

Abstract
Ab-producing plasma cells (PCs) serve as key participants in countering pathogenic challenges as well as being contributors to autoimmune and malignant disorders. Thus far, only a limited number of PC–specific markers have been identified. The characterization of the unique variable lymphocyte receptor (VLR) Abs that are made by evolutionarily distant jawless vertebrates prompted us to investigate whether VLR Abs could detect novel PC antigens that have not been recognized by conventional Abs. Here, we describe a monoclonal lamprey Ab, VLRB MM3, that was raised against primary multiple myeloma cells. VLRB MM3 recognizes a unique epitope of the CD38 ectoenzyme that is present on plasmablasts and PCs from healthy individuals and on most, but not all, multiple myelomas. Binding by the VLRB MM3 Ab coincides with CD38 dimerization and NAD glycohydrolase activity. Our data demonstrate that the lamprey VLRB MM3 Ab is a unique reagent for the identification of plasmablasts and PCs, with potential applications in the diagnosis and therapeutic intervention of PC or autoimmune disorders.
Introduction
Ab-secreting plasma cells (PCs) represent the final stage of B cell differentiation and are key components of the adaptive immune response to pathogenic challenges. In addition to producing protective Abs in the context of natural infection or vaccination (1), PCs also contribute to the pathogenesis of various auto-immune and neoplastic disorders (2–5). The terminal differentiation of B lymphocytes into PCs with a distinctive secretory ultrastructure is accompanied by pronounced changes in the transcriptome and proteome. PC differentiation is governed by the downregulation of the PAX5 transcription factor, considered a master regulator for B lineage commitment (6), and the concomitant upregulation of BLIMP1, IRF4, and XBP1 transcription factors, the latter of which is a key regulator of the unfolded protein response (7). Originally considered to be short-lived cells (8), more recent studies indicate that PCs residing in BM are often long lived (9–12). Initially identified by their characteristic morphology and abundant intracellular Ig, cell-surface markers such as the CD38 and CD138 (Syndecan-1) antigens are often used currently to identify PCs. However, these glycoproteins are also present on other B- and non–B-lineage cells (13). While combinations of mAbs against different cell-surface antigens have been used recently to characterize different subpopulations of PCs in BM and blood (12, 14–18), the lack of specific surface markers for PCs hampers the investigation of their contributions to immune responses and the development of therapeutic reagents targeting PCs involved in immune system disorders.
Until recently, the adaptive immune system was considered to have evolved in jawed vertebrates after their separation from the jawless vertebrate lineage approximately 500 million years ago. However, early studies indicated that the surviving jawless vertebrate representatives, sea lampreys (19) and hagfish (20), produce specific hemagglutinins in response to immunization with heterologous erythrocytes. More recent studies revealed that these jawless vertebrates have an alternative adaptive immune system, in which entirely different lymphocyte receptors and Ab-like proteins are used for antigen recognition (21). Instead of the Ig-based T cell receptors (TCRs) and B cell receptors (BCRs) that jawed vertebrates use for antigen recognition, lampreys and hagfish use variable lymphocyte receptors (VLRs) containing leucine-rich repeat (LRR) sequences for the same purpose (22). The VLRs have a signal peptide, a capping N-terminal LRR followed by a conserved LRR1, a variable number of highly diverse LRRv units, a short connecting peptide, a C-terminal capping peptide, and an invariant stalk region (21). Three distinct VLR genes, VLRA, VLRB, and VLRC, have been described; they are all incomplete in their germline configuration, in that they have coding sequences for the signal peptide, portions of the N-terminal and C-terminal capping LRRs, and a stalk region, separated by intervening noncoding sequences and flanked by hundreds of different LRR-encoding cassettes. These LRR cassettes serve as template donors in a serial piece-wise assembly process to form complete or mature VLR genes using a gene conversion–like mechanism to yield an estimated potential repertoire of 1014 to 1017 antigen receptors of each type. VLRA and VLRC molecules are expressed as cell-surface receptors on lymphocytes sharing characteristics with the α/β and γ/δ T cell lineages of jawed vertebrates (23, 24). The VLRB receptors are expressed on cells resembling B cells and are secreted by their PC progeny as multimeric antigen-binding proteins (25). Structural analyses of monoclonal VLRB Abs in complex with their cognate antigens have shown that the antigen interacts with residues located at the inner concave surface of the solenoid-shaped VLR Ab as well as with residues in a flexible, highly variable loop that protrudes from the capping C-terminal LRR (26, 27). We reasoned that the unique protein architecture of VLR Abs and the great evolutionary distance between lampreys and humans would allow the production of novel VLRB Abs against biomedically relevant antigens against which conventional Abs are not readily produced because of structural or tolerogenic constraints. Here, we report the generation and characterization of the monoclonal VLRB MM3 Ab as a reagent for the specific detection of PCs in both humans and nonhuman primates (NHPs).
