

Abstract
Aim
We investigated the effect of cell seeding dose and incubation time on tissue-engineered vascular graft (TEVG) patency.
Materials & methods
Various doses of bone marrow-derived mononuclear cells (BM-MNCs) were seeded onto TEVGs, incubated for 0 or 12 h, and implanted in C57BL/6 mice. Different doses of human BM-MNCs were seeded onto TEVGs and measured for cell attachment.
Results
The incubation time showed no significant effect on TEVG patency. However, TEVG patency was significantly increased in a dose-dependent manner. In the human graft, more bone marrow used for seeding resulted in increased cell attachment in a dose-dependent manner.
Conclusion
Increasing the BM-MNC dose and reducing incubation time is a viable strategy for improving the performance and utility of the graft.
Keywords: BM-MNC, bone marrow mononuclear cells, C57BL/6 mice, congenital heart defect, Fontan operation, regenerative medicine, TEVG, tissue engineering
We developed the first tissue-engineered vascular graft (TEVG) with growth potential, designed specifically for use in congenital heart surgery [1–3]. Results from our first human clinical trial confirmed the growth capacity of the TEVG and demonstrated no graft-related deaths or graft failures [4]. However, results of this study also demonstrated that stenosis was the primary graft-related complication effecting nearly 25% of graft recipients, with 16% of recipients developing critical stenosis (>75% decrease in luminal diameter) [5]. Before routine clinical use of the TEVG can be recommended, the assembly of the TEVG must be optimized to help inhibit the formation of TEVG stenosis and minimize the time required to make the graft and improve its overall utility [6].
To optimize the TEVG, we developed a murine model to investigate the cellular and molecular mechanisms underlying vascular neotissue formation in the TEVG [7]. It was discovered that vascular neotissue arises from the ingrowth of endothelial cells (ECs) and smooth muscle cells from the neighboring blood vessel wall and that, contrary to the classic tissue engineering paradigm, seeded cells do not contribute to vascular neotissue, but instead rapidly disappear after implantation [7,8]. We also found that vascular neotissue formation is a host macrophage-mediated regenerative process and that cell seeding is not essential for vascular neotissue formation [9]. Conversely, we previously discovered that the degree of host macrophage infiltration is directly proportional to the incidence of TEVG stenosis [9] and that bone marrow-derived mononuclear cell (BM-MNC) seeding inhibits the formation of TEVG stenosis via a TGF-β immune-mediated paracrine effect [10]. So, although BM-MNC seeding does not contribute to neotissue formation, it is critical in preventing stenosis and improving patency. These results highlight the importance of BM-MNC seeding, but we have not mechanistically studied the optimal dose of cell seeding to effectively prevent TEVG stenosis.
To insure the best clinical outcome using TEVG technology, it is also important to optimize the time for TEVG assembly in order to minimize the time for the surgical procedure, decrease the potential for contamination, and thereby decrease the risk associated with using this technology. Assembling a TEVG used in our clinical trial involves a three step process, first, cell harvest and isolation, second, cell seeding onto the scaffold, and third, incubating the seeded graft for a period of time [4]. Previously we tried to reduce the time to assemble the TEVG by using a closed disposable system and were able to reduce the time for cell isolation and seeding without affecting TEVG patency [11,12]. However, to date, we have not studied the optimal incubation time after cell seeding onto the scaffold. In the initial human clinical trial, the 2 h incubation time was selected arbitrarily, therefore, in order to optimize the TEVG assembly process, without affecting patency of the TEVG and improving safety of the patients, we need to ascertain the optimal incubation time.
Herein, we attempted to refine the assembly of the TEVG with a focus on cell dosing and optimal incubation time. The effects of cell dose and duration of incubation on the formation of TEVG stenosis were investigated using a murine model and correlated our results with the degree of macrophage infiltration in order to provide insight into the mechanism underlying this process.
Material & methods
Bone marrow harvesting & seeding for mouse graft
All animal experiments were approved by the Nationwide Children's Hospital institutional guidelines for the use and care of animals (IACUC). Bone marrow was collected from the femurs and tibias of syngeneic C57BL/6 mice and mononuclear cells were isolated using the density centrifugation method [13]. In order to determine the optimal cell incubation time, 1 × 106 cells/graft was seeded statically onto biodegradable scaffolds (polyglycolic acid sheet with a co-polymer sealant solution of poly-l-lactide and -ε-caprolactone, 0.82 mm in inner diameter and 3 mm in length, provided from Gunze Co. Ltd, Kyoto, Japan) and incubated for 0, 0.5, 2, and 12 h in RPMI 1640 (Sigma, MO, USA) at 37°C in a CO2 incubator. In order to determine the optimal cell dose, four different doses of isolated mononuclear cells were seeded statically, 0.0, 0.1, 1, 10 × 106 cells/graft, and incubated overnight. After the incubation, grafts were used either for cell counting using a DNA assay (n = 6/group) to quantify cell attachment to the scaffold or implanted onto mice as an inferior vena cava (IVC) interposition graft (n = 25/group) [13].
