controls. Arch Gen Psychiatry. 1987;44(3):211–8. doi: 10.1001/archpsyc.1987.01800150017003. [DOI] [PubMed] [Google Scholar]
- 36.Baxter LR, Jr, Schwartz JM, Mazziotta JC, et al. Cerebral glucose metabolic rates in nondepressed patients with obsessive-compulsive disorder. Am J Psychiatry. 1988;145(12):1560–3. doi: 10.1176/ajp.145.12.1560. [DOI] [PubMed] [Google Scholar]
- 37.Swedo SE, Schapiro MB, Grady CL, et al. Cerebral glucose metabolism in childhood-onset obsessive-compulsive disorder. Arch Gen Psychiatry. 1989;46(6):518–23. doi: 10.1001/archpsyc.1989.01810060038007. [DOI] [PubMed] [Google Scholar]
- 38.Sawle GV, Hymas NF, Lees AJ, Frackowiak RS. Obsessional slowness. Functional studies with positron emission tomography. Brain. 1991;114(Pt 5):2191–202. doi: 10.1093/brain/114.5.2191. [DOI] [PubMed] [Google Scholar]
- 39.Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62–70. doi: 10.1001/archpsyc.1994.03950010062008. [DOI] [PubMed] [Google Scholar]
- 40.McGuire PK, Bench CJ, Frith CD, Marks IM, Frackowiak RS, Dolan RJ. Functional anatomy of obsessive-compulsive phenomena. Br J Psychiatry. 1994;164(4):459–68. doi: 10.1192/bjp.164.4.459. [DOI] [PubMed] [Google Scholar]
- 41.Benkelfat C, Nordahl TE, Semple WE, King AC, Murphy DL, Cohen RM. Local cerebral glucose metabolic rates in obsessive-compulsive disorder. Patients treated with clomipramine. Arch Gen Psychiatry. 1990;47(9):840–8. doi: 10.1001/archpsyc.1990.01810210048007. [DOI] [PubMed] [Google Scholar]
- 42.Swedo SE, Pietrini P, Leonard HL, et al. Cerebral glucose metabolism in childhood-onset obsessive-compulsive disorder. Revisuali-zation during pharmacotherapy. Arch Gen Psychiatry. 1992;49(9):690–4. doi: 10.1001/archpsyc.1992.01820090018003. [DOI] [PubMed] [Google Scholar]
- 43.Baxter LR, Jr, Schwartz JM, Bergman KS, et al. Caudate glucose metabolic rate changes with both drug and behavior therapy for obsessive-compulsive disorder. Arch Gen Psychiatry. 1992;49(9):681–9. doi: 10.1001/archpsyc.1992.01820090009002. [DOI] [PubMed] [Google Scholar]
- 44.Perani D, Colombo C, Bressi S, et al. [18F]FDG PET study in obsessive-compulsive disorder. A clinical/metabolic correlation study after treatment. Br J Psychiatry. 1995;166(2):244–50. doi: 10.1192/bjp.166.2.244. [DOI] [PubMed] [Google Scholar]
- 45.Saxena S, Brody AL, Maidment KM, et al. Localized orbitofrontal and subcortical metabolic changes and predictors of response to paroxetine treatment in obsessive-compulsive disorder. Neuropsychopharmacology. 1999;21(6):683–93. doi: 10.1016/S0893-133X(99)00082-2. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz JM, Stoessel PW, Baxter LR, Jr, Martin KM, Phelps ME. Systematic changes in cerebral glucose metabolic rate after successful behavior modification treatment of obsessive-compulsive disorder. Arch Gen Psychiatry. 1996;53(2):109–13. doi: 10.1001/archpsyc.1996.01830020023004. [DOI] [PubMed] [Google Scholar]
- 47.Mindus P, Nyman H. Normalization of personality characteristics in patients with incapacitating anxiety disorders after capsulotomy. Acta Psychiatr Scand. 1991;83(4):283–91. doi: 10.1111/j.1600-0447.1991.tb05541.x. [DOI] [PubMed] [Google Scholar]
- 48.Ebert D, Speck O, Konig A, Berger M, Hennig J, Hohagen F. 1H-magnetic resonance spectroscopy in obsessive-compulsive disorder: evidence for neuronal loss in the cingulate gyrus and the right striatum. Psychiatry Res. 1997;74(3):173–6. doi: 10.1016/s0925-4927(97)00016-4. [DOI] [PubMed] [Google Scholar]