Results
Isolation of the monoclonal VLRB MM3 Ab
In an effort to generate lamprey Abs that detect malignant PCs, we immunized 2 lamprey larvae with BM aspirates obtained from 1 multiple myeloma patient and tested 153 monoclonal VLRB Abs isolated from VLRB libraries established from the immunized lampreys. The monoclonal VLRB Ab MM3 was selected, because it reacted with all malignant (CD38++/CD45−) and nonmalignant (CD38++/CD45+) PCs and not with other cells from the BM aspirate used for immunization (Figure 1A). To facilitate purification and manipulation of this reagent, we engineered HA- and 6xHis-tags in the conserved stalk region of the molecule, as illustrated in Figure 1B and ref. 28, and deleted the C-terminal 42 residues to generate a secreted, monomeric form of the VLRB MM3 Ab (Figure 1C).
Figure 1. Isolation of monoclonal VLRB MM3.

(A) BM aspirate from a patient with multiple myeloma was stained with VLRB MM3 containing culture supernatant from transiently transfected HEK293T cells. The cells were costained for CD38 and CD45 expression and separated into non-PCs (gate I, CD38−/lo), nonmalignant PCs (gate II, CD45+/CD38++), and malignant PCs (gate III, CD45−/lo/CD38++). Histograms depict VLRB MM3 reactivity within the indicated gates. VLRB MM3 signals are indicated by solid black lines, and negative control VLR4 signals are shown as solid gray histograms. (B) Structural characteristics of VLRB MM3. Individual VLRB MM3 units consist of a signal peptide (SP), N-terminal LRR (LRR NT), LRR-1, three variable LRRv units, a connecting peptide, C-terminal capping LRR (LRR CT), and the invariable stalk region. Location of engineered sequences encoding the HA- and 6xHis epitope tags (H/H) into the stalk region are indicated. (C) Western blot (WB) analysis of WCLs and supernatants of HEK293T cells transiently transfected with VLRB MM3 and the multimerization-deficient mutant VLRB MM3ΔC. The samples were separated by SDS-PAGE under reducing and nonreducing conditions, and recombinant VLRB protein was detected with Abs specific to the HA-epitope tag. LLR, leucine-rich repeat; PCs, plasma cells; VLRB, variable lymphocyte receptor B; WCLs, whole-cell lysates.
Monoclonal VLRB MM3 Ab specifically recognizes human and NHP PCs
The staining pattern of VLRB MM3 of the BM aspirate used for immunization of the lamprey larvae suggested PC specificity of this reagent. To test this possibility, we examined the reactivity of the VLRB MM3 Ab with cells in human tonsil, spleen, and BM samples by immunofluorescence flow cytometry. VLRB MM3 detected PCs exclusively in all of these tissues, and, in tonsils, it reacted with both the major IgD− and minor IgD+ PC subpopulations (Figure 2A, top 2 rows). In blood, where circulating plasmablasts are normally very low in frequency, VLRB MM3-positive cell numbers were not easily detectable (Figure 2A, bottom row). However, a readily detectable population of VLRB MM3-positive cells was found in blood 7 days after influenza vaccination (Figure 2A, bottom row, right panel), a time at which the circulating plasmablast response reaches its peak (14).