Cell source & seeding for human graft
Human bone marrow was purchased from Lonza (Lonza Walkersville Inc., MD, USA). Three different doses of bone marrow were used in this study, 12.5, 25, and 50 ml/graft (n = 5, 7, 8, respectively). After filtering BM through 100 μm filters, the BM-MNCs were isolated by a density centrifugation method [14]. After counting the total number of isolated BM-MNCs, the cells were then seeded onto biodegradable scaffold (18 mm in diameter and ~120 mm in length, provided from Gunze, Kyoto, Japan) using the vacuum seeding method [14] and incubated for 2 h similarly to currently ongoing clinical trial (IDE 14127). The scaffold was cut into 5 mm2 sections and cell attachment was quantified using a DNA assay.
In vitro DNA assay
Cell attachment onto the TEVG scaffolds was determined by measuring DNA content using the fluorimetric Quant-iT™ PicoGreen® dsDNA Assay Kit (Life Technologies, NY, USA) following the manufacturer's instructions. The Quant-iT PicoGreen dsDNA reagent is a fluorescent nucleic acid stain for the quantification of double stranded DNA in solution. After lysing the cells attached to the scaffold, the bound stain in dsDNA was detected using a fluorescence microplate reader. A standard curve of cell number as a function of fluorescence was generated using known quantities of human bone marrow cells, the cells seeded onto each scaffolds is determined. The number of attached cells/area was calculated by dividing the total attached cell numbers over the luminal surface area and was expressed as 103 cells/mm2 area. The seeding efficiency was calculated by dividing the attached cells/area by the seeded cells/area [14].
Surgical implantation
The grafts were implanted as IVC interposition grafts onto 6–8-week-old female C57BL/6 mice as described previously [13]. Briefly, after induction of anesthesia (Ketamine, 100 mg/kg and xylazine 10 mg/kg; ketoprofen 5 mg/kg as analgesic, intraperitoneal [ip.]) a midline laparotomy incision was made and the aorta and IVC were bluntly separated. Two microclamps were placed on both sides of the aorta and IVC, and then the IVC was transected. The TEVG was implanted as an end-to-end IVC interposition graft using 10.0 prolene sutures. The TEVGs were harvested 2 weeks after implantation. Freshly harvested samples were perfusion fixed with 10% formalin and used for histology and immunohistochemistry.
Histology & immunohistochemistry
Explanted TEVGs were fixed in 10% neutral buffered formalin overnight, embedded in paraffin, and sectioned (4 μm thick sections) as described previously [7]. Sections were stained with hematoxylin and eosin (H&E), Hart's, Masson's Trichrome, and Alcian blue stainings. Macrophages, MMP-2, and MMP-9 were identified via immunohistochemistry. After deparaffinization, rehydration, and blocking for endogenous peroxidase activity and nonspecific background staining, sections were incubated with the following primary antibodies: rat anti-F4/80 (1:1000; AbD Serotec, Oxford, UK), rabbit anti-MMP-2 (1:200; Abcam, Cambridge, UK), and rabbit anti-MMP-9 (1:200; Abcam). Primary antibody binding was detected by incubation with species appropriate biotinylated secondary antibodies, namely, goat antirat IgG (1:200; Vector Laboratories, CA, USA), and goat antirabbit IgG (1:200; Vector) respectively, followed by binding of horseradish peroxidase streptavidin (Vector) and subsequent chromogenic development with 3,3-diaminobenzidine (Vector). Nuclei were counterstained with hematoxylin (Gill's Formula, Vector). Light field images were obtained with a Zeiss Axio Imager.A2 microscope. Immunofluorescent staining was used to identify EC and smooth muscle cells (SMC). Slides were incubated overnight with a primary antibody solution of rabbit anti-CD31 (1:50, Abcam) and mouse antihuman smooth muscle actin (α-SMA, 1:500, Dako, CA, USA) which cross-reacts with mouse α-SMA. Antibody binding was detected with Alexa Fluor 488® goat antirabbit IgG (1:300; Invitrogen, CA, USA) and Alexa Fluor 647® goat antimouse IgG (1:300; Invitrogen) secondary antibodies. Cell nuclei were identified by subsequent counterstaining with 4′, 6-diamidini-2-phenylindole (DAPI; Invitrogen). Fluorescent images were obtained with an Olympus IX51 inverted microscope and exposure time was informed by appropriate negative controls.