- 49.Bartha R, Stein MB, Williamson PC, et al. A short echo 1H spectroscopy and volumetric MRI study of the corpus striatum in patients with obsessive-compulsive disorder and comparison subjects. Am J Psychiatry. 1998;155(11):1584–91. doi: 10.1176/ajp.155.11.1584. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg DR, Keshavan MS. A.E. Bennett Research Award. Toward a neurodevelopmental model of of obsessive--compulsive disorder. Biol Psychiatry. 1998;43(9):623–40. doi: 10.1016/s0006-3223(97)00443-5. [DOI] [PubMed] [Google Scholar]
- 51.Pittenger C, Krystal JH, Coric V. Glutamate-modulating drugs as novel pharmacotherapeutic agents in the treatment of obsessive-compulsive disorder. NeuroRx. 2006;3(1):69–81. doi: 10.1016/j.nurx.2005.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baxter LR, Jr, Saxena S, Brody AL, et al. Brain Mediation of Obsessive-Compulsive Disorder Symptoms: Evidence From Functional Brain Imaging Studies in the Human and Nonhuman Primate. Semin Clin Neuropsychiatry. 1996;1(1):32–47. doi: 10.1053/SCNP00100032. [DOI] [PubMed] [Google Scholar]
- 53.Modell JG, Mountz JM, Curtis GC, Greden JF. Neurophysiologic dysfunction in basal ganglia/limbic striatal and thalamocortical circuits as a pathogenetic mechanism of obsessive-compulsive disorder. J Neuropsychiatry Clin Neurosci. 1989;1(1):27–36. doi: 10.1176/jnp.1.1.27. [DOI] [PubMed] [Google Scholar]
- 54.Insel TR, Winslow JT. Neurobiology of obsessive compulsive disorder. Psychiatr Clin North Am. 1992;15(4):813–24. [PubMed] [Google Scholar]
- 55.Graybiel AM. The basal ganglia and chunking of action repertoires. Neurobiol Learn Mem. 1998;70(12):119–36. doi: 10.1006/nlme.1998.3843. [DOI] [PubMed] [Google Scholar]
- 56.Menzies L, Chamberlain SR, Laird AR, Thelen SM, Sahakian BJ, Bullmore ET. Integrating evidence from neuroimaging and neuropsychological studies of obsessive-compulsive disorder: the orbitofronto-striatal model revisited. Neurosci Biobehav Rev. 2008;32(3):525–49. doi: 10.1016/j.neubiorev.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aouizerate B, Guehl D, Cuny E, et al. Pathophysiology of obsessive-compulsive disorder: a necessary link between phenomenology, neuropsychology, imagery and physiology. Prog Neurobiol. 2004;72(3):195–221. doi: 10.1016/j.pneurobio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 58.Mindus, Jenike MA. Neurosurgical treatment of malignant obsessive compulsive disorder. Psychiatr Clin North Am. 1992;15(4):921–38. [PubMed] [Google Scholar]
- 59.Nuttin BJ, Gabriels LA, Cosyns PR, et al. Long-term electrical capsular stimulation in patients with obsessive-compulsive disorder. Neurosurgery. 2003;52(6):1263–72. doi: 10.1227/01.neu.0000064565.49299.9a. discussion 72-4. [DOI] [PubMed] [Google Scholar]
- 60.Abelson JL, Curtis GC, Sagher O, et al. Deep brain stimulation for refractory obsessive-compulsive disorder. Biol Psychiatry. 2005;57(5):510–6. doi: 10.1016/j.biopsych.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 61.Sturm V, Lenartz D, Koulousakis A, et al. The nucleus accumbens: a target for deep brain stimulation in obsessive-compulsive- and anxiety-disorders. J Chem Neuroanat. 2003;26(4):293–9. doi: 10.1016/j.jchemneu.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 62.Aouizerate B, Martin-Guehl C, Cuny E, et al. Deep brain stimulation for OCD and major depression. Am J Psychiatry. 2005;162(11):2192. doi: 10.1176/appi.ajp.162.11.2192. [DOI] [PubMed] [Google Scholar]
- 63.McCracken CB, Grace AA. High-frequency deep brain stimulation of the nucleus accumbens region suppresses neuronal activity and selectively modulates afferent drive in rat orbitofrontal cortex in vivo. J Neurosci. 2007;27(46):12601–10. doi: 10.1523/JNEUROSCI.3750-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laplane D, Levasseur M, Pillon B, et al. Obsessive-compulsive and other behavioural changes with bilateral basal ganglia lesions. A neuropsychological, magnetic resonance imaging and positron tomography study. Brain. 1989;112(Pt 3):699–725. doi: 10.1093/brain/112.3.699. [DOI] [PubMed] [Google Scholar]