Figure 2. PC specificity of VLRB MM3.

(A) Human tonsillar lymphocytes were separated into the following subpopulations: IgD− PCs (CD19+/IgD−/CD38++); IgD+ PCs (CD19+/IgD+/CD38++); naive B cells (CD19+/IgD+/CD38−); memory B cells (CD19+/IgD−/CD38−); pregerminal center B cells (CD19+/IgD+/CD38+); germinal center B cells (CD19+/IgD−/CD38+); non–B/T cells (CD19−/CD3−); and T cells (CD19−/CD3+). Shown is a representative of 15 examined tonsil tissues. Human splenic cells were separated into the indicated cell subpopulations described for tonsillar cells, with non–B cells encompassing all CD19− cells. Human BM was separated into PCs (CD38++/CD138+) and non-PCs (CD38−/CD138−). Shown is a representative of 3 analyzed BM tissue samples. Note that the BM sample was stained with Fc-MM3 fusion proteins. Human PBMCs were separated into naive B cells (CD19+/IgD+/CD27−), memory B cells (CD19+/IgD−/ CD27+), marginal zone equivalent cells (CD19+/IgD+/CD27+), transitional B cells (CD19+/IgD+/CD27−/CD10+/CD24+), non–B/T cells (CD19−/CD3−), T cells (CD19−/ CD3+), and monocytes (FSC/SSC characteristics). Shown is a representative example of 6 PBMC samples. Plasmablasts from influenza vaccine–immunized individuals were identified as CD19+/CD3−/IgD−/CD38++/CD27+. Shown is a representative example of 8 analyzed blood samples from day 7 after vaccination. VLRB MM3 signals are indicated by solid black lines and negative control VLR4 signals are shown as solid gray histograms. (B) NHP BM samples were stained with VLRB MM3 and gated on the lymphocyte gate (FSC/SSC). VLRB MM3–positive cells were FACS purified and analyzed by transmission electron microscopy (original magnification, ×4,000). Scale bar: 1 μm. In all histograms, VLRB MM3 reactivity is indicated by solid black lines, and negative control VLR4 signals are indicated by filled gray histograms. FSC/SSC, forward scatter/side scatter; GC, germinal center; NHP, nonhuman primate; Non-B, non–B cells; PBMCs, peripheral blood mononuclear cells; PCs, plasma cells; T, T cells; VLRB, variable lymphocyte receptor B.
We also investigated the reactivity of VLRB MM3 with NHP PCs, which are not readily identified by the cell-surface markers available for human PC detection (29). VLRB MM3 reacted with a subpopulation of the BM cells in 2 frequently studied NHP species, sooty mangabeys and rhesus macaques (Figure 2B). Transmission electron microscopy of purified VLRB MM3-reactive cells revealed typical ultrastructural PC characteristics, including greatly extended ER with abundant ribosomes. These results indicate that the VLRB MM3-reactive antigen is selectively expressed on the cell surface of normal plasmablasts and PCs in humans and NHPs. However, we found that VLRB MM3 did not recognize murine PCs (data not shown), thus eliminating mice as a suitable model species for studying the VLRB MM3 antigen.
Heterogeneity of human PCs has been demonstrated in recent studies (30, 31). Four PC subpopulations (IgD−/CD38++) can be distinguished by differential expression of the CD19 and CD138 cell-surface antigens: population “A” (CD19+/CD138−); population “B” (CD19+/CD138+); population “C” (CD19−/ CD138−); and population “D” (CD19−/CD138+) (12). Using these distinguishing criteria, we determined that VLRB MM3 was reactive with each of these PC populations in BM and blood (Figure 3, top row). Moreover, following vaccination, both CD138+ and CD138− Ab-secreting PC or plasmablast populations, as defined by the immunophenotype CD19+/CD27++/CD38++, stained positive for VLRB MM3. These data indicate that the antigenic determinant recognized by the lamprey VLRB MM3 Ab is present on all of the presently defined populations of human PCs.