Quantitative immunohistochemistry
The number of macrophages infiltrating the TEVG was quantified for each explanted scaffold. Nuclei with positive F4/80 expression were imaged in five equally spaced regions of each section at 400× high power filed (HPF) and counted and totaled using ImageJ software (NIH, MD, USA) [9].
TEVG morphometry
Graft luminal diameter was measured from H&E stained slides using ImageJ software. The luminal diameter was determined by dividing the circumference of the lumen by π. The stenosis rate in the graft was calculated by the ratio between measured luminal diameter and original graft luminal diameter. Critical stenosis was defined as more than a 75% decrease in luminal diameter [9].
Statistical analysis
One-way analysis of variance (ANOVA) was used to determine differences in cell attachment. Fisher's exact test was used to compare stenosis rate of the grafts. p-values less than 0.05 indicated statistical significance. Numeric values are listed as mean ± standard deviation.
Results
Natural history of neotissue formation in the murine model
In our previous studies we demonstrated that TEVG stenosis typically occurs within the first 2 weeks after implantation [9]. At gross observation, the TEVG was partially degraded and significant neotissue formation had occurred at 2 weeks after implantation. Figure 1A & J shows the representative histological images of patent and occluded TEVGs. Patent TEVGs developed a confluent EC layer along the luminal surface and underlying SMCs appeared (Figure 1B–D). By contrast, occluded grafts showed disorganized neotissue formation composed primarily of SMCs embedded in collagen (Figure 1K–M) with either an occluded lumen or a narrowed endothelial lined lumen. Both patent and occluded grafts showed early development of elastin, collagen, and glycosaminoglycans, mainly around the remaining grafts (Figure 1E–G & N–P). MMP-2 and MMP-9 activities were also observed in the neotissue close to the residual scaffold material suggesting ongoing tissue remodeling (Figure 1H–I & Q–R).
Figure 1. Histological images of patent and occluded grafts.
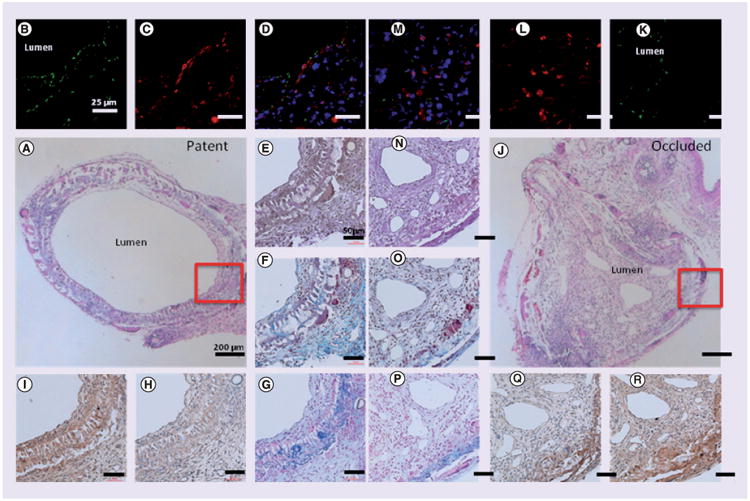
Explanted grafts stained with hematoxylin and eosin (A & J), CD31 for endothelial cells (B & K), smooth muscle actin for smooth muscle cells (C & L), superimposed images of CD31 and smooth muscle actin (D & M), Hart's for elastin (E & N), Masson's Trichrome for collagen (F & O), Alcian Blue for glycosaminoglycans (G & P), MMP-2 (H & Q), and MMP-9 (I & R).
Duration of incubation does not affect TEVG patency
To assess the effect of duration of incubation time on TEVG stenosis, we implanted the 1 × 106 BM-MNCs seeded TEVG with 0 and 12 h incubations (n = 25/group). The TEVGs were harvested 2 weeks after implantation and the graft patency was compared. The critical stenosis rate was significantly higher in the unseeded group than that of both 0 and 12-h incubation (84 vs 12% and 20%; p < 0.0001; Figure 2A). Between the 0 and 12 h groups, there was no statistical difference (Figure 2A).