- 65.Chacko RC, Corbin MA, Harper RG. Acquired obsessive-compulsive disorder associated with basal ganglia lesions. J Neuropsychiatry Clin Neurosci. 2000;12(2):269–72. doi: 10.1176/jnp.12.2.269. [DOI] [PubMed] [Google Scholar]
- 66.Snider LA, Swedo SE. PANDAS: current status and directions for research. Mol Psychiatry. 2004;9(10):900–7. doi: 10.1038/sj.mp.4001542. [DOI] [PubMed] [Google Scholar]
- 67.Swedo SE, Rapoport JL, Cheslow DL, et al. High prevalence of obsessive-compulsive symptoms in patients with Sydenham's chorea. Am J Psychiatry. 1989;146(2):246–9. doi: 10.1176/ajp.146.2.246. [DOI] [PubMed] [Google Scholar]
- 68.Asbahr FR, Negrao AB, Gentil V, et al. Obsessive-compulsive and related symptoms in children and adolescents with rheumatic fever with and without chorea: a prospective 6-month study. Am J Psychiatry. 1998;155(8):1122–4. doi: 10.1176/ajp.155.8.1122. [DOI] [PubMed] [Google Scholar]
- 69.Hounie AG, Pauls DL, Mercadante MT, et al. Obsessive-compulsive spectrum disorders in rheumatic fever with and without Sydenham's chorea. J Clin Psychiatry. 2004;65(7):994–9. doi: 10.4088/jcp.v65n0717. [DOI] [PubMed] [Google Scholar]
- 70.Swedo SE, Leonard HL, Garvey M, et al. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: clinical description of the first 50 cases. Am J Psychiatry. 1998;155(2):264–71. doi: 10.1176/ajp.155.2.264. [DOI] [PubMed] [Google Scholar]
- 71.Szeszko PR, Robinson D, Alvir JM, et al. Orbital frontal and amygdala volume reductions in obsessive-compulsive disorder. Arch Gen Psychiatry. 1999;56(10):913–9. doi: 10.1001/archpsyc.56.10.913. [DOI] [PubMed] [Google Scholar]
- 72.van den Heuvel OA, Veltman DJ, Groenewegen HJ, et al. Amygdala activity in obsessive-compulsive disorder with contamination fear: a study with oxygen-15 water positron emission tomography. Psychiatry Res. 2004 Dec 30;132(3):225–37. doi: 10.1016/j.pscychresns.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 73.Cannistraro PA, Wright CI, Wedig MM, et al. Amygdala responses to human faces in obsessive-compulsive disorder. Biol Psychiatry. 2004;56(12):916–20. doi: 10.1016/j.biopsych.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 74.Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17(5):208–14. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 75.LeDoux J. Fear and the brain: where have we been, and where are we going? Biol Psychiatry. 1998;44(12):1229–38. doi: 10.1016/s0006-3223(98)00282-0. [DOI] [PubMed] [Google Scholar]
- 76.Husted DS, Shapira NA, Goodman WK. The neurocircuitry of obsessive-compulsive disorder and disgust. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(3):389–99. doi: 10.1016/j.pnpbp.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 77.Hemmings SM, Stein DJ. The current status of association studies in obsessive-compulsive disorder. Psychiatr Clin North Am. 2006;29(2):411–44. doi: 10.1016/j.psc.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 78.Chakrabarty K, Bhattacharyya S, Christopher R, Khanna S. Glutamatergic dysfunction in OCD. Neuropsychopharmacology. 2005;30(9):1735–40. doi: 10.1038/sj.npp.1300733. [DOI] [PubMed] [Google Scholar]
- 79.Arnold PD, Rosenberg DR, Mundo E, Tharmalingam S, Kennedy JL, Richter MA. Association of a glutamate (NMDA) subunit receptor gene (GRIN2B) with obsessive-compulsive disorder: a preliminary study. Psychopharmacology (Berl) 2004;174(4):530–8. doi: 10.1007/s00213-004-1847-1. [DOI] [PubMed] [Google Scholar]