Figure 3. Reactivity of VLRB MM3 with distinct subpopulations of human PCs and plasmablasts.
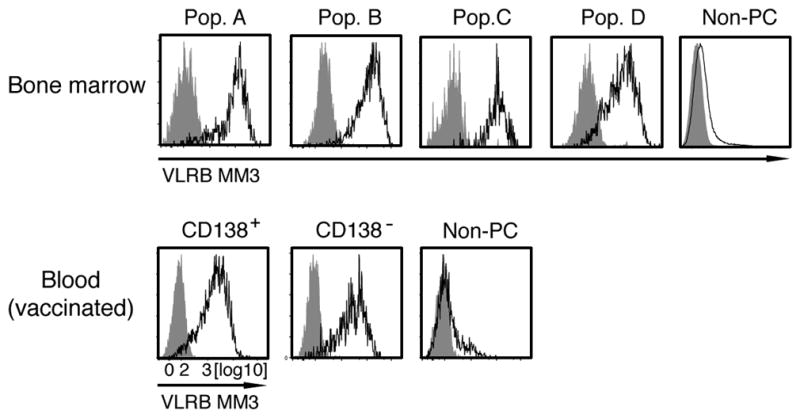
Top row: Human BM PCs were gated on IgD−/CD38++ cells and further subdivided into population “A” (CD19+/CD138−); population “B” (CD19+/ CD138+); population “C” (CD19−/CD138−); and population “D” (CD19−/CD138+). The various subpopulations were analyzed for reactivity with VLRB MM3 (black histogram) or negative control VLR4 (gray histogram). Shown is a representative of 3 BM samples. Bottom row: Plasmablasts from PBMCs collected on day 7 following vaccination were divided on the basis of CD138 expression and analyzed for reactivity with VLRB MM3 (black histogram) or negative control VLR4 (gray histogram). Shown is a representative of 8 vaccinated PBMC samples. PBMCs, peripheral blood mononuclear cells; PCs, plasma cells; Pop., population; VLR, variable lymphocyte receptor.
Monoclonal VLRB MM3 recognizes the CD38 antigen
The unprecedented PC specificity of the VLRB MM3 Ab raised the issue of the nature of the target antigen. Initially, we screened a large panel of cell lines in search of VLRB MM3-reactive immortalized cells, the study of which could facilitate identification and characterization of the target antigen. As expected, no human cell lines of non–B lineage were reactive with the VLRB MM3 Ab. To our surprise, VLRB MM3 did not react with the 9 plasmacytoma cell lines that we examined (Figure 4A, top left panel), all of which express the CD138 antigen that is commonly used as a PC marker, although VLRB MM3 consistently recognized PCs in all of the tissue samples obtained by surgical tonsillectomy (Figure 4A, top right panel). Among the human cell lines examined, we found that VLRB MM3 reacted strongly with the Daudi cell line of Burkitt’s lymphoma origin (Figure 4A, top left panel), thereby providing a model cell line for studying the VLRB MM3 target antigen. Treatment of Daudi cells with proteinase K prior to staining with VLRB MM3 and flow cytometric analysis greatly reduced binding of the VLRB Ab (data not shown), suggesting that the epitope recognized by VLRB MM3 could be located on a cell-surface (glyco)protein. We then used the VLRB MM3 Ab as an affinity reagent to isolate and purify the predicted protein antigen from Daudi cells, followed by molecular identification of the antigen by tandem mass spectrometry (Table 1). The most prominent signal obtained from mass spectrometric analyses of the VLRB MM3 IPs indicated the CD38 molecule, a multifunctional ectoenzyme with ADP-ribosyl cyclase, NAD+ nucleosidase, and cyclic ADP-ribose hydrolase activity (32). The credence of this result was supported by its concordance with the relatively high levels of CD38 expression by the Daudi B cells and primary PCs (Figure 4A, bottom panels).