Figure 2. The effect of graft incubation time and cell seeding dose on tissue-engineered vascular graft stenosis.

There was no significant difference in the critical stenosis rate for the grafts that were incubated for either 0 or 12 h. However, both showed a significantly lower critical stenosis rate in comparison to the unseeded control group ([A] ****p < 0.0001). Increasing the dose of cells seeded onto the scaffold decreases the incidence of tissue-engineered vascular graft stenosis. The incidence of critical stenosis rate was significantly lower in both 1 × 106 and 10 × 106 seeded groups in comparison to unseeded control or 0.1 × 106 seeded groups ([B] ****p < 0.0001).
Cell seeding inhibits the formation of TEVG critical stenosis in a dose-dependent manner
To further investigate the effect of cell seeding dose on the formation of TEVG stenosis, 0.1, 1, 10 × 106 cell seeded grafts alongside with unseeded control were implanted as an IVC interposition graft (n = 25/group). After 2 weeks implantation, there was a significant reduction in the stenosis rate in both 1 and 10 × 106 cell seeded groups than the unseeded control and 0.1 × 106 cell seeded groups (20 and 5% vs 84 and 64%; p < 0.0001; Figure 2B).
Macrophage infiltration in TEVG occurs with a cell dose-dependent manner
In order to determine whether cell dose affects macrophage infiltration into the TEVG, the infiltrating macrophages were evaluated by quantitative histological morphometric analysis. The unseeded control and 0.1 × 106 seeded group showed no statistical differences in infiltrated macrophages between them, however, the number of macrophages was significantly decreased in both the 1 × 106 and 10 × 106 groups (159.05 ± 11.59 and 159.17 ± 16.64 versus 122.02 ± 14.76 and 120.68 ± 22.91 cells/HPF; p < 0.0001; Figure 3).
Figure 3. Cell seeding reduces macrophage infiltration in a dose-dependent fashion.
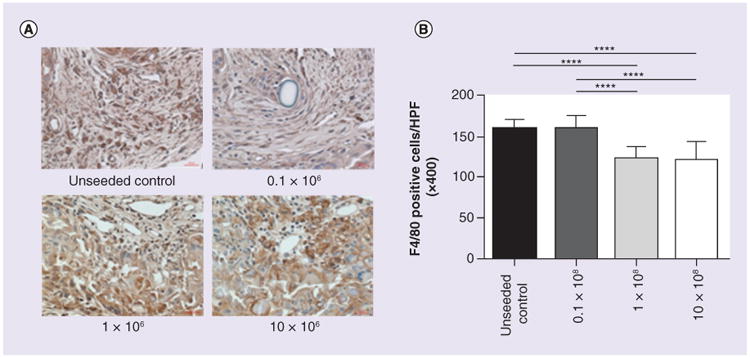
(A) Immunohistological images of F4/80 staining and (B) the number of F4/80 positive cells per HPF (×400). The macrophage infiltration into the tissue-engineered vascular graft in the unseeded control and 0.1 × 106 group significantly increased compare to the 1 × 106 and 10 × 106 groups (159.05 ± 11.59 and 159.17 ± 16.64 vs 122.02 ± 14.76 and 120.68 ± 22.91 cells/HPF; ****p < 0.0001).
HPF: High power field.
Duration of incubation does not affect cell attachment & seeding efficiency
To assess the effect of duration of incubation time on cell attachment and seeding efficiency, 1 × 106 BM-MNCs were seeded onto the biodegradable scaffold and incubated for 0, 0.5, 2, and 12 h (n = 6/group). An in vitro DNA assay for cell counting showed no statistical difference in the number of seeded cells and seeding efficiency (Figure 4A & B).
Figure 4. In vitro cell attachment and cell seeding efficiency for murine grafts.

(A) The duration of incubation did not significantly affect either cell attachment or (B) cell seeding efficiency. Cell attachment significantly increased with increase in cell seeding dose ([C] ***p < 0.001; ****p < 0.0001). However, the cell seeding efficiency was highest in 1 × 106 cell seeded group, then decreased in the 10 × 106 cell seeded group.