- 80.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11(3):327–35. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 81.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8(6):413–26. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 82.Rauch SL, Savage CR, Alpert NM, et al. Probing striatal function in obsessive-compulsive disorder: a PET study of implicit sequence learning. J Neuropsychiatry Clin Neurosci. 1997;9(4):568–73. doi: 10.1176/jnp.9.4.568. [DOI] [PubMed] [Google Scholar]
- 83.Arnold PD, Sicard T, Burroughs E, Richter MA, Kennedy JL. Glutamate transporter gene SLC1A1 associated with obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63(7):769–76. doi: 10.1001/archpsyc.63.7.769. [DOI] [PubMed] [Google Scholar]
- 84.Dickel DE, Veenstra-VanderWeele J, Cox NJ, et al. Association testing of the positional and functional candidate gene SLC1A1/EAAC1 in early-onset obsessive-compulsive disorder. Arch Gen Psychiatry. 2006;63(7):778–85. doi: 10.1001/archpsyc.63.7.778. [DOI] [PubMed] [Google Scholar]
- 85.Hanna GL, Veenstra-VanderWeele J, Cox NJ, et al. Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. Am J Med Genet. 2002;114(5):541–52. doi: 10.1002/ajmg.10519. [DOI] [PubMed] [Google Scholar]
- 86.Willour VL, Yao Shugart Y, Samuels J, et al. Replication study supports evidence for linkage to 9p24 in obsessive-compulsive disorder. Am J Hum Genet. 2004;75(3):508–13. doi: 10.1086/423899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanai Y, Hediger MA. The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 2004;447(5):469–79. doi: 10.1007/s00424-003-1146-4. [DOI] [PubMed] [Google Scholar]
- 88.Rothstein JD, Martin L, Levey AI, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13(3):713–25. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 89.Stewart SE, Fagerness JA, Platko J, et al. Association of the SLC1A1 glutamate transporter gene and obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(8):1027–33. doi: 10.1002/ajmg.b.30533. [DOI] [PubMed] [Google Scholar]
- 90.Veenstra-VanderWeele J, Kim SJ, Gonen D, Hanna GL, Leventhal BL, Cook EH., Jr Genomic organization of the SLC1A1/EAAC1 gene and mutation screening in early-onset obsessive-compulsive disorder. Mol Psychiatry. 2001;6(2):160–7. doi: 10.1038/sj.mp.4000806. [DOI] [PubMed] [Google Scholar]
- 91.Delorme R, Krebs MO, Chabane N, et al. Frequency and transmission of glutamate receptors GRIK2 and GRIK3 polymorphisms in patients with obsessive compulsive disorder. Neuroreport. 2004;15(4):699–702. doi: 10.1097/00001756-200403220-00025. [DOI] [PubMed] [Google Scholar]
- 92.Shuang M, Liu J, Jia MX, et al. Family-based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am J Med Genet B Neuropsychiatr Genet. 2004;131B(1):48–50. doi: 10.1002/ajmg.b.30025. [DOI] [PubMed] [Google Scholar]
- 93.Jamain S, Betancur C, Quach H, et al. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry. 2002;7(3):302–10. doi: 10.1038/sj.mp.4000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim SA, Kim JH, Park M, Cho IH, Yoo HJ. Family-based association study between GRIK2 polymorphisms and autism spectrum disorders in the Korean trios. Neurosci Res. 2007;58(3):332–5. doi: 10.1016/j.neures.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 95.Welch JM, Wang D, Feng G. Differential mRNA expression and protein localization of the SAP90/PSD-95-associated proteins (SA-PAPs) in the nervous system of the mouse. J Comp Neurol. 2004;472(1):24–39. doi: 10.1002/cne.20060. [DOI] [PubMed] [Google Scholar]