Figure 4. Recognition of CD38 by VLRB MM3.

(A) Reactivity of panels of human cell lines (left) and primary human tonsillar lymphocyte populations (right) with VLRB MM3 (top row) and conventional anti-CD38 Abs (bottom row). Shown are mean the fluorescence intensities ± SD for cell lines (n = 3) and the median fluorescence intensities (n = 16) for primary cells normalized to negative control VLR4 or isotype control Abs, respectively. (B) Daudi B cells were transiently transfected with siRNA targeting CD38 or scrambled control siRNA and GFP expression plasmids and stained with VLRB MM3 and anti-CD38, respectively. In control experiments, cells were stained with VLRB EHT46 and anti-CD20, respectively. Shown are the mean fluorescent intensities normalized to the indicated controls ± SD (n = 5) gated on GFP-positive, propidium iodide–negative cell populations. *P < 0.05, by Student’s t test. (C) Ablation of VLRB MM3 reactivity by targeting CD38 in Daudi B cells using CRISPR/Cas9. Daudi B cells transfected with CRISPR/Cas9 constructs targeting CD38 or GFP as negative control were stained with VLRB MM3 and anti-CD38 Abs. Similar results were obtained with a second, independent CRISPR/Cas9 construct targeting CD38. BL, B lymphoid leukemia; DLBL, diffuse large B cell lymphoma; GC, germinal center; MFI, mean fluorescence intensity; Non-B/T, non–B/T cell; norm., normalized; VLRB, variable lymphocyte receptor B.
Table 1.
Protein antigens identified in LC-MS/MS analyses of VLRB MM3 IPs
| Protein ID | Gene name | Protein name/function | MS signal Control #1 | MS signal Control #2 | MS signal MM3 #1 | MS signal MM3 #2 |
|---|---|---|---|---|---|---|
| P28907 | CD38 | ADP-ribosyl cyclase | 0 | 0 | 68985 | 111980 |
| P62266 | RPS23 | 40S ribosomal protein S23 | 0 | 33028 | 88750 | 51479 |
| Q9HBD1 | RC3H2 | RING finger and CCCH-type zinc finger domain containing protein 2 | 0 | 127130 | 78994 | 189860 |
| P62753 | RPS6 | 40S ribosomal protein S6 | 0 | 35072 | 23043 | 47566 |
| Q00839 | HNRNPU | Heterogeneous nuclear ribonucleoprotein U | 0 | 34950 | 41117 | 42991 |
The top 5 protein antigens from 2 independent VLRB MM3 IPs and their respective negative controls (no VLRB) are shown. Values indicate label-free quantification (LFQ) intensities. LC-MS/MS, liquid chromatography–tandem mass spectrometry; MS, mass spectrometry; VLRB, variable lymphocyte receptor B.
In order to verify the specificity of VLRB MM3 for CD38, we used siRNA and CRISPR/Cas9 approaches to selectively suppress CD38 expression on the Daudi B cells. Reduction of CD38 cell-surface expression by siRNA treatment resulted in reduced reactivity with conventional anti-CD38 Abs and with VLRB MM3 (Figure 4B). In a control experiment, the CD38 siRNA treatment did not affect recognition of Daudi cells by a lamprey mAb, VLRB EHT46, which recognizes a ubiquitously expressed cell-surface antigen. Similarly, staining of the transfected Daudi cells with conventional anti-CD20 Abs was unaffected, demonstrating specificity of the experimental approach. When CD38 expression on Daudi B cells was reduced using CRISPR/ Cas9 constructs targeting CD38, we observed a concomitant reduction of VLRB MM3 reactivity, whereas no reduction in VLRB MM3 reactivity was observed in control GFP-targeting experiments (Figure 4C).