Cell attachment & seeding efficiency increase in a cell seeding dose-dependent manner
To determine whether cell seeding dose has an effect on cell attachment and seeding efficiency, 0.1, 1 and 10 × 106 BM-MNCs were seeded onto the biodegradable scaffold and incubated overnight (n = 6/group). The 1 × 106 cell seeded group showed over a 12-fold increase in cell attachment than the 0.1 × 106 seeded group (19.09 ± 13.03 × 103 vs 1.56 ± 1.23 × 103 cells/mm2; p < 0.001; Figure 4C). The 10 × 106 cell seeded group showed significant increase in cell attachment compared with the 1 × 106 cell seeded group, even though ten times more cells were seeded (95.85 ± 48.39 × 103 vs 19.09 ± 13.03 × 103 cells/mm2; p < 0.001; Figure 4C). However, the corresponding cell seeding efficiency in the 10 × 106 group was decreased, though not significantly, than both 0.1 or 1 × 106 cell seeded groups (9.16 ± 4.09 vs 14.8 ± 11.69 and 18.00 ± 11.71%; Figure 4D).
Cell attachment in human graft is also dose-dependent similarly to the mouse graft
To compare the cell attachment and seeding efficiency from this mouse study to the human clinical study, three different doses of human bone marrow, 12.5, 25 and 50 ml/grafts (n = 5, 7 and 8, respectively), were seeded according to the methods used for the human clinical trial [14]. The total BM-MNCs isolated from the 12.5, 25, and 50 ml group were 57 ± 21, 91 ± 26, and 165 ± 96 × 106 cells (Figure 5A). After cell seeding, the attached cells/area was increased in dose-dependent manner. Especially, cell attachment in 50 ml group was significantly more than 12.5 ml group (2.83 ± 1.67 vs 0.88 ± 0.21 × 103 cells/mm2; p < 0.05; Figure 5B). The seeding efficiency for all three groups was comparable to that of the mouse graft, which was between 10 and 20%. Our results suggest that increasing the amount of bone marrow harvested from patients would increase BM-MNC attachment in the graft in a dose-dependent manner similar to the mouse graft.
Figure 5. Validation study: human bone marrow-derived mononuclear cells attach to the tissue-engineered vascular graft scaffold in a dose-dependent fashion similar to the murine bone marrow-derived mononuclear cells.

Increase in bone marrow volume (12.5, 25, and 50 ml) increased (A) total seeded cells and (B) attached cell numbers. (C) Cell seeding efficiency remained same in all groups.
Discussion
Despite promising results using TEVGs in treating patients with congenital heart disease, the high incidence of graft stenosis in clinical applications hinders wide spread use of this technology [15–17]. Therefore, to further optimize the TEVG assembly and increase the patency of the TEVG, we focused this research on optimizing cell seeding dose and graft incubation time after seeding. Our results demonstrated that cell seeding increases TEVG patency in a dose-dependent manner. However, duration of incubation time showed minimal effect on TEVG patency.
In this study, we showed that when more cells were seeded, the TEVG patency was improved. Moreover, BM-MNC number and attachment was increased by increasing the amount of bone marrow used for seeding. Based on the results of this study, it appears that it would require supra-physiological doses of BM-MNC (more BM-MNC than can be harvested from a single donor) in order to saturate the scaffold to the point that additional cell seeding would not significantly increase the number of cells attached to the scaffold (Figure 5). The original dose of cells selected for making the TEVG in human clinical trial was chosen empirically and represented the number of BM-MNC that could be isolated from 5 ml/kg of bone marrow. From a clinical perspective, up to 20 ml/kg of bone marrow can be harvested from an individual without incurring significant adverse effects and is routinely used for harvesting bone marrow for bone marrow transplantations [18]. By increasing bone marrow harvested from 5 ml/kg (12.5 ml) to 20 ml/kg (50 ml) from a 2.5 kg patient, we would estimate a three- to four-fold increase of BM-MNCs and would increase the number of cells attached to the scaffold by approximately 300–400% (Figure 5). Assuming the results of the murine study are relevant to the human condition, we estimate that a 400% increase in cell attachment would result in a 40–80% reduction in incidence of stenosis with the new cell dosing strategy. Further increase in the cell dose is possible, but its use would need to be measured against the increased risk of adverse hemodynamic consequences and the need for transfusion that would be associated with harvesting >20 ml/kg of bone marrow [18]. Use of growth factors to stimulate bone marrow growth represents an additional strategy for increasing the yield of BM-MNC, but to date has not been explored for this particular application.