- 96.Welch JM, Lu J, Rodriguiz RM, et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature. 2007;448(7156):894–900. doi: 10.1038/nature06104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Züchner SWJ, Ashley-Koch AE, Collins AL, et al. Multiple SAPAP3 missense variants in trichotillimania and OCD. Mol Psychiatry. 2008 doi: 10.1038/mp.2008.83. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bienvenu OJ, W Y, Shugart YY, Welch JW, et al. Sapap3 gene appears associated with pathological grooming in humans. Am J Med Genet Part B Neuropsychiatr Genet. 2008 doi: 10.1002/ajmg.b.30897. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joel D. Current animal models of obsessive compulsive disorder: a critical review. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(3):374–88. doi: 10.1016/j.pnpbp.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 100.Korff S, Harvey BH. Animal models of obsessive-compulsive disorder: rationale to understanding psychobiology and pharmacology. Psychiatr Clin North Am. 2006;29(2):371–90. doi: 10.1016/j.psc.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 101.Campbell KM, de Lecea L, Severynse DM, et al. OCD-Like behaviors caused by a neuropotentiating transgene targeted to cortical and limbic D1+ neurons. J Neurosci. 1999;19(12):5044–53. doi: 10.1523/JNEUROSCI.19-12-05044.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.McGrath MJ, Campbell KM, Veldman MB, Burton FH. Anxiety in a transgenic mouse model of cortical-limbic neuro-potentiated compulsive behavior. Behav Pharmacol. 1999;10(5):435–43. doi: 10.1097/00008877-199909000-00001. [DOI] [PubMed] [Google Scholar]
- 103.Nordstrom EJ, Burton FH. A transgenic model of comorbid Tourette's syndrome and obsessive-compulsive disorder circuitry. Mol Psychiatry. 2002;7(6):617–25. 524. doi: 10.1038/sj.mp.4001144. [DOI] [PubMed] [Google Scholar]
- 104.Cohen DJ, Young JG, Nathanson JA, Shaywitz BA. Clonidine in Tourette's syndrome. Lancet. 1979;2(8142):551–3. doi: 10.1016/s0140-6736(79)91614-3. [DOI] [PubMed] [Google Scholar]
- 105.Capecchi MR. Hox genes and mammalian development. Cold Spring Harb Symp Quant Biol. 1997;62:273–81. [PubMed] [Google Scholar]
- 106.Greer JM, Capecchi MR. Hoxb8 is required for normal grooming behavior in mice. Neuron. 2002;33(1):23–34. doi: 10.1016/s0896-6273(01)00564-5. [DOI] [PubMed] [Google Scholar]
- 107.Tecott LH, Sun LM, Akana SF, et al. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature. 1995;374(6522):542–6. doi: 10.1038/374542a0. [DOI] [PubMed] [Google Scholar]
- 108.Chou-Green JM, Holscher TD, Dallman MF, Akana SF. Compulsive behavior in the 5-HT2C receptor knockout mouse. Physiol Behav. 2003;78(45):641–9. doi: 10.1016/s0031-9384(03)00047-7. [DOI] [PubMed] [Google Scholar]
- 109.Zhuang X, Oosting RS, Jones SR, et al. Hyperactivity and impaired response habituation in hyperdopaminergic mice. Proc Natl Acad Sci USA. 2001;98(4):1982–7. doi: 10.1073/pnas.98.4.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berridge KC, Aldridge JW, Houchard KR, Zhuang X. Sequential super-stereotypy of an instinctive fixed action pattern in hyperdopaminergic mutant mice: a model of obsessive compulsive disorder and Tourette's. BMC Biol. 2005;3:4. doi: 10.1186/1741-7007-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ralph-Williams RJ, Paulus MP, Zhuang X, Hen R, Geyer MA. Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biol Psychiatry. 2003;53(4):352–9. doi: 10.1016/s0006-3223(02)01489-0. [DOI] [PubMed] [Google Scholar]
- 112.Pecina S, Cagniard B, Berridge KC, Aldridge JW, Zhuang X. Hyperdopaminergic mutant mice have higher “wanting” but not “liking” for sweet rewards. J Neurosci. 2003;23(28):9395–402. doi: 10.1523/JNEUROSCI.23-28-09395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu N, Cepeda C, Zhuang X, Levine MS. Altered corticostriatal neurotransmission and modulation in dopamine transporter knockdown mice. J Neurophysiol. 2007;98(1):423–32. doi: 10.1152/jn.00971.2006. [DOI] [PubMed] [Google Scholar]