As another means of testing the specificity of the VLRB MM3 Ab to an epitope on the CD38 molecule, we generated BJAB 5–38 cells, a BJAB transfectant that constitutively expresses the CD5-GFP and CD38-GFP fusion proteins at the cell surface. The cells were incubated with monoclonal VLR32 (specific to the human CD5 antigen, ref. 23) VLRB MM3, or VLR4 (specific to the control BclA antigen of the exosporium of Bacillus anthracis, ref. 29), followed by cell lysis and IP of the cell lysates with Abs directed against the HA-epitope tag located in the invariant VLRB stalk region. Whereas the CD5-GFP fusion protein was detected in the VLR32 IPs, the CD38-GFP fusion protein was detected in the VLRB MM3 IPs, further evidence for the CD38 specificity of the VLRB MM3 Ab (Supplemental Figure 1).
VLRB MM3 recognition of CD38 correlates with CD38 enzymatic activity
Our observation of VLRB MM3 reactivity with some CD38-expressing cells and not with others raised the possibility that relatively high levels of CD38 expression are required for VLRB MM3 recognition. In experiments in which we transiently transfected BJAB cells with plasmids encoding CD38-EGFP fusion proteins before flow cytometric analysis for binding by VLRB MM3 or a conventional anti-CD38 Ab, we observed that VLRB MM3 recognized only the cells expressing the highest levels of CD38-EGFP (Figure 5A, top row, left panel). Once an apparent threshold of CD38 expression levels was crossed, the VLRB MM3 staining intensity increased dramatically. In contrast, staining of the CD38-EGFP transfectants with conventional anti-CD38 Abs showed an expected direct correlation of staining intensity with CD38-EGFP expression levels. In control experiments, we transfected BJAB cells with plasmids encoding CD5-GFP fusion constructs and analyzed them by flow cytometry using conventional anti-CD5 Abs or with the lamprey CD5-specific VLR32 Ab (28). These experiments also revealed the expected direct correlation of cell-surface staining with increasing amounts of CD5-EGFP fusion protein expression (Figure 5A, right panels), indicating that the threshold-based recognition of CD38 by VLRB MM3 was not a binding characteristic inherent to all VLRB Abs. Similar results were obtained when these experiments were repeated in the OCI-my5 plasmacytoma cell line (data not shown).
Figure 5. Dimerization of CD38 increases recognition by VLRB MM3.

(A) BJAB cells were transiently transfected with the indicated CD38-EGFP or CD5-EGFP fusion constructs and stained with VLRB MM3 (specific to CD38), VLR32 (specific for CD5), conventional anti-CD38 or anti-CD5 Abs (top row) or with the negative control VLR4 or isotype-matched conventional Ab (bottom row). Ab reactivity was assessed 48 hours after transfection (gated on propidium iodide–negative cells). Shown is a representative of at least 6 independently performed analyses. Note the apparent threshold-based recognition of CD38 by VLRB MM3. (B) Schematic of CD38-EGFP-GyrB fusion constructs, permitting inducible dimerization following addition of coumermycin. (C) BJAB cells transiently transfected with CD38-EGFP-GyrB constructs were stained with VLRB MM3 or conventional anti-CD38 Abs following incubation with coumermycin for 20 minutes at the indicated concentrations. Median fluorescence intensities for reactivity of VLRB MM3 and anti-CD38 Abs normalized to VLR4 and isotype control Abs, respectively, were assessed for the indicated gates on the basis of EGFP expression levels. Depicted are mean fluorescence intensity values ± SD (n = 4). *P < 0.05, by Student’s t test. Cm, coumermycin; MFI, mean fluorescence intensity; VLRB, variable lymphocyte receptor B.