An additional obstacle hindering the translation of this technology is the amount of time required to assemble the TEVG. Minimizing the amount of time needed to assemble the TEVG is critical and improves its utility. Assembly refers to first, the process of isolating BM-MNC, second, seeding the scaffold, and third, incubating the seeded construct. Using our current methodology based on isolating the BM-MNC using density centrifugation in Ficoll followed by a 2-h incubation approximately 268 ± 9 min are required to assemble the TEVG [4]. In an attempt to reduce the time required for TEVG assembly, we previously developed a closed disposable system using a filtration/elusion system to minimize time for the first two parts of the TEVG assembly, isolating BM-MNC and seeding [12]. Using the system, we could be able to drastically minimize the time for cell isolation and seeding without impacting the patency using a sheep model [11]. However, prior to the current study, the optimal incubation time was not determined mechanistically. Results of this study showed that the incubation time did not affect the TEVG patency, and the number of cells attached to the scaffold actually decreases with increased periods of incubation. Thus by using the closed apparatus and the reduced incubation period we could decrease the time required to assemble the TEVG from 268 ± 9 to 17 ± 2 min, further increasing its safety and clinical utility [11].
Conclusion
Stenosis is a primary graft-related complication. Our previous work suggests that graft stenosis is caused by excessive macrophage infiltration and more importantly, cell seeding reduced monocyte/macrophage infiltration and their expression of proinflammatory markers [9]. In this study we demonstrated that increasing the cell dose reduced the degree of macrophage infiltration, suggesting that the mechanism of action underlying this beneficial effect is immune mediated. These findings are consistent with our previous studies demonstrating that cell seeding reduces the degree of host macrophage infiltration and inhibits stenosis compared with unseeded controls. We have also shown that the time for TEVG incubation did not affect the TEVG patency, which can be a way to minimize the time required for TEVG assembly. In this investigation we studied the role of cell dose and duration of incubation and were able to identify strategies for improving the safety and clinical utility of the TEVG.
Future perspective
BM-MNC seeding prevents stenosis via a paracrine effect [19,20]. The more cells seeded onto a scaffold, the less stenosis occurrence. However, to have a one day surgical procedure, the amount of cells seeded is limited by the amount of bone marrow aspirated on the day of surgery. Optimization through rational mechanism-based design is essential for the safe translation of this technology to the clinic. One potential route for evolution in regenerative implants is in drugs or materials that mimic seeded bone marrow cells. For instance, if there is a drug that can mimic the effect of supraphysiological levels of cells, then can stenosis be stopped all together? While inspired by tissue engineering, this would represent a trip ‘back to the future’ and revival of the pioneering work developing vascular grafts from biodegradable materials [21–25]. There are many benefits to this approach: less time under anesthesia for the patient, less chance of contamination, less chance of a negative immune response, off-the-shelf availability for the implant, and simpler regulatory pathway. However, despite the advantages of cell-free tissue engineering, we contend that there are ongoing advantages to the cell seeded approach too. One advantage of cell seeding is the smart cell delivery system. The seeded cells, especially autologous cells, release signals in response to the body's feedback mechanism unlike the drug eluting scaffolds which release the drug regardless of the feedback system. Thus, both approaches should be rigorously explored and utilized in our attempt to develop an improved vascular graft with growth capacity for use in children.
Ethical conduct of research.
All animal experiments were approved by the Nationwide Children's Hospital institutional guidelines for the use and care of animals (IACUC). The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved
Executive summary.
Duration of incubation does not affect tissue-engineered vascular graft patency
The patency rate in both 0 and 12 h incubation groups was significantly higher than that of the unseeded control group. Between the 0 and 12 h groups, there was no significant difference in patency. Therefore, duration of incubation after cell seeding does not affect tissue-engineered vascular graft (TEVG) patency.
Cell seeding inhibits the formation of TEVG stenosis in a dose-dependent manner
There was a significant reduction in the stenosis rate in both 1 and 10 × 106 cell seeded groups than the unseeded control and 0.1 × 106 cell seeded groups.
TEVGs that had 10 × 106 cells seeded, further decreased the stenosis rate compare to 1 × 106 cells seeded, but not significantly.
Macrophage infiltration in TEVG occurs with a cell dose-dependent manner
Similar to the TEVG patency, the numbers of infiltrating macrophages in the unseeded control and 0.1 × 106 cell seeded group were significantly higher than in both the 1 × 106 and 10 × 106 groups.
Duration of incubation does not affect cell attachment & seeding efficiency
An in vitro DNA assay for cell counting showed that the number of seeded cells and seeding efficiency decreased over the duration of incubation time, however, not significantly.
Cell attachment & seeding efficiency increase in a cell seeding dose-dependent manner
The 10 × 106 cell seeded group showed a fivefold increase in cell attachment compared with the 1 × 106 cell seeded group, even though tentimes more cells were seeded.