- 114.Hill RA, McInnes KJ, Gong EC, Jones ME, Simpson ER, Boon WC. Estrogen deficient male mice develop compulsive behavior. Biol Psychiatry. 2007;61(3):359–66. doi: 10.1016/j.biopsych.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 115.Kopin IJ. Monoamine oxidase and catecholamine metabolism. J Neural Transm Suppl. 1994;41:57–67. doi: 10.1007/978-3-7091-9324-2_7. [DOI] [PubMed] [Google Scholar]
- 116.Kruk MR, Westphal KG, Van Erp AM, et al. The hypothalamus: cross-roads of endocrine and behavioural regulation in grooming and aggression. Neurosci Biobehav Rev. 1998;23(2):163–77. doi: 10.1016/s0149-7634(98)00018-9. [DOI] [PubMed] [Google Scholar]
- 117.Rhodes JS, Garland T, Jr, Gammie SC. Patterns of brain activity associated with variation in voluntary wheel-running behavior. Behav Neurosci. 2003;117(6):1243–56. doi: 10.1037/0735-7044.117.6.1243. [DOI] [PubMed] [Google Scholar]
- 118.Karayiorgou M, Altemus M, Galke BL, et al. Genotype determining low catechol-O-methyltransferase activity as a risk factor for obsessive-compulsive disorder. Proc Natl Acad Sci USA. 1997;94(9):4572–5. doi: 10.1073/pnas.94.9.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karayiorgou M, Sobin C, Blundell ML, et al. Family-based association studies support a sexually dimorphic effect of COMT and MAOA on genetic susceptibility to obsessive-compulsive disorder. Biol Psychiatry. 1999;45(9):1178–89. doi: 10.1016/s0006-3223(98)00319-9. [DOI] [PubMed] [Google Scholar]
- 120.Pooley EC, Fineberg N, Harrison PJ. The met(158) allele of catechol-O-methyltransferase (COMT) is associated with obsessive-compulsive disorder in men: case-control study and meta-analysis. Mol Psychiatry. 2007;12(6):556–61. doi: 10.1038/sj.mp.4001951. [DOI] [PubMed] [Google Scholar]
- 121.Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. SA-PAPs. A family of PSD-95/SAP90-associated proteins localized at postsynaptic density. J Biol Chem. 1997;272(18):11943–51. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- 122.Kim E, Naisbitt S, Hsueh YP, et al. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol. 1997;136(3):669–78. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Satoh K, Yanai H, Senda T, et al. DAP-1, a novel protein that interacts with the guanylate kinase-like domains of hDLG and PSD-95. Genes Cells. 1997;2(6):415–24. doi: 10.1046/j.1365-2443.1997.1310329.x. [DOI] [PubMed] [Google Scholar]
- 124.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5(10):771–81. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 125.Lacomblez L, Bensimon G, Leigh PN, et al. Long-term safety of riluzole in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3(1):23–9. doi: 10.1080/146608202317576507. [DOI] [PubMed] [Google Scholar]
- 126.Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci. 2000;12(10):3567–74. doi: 10.1046/j.1460-9568.2000.00242.x. [DOI] [PubMed] [Google Scholar]
- 127.Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes) Neuroscience. 2004;125(1):191–201. doi: 10.1016/j.neuroscience.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 128.Frizzo ME, Dall'Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24(1):123–8. doi: 10.1023/B:CEMN.0000012717.37839.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coric V, Milanovic S, Wasylink S, Patel P, Malison R, Krystal JH. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with obsessive-compulsive disorder and major depressive disorder. Psychopharmacology (Berl) 2003;167(2):219–20. doi: 10.1007/s00213-003-1396-z. [DOI] [PubMed] [Google Scholar]
- 130.Coric V, Taskiran S, Pittenger C, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424–8. doi: 10.1016/j.biopsych.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 131.Pittenger C, Kelmendi B, Wasylink S, Bloch MH, Coric V. Riluzole augmentation in treatment-refractory obsessive-compulsive disorder: a series of 13 cases, with long-term follow-up. J Clin Psychopharmacol. 2008;28(3):363–7. doi: 10.1097/JCP.0b013e3181727548. [DOI] [PubMed] [Google Scholar]
- 132.Grant P, Lougee L, Hirschtritt M, Swedo SE. An open-label trial of riluzole, a glutamate antagonist, in children with treatment-resistant obsessive-compulsive disorder. J Child Adolesc Psychopharmacol. 2007;17(6):761–7. doi: 10.1089/cap.2007.0021. [DOI] [PubMed] [Google Scholar]
- 133.Sasso DA, Kalanithi PS, Trueblood KV, et al. Beneficial effects of the glutamate-modulating agent riluzole on disordered eating and pathological skin-picking behaviors. J Clin Psychopharmacol. 2006;26(6):685–7. doi: 10.1097/01.jcp.0000245567.29531.d6. [DOI] [PubMed] [Google Scholar]
- 134.Coric V, Kelmendi B, Pittenger C, Wasylink S, Bloch MH, Green J. Beneficial effects of the antiglutamatergic agent riluzole in a patient diagnosed with trichotillomania. J Clin Psychiatry. 2007;68(1):170–1. doi: 10.4088/jcp.v68n0123f. [DOI] [PubMed] [Google Scholar]
- 135.Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. 2008;22(9):761–86. doi: 10.2165/00023210-200822090-00004. [DOI] [PubMed] [Google Scholar]
- 136.Angehagen M, Ronnback L, Hansson E, Ben-Menachem E. Topiramate reduces AMPA-induced Ca(2+) transients and inhibits GluR1 subunit phosphorylation in astrocytes from primary cultures. J Neurochem. 2005;94(4):1124–30. doi: 10.1111/j.1471-4159.2005.03259.x. [DOI] [PubMed] [Google Scholar]
- 137.Van Ameringen M, Mancini C, Patterson B, Bennett M. Topiramate augmentation in treatment-resistant obsessive-compulsive disorder: a retrospective, open-label case series. Depress Anxiety. 2006;23(1):1–5. doi: 10.1002/da.20118. [DOI] [PubMed] [Google Scholar]
- 138.Hollander E, Dell'Osso B. Topiramate plus paroxetine in treatment-resistant obsessive-compulsive disorder. Int Clin Psychopharmacol. 2006;21(3):189–91. doi: 10.1097/01.yic.0000199453.54799.cc. [DOI] [PubMed] [Google Scholar]
- 139.Kumar TC, Khanna S. Lamotrigine augmentation of serotonin reuptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527–8. doi: 10.1080/j.1440-1614.2000.0751c.x. [DOI] [PubMed] [Google Scholar]
- 140.Ozkara C, Ozmen M, Erdogan A, Yalug I. Topiramate related obsessive-compulsive disorder. Eur Psychiatry. 2005;20(1):78–9. doi: 10.1016/j.eurpsy.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 141.Alkin T, Onur E, Ozerdem A. Co-occurence of blepharospasm, tourettism and obsessive-compulsive symptoms during lamotrigine treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1339–40. doi: 10.1016/j.pnpbp.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 142.Kuloglu M, Caykoylu A, Ekinci O, Yilmaz E. Lamotrigine-induced obsessional symptoms in a patient with bipolar II disorder: a case report. J Psychopharmacol. 2008 doi: 10.1177/0269881108095082. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 143.Kemp DE, Gilmer WS, Fleck J, Dago PL. An association of intrusive, repetitive phrases with lamotrigine treatment in bipolar II disorder. CNS Spectrums. 2007;12(2):106–11. doi: 10.1017/s1092852900020617. [DOI] [PubMed] [Google Scholar]
- 144.Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299(1):12–20. [PubMed] [Google Scholar]
- 145.Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25(27):6389–93. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lafleur DL, Pittenger C, Kelmendi B, et al. N-acetylcysteine augmentation in serotonin reuptake inhibitor refractory obsessive-compulsive disorder. Psychopharmacology (Berl) 2006;184(2):254–6. doi: 10.1007/s00213-005-0246-6. [DOI] [PubMed] [Google Scholar]