The corresponding cell seeding efficiency also showed a nearly twofold decrease in the 10 × 106 group than the 0.1 or 1 × 106 cell seeded groups.
Cell attachment in human graft is also dose-dependent similarly to the mouse graft
Increasing the amount of bone marrow harvested from patients' increases bone marrow-derived mononuclear cell attachment in the graft in a dose-dependent manner, similar to the mouse graft.
Conclusion
In this investigation we studied the role of cell seeding dose and duration of incubation and were able to identify strategies for improving the safety and clinical utility of the TEVG.
Increasing the cell dose reduced the degree of macrophage infiltration, suggesting that the mechanism of action underlying this beneficial effect is immune mediated.
Cell incubation did not affect TEVG patency, which can be a way to minimize the time required for TEVG assembly.
Acknowledgments
This research was supported by R01-HL098228 (C Breuer). C Breuer and T Shinoka have received grant support from the Pall Corporation and Gunze Limited. All authors have read the journals policy on conflict of interest. C Breuer and T Shinoka have patents related to the TEVG.
Footnotes
Financial & competing interests disclosure: The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript
References
Papers of special note have been highlighted as:
•• of considerable interest
- 1.Brennan MP, Dardik A, Hibino N, et al. Tissue-engineered vascular grafts demonstrate evidence of growth and development when implanted in a juvenile animal model. Ann Surg. 2008;248(3):370–377. doi: 10.1097/SLA.0b013e318184dcbd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isomatsu Y, Shin'oka T, Matsumura G, et al. Extracardiac total cavopulmonary connection using a tissue-engineered graft. J Thorac Cardiovasc Surg. 2003;126(6):1958–1962. doi: 10.1016/s0022-5223(03)01042-0. [DOI] [PubMed] [Google Scholar]
- 3.Shin'oka T, Imai Y, Ikada Y. Transplantation of a tissue-engineered pulmonary artery. N Engl J Med. 2001;344(7):532–533. doi: 10.1056/NEJM200102153440717. [DOI] [PubMed] [Google Scholar]
- 4••.Shin'oka T, Matsumura G, Hibino N, et al. Midterm clinical result of tissue-engineered vascular autografts seeded with autologous bone marrow cells. J Thorac Cardiovasc Surg. 2005;129(6):1330–1338. doi: 10.1016/j.jtcvs.2004.12.047. Describes the first tissue-engineered vascular graft (TEVG) implanted in a patients' body. [DOI] [PubMed] [Google Scholar]
- 5••.Hibino N, Mcgillicuddy E, Matsumura G, et al. Late-term results of tissue-engineered vascular grafts in humans. J Thorac Cardiovasc Surg. 2010;139(2):431–436. doi: 10.1016/j.jtcvs.2009.09.057. Accounts the late-term results of TEVG implanted in 25 human subjects over the course of several years (mean follow-up 5.8 years) [DOI] [PubMed] [Google Scholar]
- 6.Patterson JT, Gilliland T, Maxfield MW, et al. Tissue-engineered vascular grafts for use in the treatment of congenital heart disease: from the bench to the clinic and back again. Regen Med. 2012;7(3):409–419. doi: 10.2217/rme.12.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7••.Roh JD, Nelson GN, Brennan MP, et al. Small-diameter biodegradable scaffolds for functional vascular tissue engineering in the mouse model. Biomaterials. 2008;29(10):1454–1463. doi: 10.1016/j.biomaterials.2007.11.041. Presented the ability to create replica sub-1 mm inner diameter biodegradable tubular scaffolds that are functional as vascular grafts in a murine model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Hibino N, Villalona G, Pietris N, et al. Tissue-engineered vascular grafts form neovessels that arise from regeneration of the adjacent blood vessel. FASEB J. 2011;25(8):2731–2739. doi: 10.1096/fj.11-182246. Discovered that cells of the neotissue in TEVGs are not derived from either the seeded cells or from bone marrow derived progenitors as has previously been suggested. They are instead derived from the neighboring blood vessel wall via a paracrine effect from seeded cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9••.Hibino N, Yi T, Duncan DR, et al. A critical role for macrophages in neovessel formation and the development of stenosis in tissue-engineered vascular grafts. FASEB J. 2011;25(12):4253–4263. doi: 10.1096/fj.11-186585. Found that the most important aspect of TEVG development is in the identification and manipulation of macrophage infiltration into the TEVG, as a logical target, for inhibiting the development of TEVG stenosis. Therefore the rational design of TEVGs should be based on developing methods for optimizing macrophage infiltration into the TEVG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan DR, Chen PY, Patterson JT, et al. Tgfbr1 inhibition blocks the formation of stenosis in tissue engineered vascular grafts. J Am Coll Cardiol. 