- 147.Warr O, Takahashi M, Attwell D. Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol. 1999;514(Pt 3):783–93. doi: 10.1111/j.1469-7793.1999.783ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4(2):131–44. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 149.Shimazaki T, Iijima M, Chaki S. Anxiolytic-like activity of MGS0039, a potent group II metabotropic glutamate receptor antagonist, in a marble-burying behavior test. Eur J Pharmacol. 2004;501(13):121–5. doi: 10.1016/j.ejphar.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 150.Pasquini M, Biondi M. Memantine augmentation for refractory obsessive-compulsive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1173–5. doi: 10.1016/j.pnpbp.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 151.Poyurovsky M, Weizman R, Weizman A, Koran L. Memantine for treatment-resistant OCD. Am J Psychiatry. 2005;162(11):2191–2. doi: 10.1176/appi.ajp.162.11.2191-a. [DOI] [PubMed] [Google Scholar]
- 152.Egashira N, Okuno R, Harada S, et al. Effects of glutamate-related drugs on marble-burying behavior in mice: implications for obsessive-compulsive disorder. Eur J Pharmacol. 2008;586(13):164–70. doi: 10.1016/j.ejphar.2008.01.035. [DOI] [PubMed] [Google Scholar]
- 153.Parsons CG, Panchenko VA, Pinchenko VO, Tsyndrenko AY, Krishtal OA. Comparative patch-clamp studies with freshly dissociated rat hippocampal and striatal neurons on the NMDA receptor antagonistic effects of amantadine and memantine. Eur J Neurosci. 1996;8(3):446–54. doi: 10.1111/j.1460-9568.1996.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 154.McGrath MJ, Campbell KM, Parks CR, Burton FH. Glutamatergic drugs exacerbate symptomatic behavior in a transgenic model of comorbid Tourette's syndrome and obsessive-compulsive disorder. Brain Res. 2000;877(1):23–30. doi: 10.1016/s0006-8993(00)02646-9. [DOI] [PubMed] [Google Scholar]
- 155.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349–52. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 156.Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77(1):309–23. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 157.Monahan JB, Handelmann GE, Hood WF, Cordi AA. D-cycloserine, a positive modulator of the N-methyl-D-aspartate receptor, enhances performance of learning tasks in rats. Pharmacol Biochem Behav. 1989;34(3):649–53. doi: 10.1016/0091-3057(89)90571-6. [DOI] [PubMed] [Google Scholar]
- 158.Watson GB, Bolanowski MA, Baganoff MP, Deppeler CL, Lanthorn TH. D-cycloserine acts as a partial agonist at the glycine modulatory site of the NMDA receptor expressed in Xenopus oocytes. Brain Res. 1990;510(1):158–60. doi: 10.1016/0006-8993(90)90745-w. [DOI] [PubMed] [Google Scholar]
- 159.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22(6):2343–51. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wilhelm S, Buhlmann U, Tolin DF, et al. Augmentation of behavior therapy with D-cycloserine for obsessive-compulsive disorder. Am J Psychiatry. 2008;165(3):335–41. doi: 10.1176/appi.ajp.2007.07050776. quiz 409. [DOI] [PubMed] [Google Scholar]
- 161.Kushner MG, Kim SW, Donahue C, et al. D-cycloserine augmented exposure therapy for obsessive-compulsive disorder. Biol Psychiatry. 2007;62(8):835–8. doi: 10.1016/j.biopsych.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 162.Storch EA, Merlo LJ, Bengtson M, et al. D-cycloserine does not enhance exposure-response prevention therapy in obsessive-compulsive disorder. Int Clin Psychopharmacol. 2007;22(4):230–7. doi: 10.1097/YIC.0b013e32819f8480. [DOI] [PubMed] [Google Scholar]
- 163.Rothbaum BO. Critical parameters for D-cycloserine enhancement of cognitive-behaviorial therapy for obsessive-compulsive disorder. Am J Psychiatry. 2008;165(3):293–6. doi: 10.1176/appi.ajp.2007.07121871. [DOI] [PubMed] [Google Scholar]