2015;65(5):512–514. doi: 10.1016/j.jacc.2014.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11••.Kurobe H, Maxfield MW, Naito Y, et al. Comparison of a closed system to a standard open technique for preparing tissue-engineered vascular grafts. Tissue Eng Part C Methods. 2015;21(1):88–93. doi: 10.1089/ten.tec.2014.0160. Closed systems demonstrated no graft-related complications. In addition, the closed system reduced the time needed to assemble the TEVG by 50%. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurobe H, Tara S, Maxfield MW, et al. Comparison of the biological equivalence of two methods for isolating bone marrow mononuclear cells for fabricating tissue-engineered vascular grafts. Tissue Eng Part C Methods. 2014;21(6):597–604. doi: 10.1089/ten.tec.2014.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee YU, Yi T, Tara S, et al. Implantation of inferior vena cava interposition graft in mouse model. J Vis Exp. 2014;(88) doi: 10.3791/51632. www.jove.com/video/51632. [DOI] [PMC free article] [PubMed]
- 14.Udelsman B, Hibino N, Villalona GA, et al. Development of an operator-independent method for seeding tissue-engineered vascular grafts. Tissue Eng Part C Methods. 2011;17(7):731–736. doi: 10.1089/ten.tec.2010.0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez CE, Achneck HE, Reichert WM, Truskey GA. Biological and engineering design considerations for vascular tissue engineered blood vessels (TEBVS) Curr Opin Chem Eng. 2014;3:83–90. doi: 10.1016/j.coche.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mcallister TN, Maruszewski M, Garrido SA, et al. Effectiveness of haemodialysis access with an autologous tissue-engineered vascular graft: a multicentre cohort study. Lancet. 2009;373(9673):1440–1446. doi: 10.1016/S0140-6736(09)60248-8. [DOI] [PubMed] [Google Scholar]
- 17.Wystrychowski W, Mcallister TN, Zagalski K, Dusserre N, Cierpka L, L'Heureux N. First human use of an allogeneic tissue-engineered vascular graft for hemodialysis access. J Vasc Surg. 2014;60(5):1353–1357. doi: 10.1016/j.jvs.2013.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Elzouki AY, Harf HA, Nazer H, Oh W, Stapleton FB, Whitley RJ. Textbook of Clinical Pediatrics. Vol. 3179. Springer; Berlin Heidelberger, Germany: 2012. [Google Scholar]
- 19.Roh JD, Sawh-Martinez R, Brennan MP, et al. Tissue-engineered vascular grafts transform into mature blood vessels via an inflammation-mediated process of vascular remodeling. Proc Natl Acad Sci USA. 2010;107(10):4669–4674. doi: 10.1073/pnas.0911465107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duncan DR, Chen PY, Patterson JT, et al. TGF β R1 inhibition blocks the formation of stenosis in tissue-engineered vascular grafts. J Am Coll Cardiol. 2015;65(5):512–514. doi: 10.1016/j.jacc.2014.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blakemore AH, Voorhees AB. The use of tubes constructed from vinyon n-cloth in bridging arterial defects – experimental and clinical. Ann Surg. 1954;140(3):324–334. doi: 10.1097/00000658-195409000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberg CB, Bell E. A blood-vessel model constructed from collagen and cultured vascular cells. Science. 1986;231(4736):397–400. doi: 10.1126/science.2934816. [DOI] [PubMed] [Google Scholar]
- 23.Bowald S, Busch C, Eriksson I. Arterial regeneration following polyglactin-910 suture mesh grafting. Surgery. 1979;86(5):722–729. [PubMed] [Google Scholar]
- 24.Vacanti JP, Morse MA, Saltzman WM, Domb AJ, Perezatayde A, Langer R. Selective cell transplantation using bioabsorbable artificial polymers as matrices. J Pediatr Surg. 1988;23(1):3–9. doi: 10.1016/s0022-3468(88)80529-3. [DOI] [PubMed] [Google Scholar]
- 25.Shinoka T, Shum-Tim D, Ma PX, et al. Creation of viable pulmonary artery autografts through tissue engineering. J Thorac Cardiov Sur. 1998;115(3):536–545. doi: 10.1016/S0022-5223(98)70315-0. [DOI] [PubMed] [Google Scholar]