The CD38 antigen is mostly monomeric but can also exist in dimeric and tetrameric conformations, the latter of which has been shown to be associated with its enzymatic activity (33, 34). The unique staining pattern obtained with VLRB MM3 could thus be related to the quarternary structure of CD38. To examine this possibility, we generated CD38-GFP-GyrB fusion constructs to allow for induced dimerization of CD38 upon addition of coumermycin (Figure 5B, Supplemental Figure 2, and ref. 35). Flow cytometric analysis of VLRB MM3 binding of BJAB B cells transiently transfected with this construct revealed that coumermycin treatment enhanced VLRB MM3 binding in a dose-dependent manner (Figure 5C, top panels). Whereas VLRB MM3 binding by the untreated cells was only observed for cells with the highest levels of CD38 expression (GFP gate II), the coumermycin treatment–related dimerization permitted VLRB MM3 recognition of cells with relatively low levels of CD38 expression, which, in the absence of coumermycin, did not bind VLRB MM3 (GFP gate I). VLRB MM3 binding was not altered by the addition of solvent controls (data not shown). At the highest concentrations of coumermycin, the VLRB MM3 staining pattern closely resembled that observed with conventional anti-CD38 Abs, in keeping with the possibility that efficient VLRB MM3 binding to CD38 requires prior dimerization of the antigen. In contrast, the binding pattern of conventional anti-CD38 Abs against the transfected cells did not change in response to coumermycin-induced dimerization (Figure 5C, bottom panels). In other studies, we tested whether glycosylation of CD38 might contribute to recognition of cells by VLRB MM3. Individual ablation by site-directed mutagenesis of each of the 4 N-linked glycosylation sites of CD38 did not affect binding of VLRB MM3 to the transfected cells (data not shown).
Ligation of cell-surface receptors frequently leads to cell activation. In the case of the CD38 ectoenzyme, Ab-mediated cross-linkage was shown to result in increase [3H] thymidine incorporation (36), in protection of germinal center cells from apoptosis (37), and in tyrosine phosphorylation of multiple intracellular proteins, including CD19 (38); it also resulted in an increase of NAD-hydrolase activity in a murine pro–B cell line (39). We therefore investigated whether VLRB MM3 binding to CD38 would correlate with CD38 enzymatic activity. Treatment of the CD38-GFP-GyrB–expressing BJAB cells with coumermycin and β-ara-2′-deoxy-2′-fluoro NAD (araF), a nonhydrolyzable NAD analog inhibitor of CD38, followed by incubation at 37°C prior to staining with VLRB MM3, resulted in a concentration-dependent reduction of VLRB MM3 binding (Figure 6A, top panel, gated on GFP-levels corresponding to gate II, indicated in Figure 5C). In contrast, treatment of the same cells with NAD did not affect VLRB MM3 binding. Recognition of CD38 by conventional anti-CD38 Abs was also unaffected in these experiments (data not shown). Assessment of NAD hydrolase activity of the CD38-GFP-GyrB–transfected cells following addition of the dimerizing agent coumermycin revealed that increased VLRB MM3 binding correlated with increased NAD hydrolase activity (Figure 6B, top left panel). Increased NAD hydrolase activity was also observed following addition of conventional anti-CD38 Abs against Daudi B cells (Figure 6B, bottom right panel). The observed increases in in NAD hydrolase activity following dimerization with coumermycin or anti-CD38 Abs are noteworthy, since not all anti-CD38 reagents are agonistic in nature (40–42). However, the enzymatic activity of exogenous CD38 following coumermycin treatment (Figure 6B, top right panel) and endogenous CD38 (Figure 6B, bottom left panel) were reduced when cells were preincubated with MM3 prior to addition of the fluorescent CD38 substrate. The residual enzymatic activity in these experiments is likely due to endogenous NAD hydrolases that are distinct from CD38, since NAD is not a CD38-specific substrate. These concordant results indicate that VLRB MM3 recognizes an epitope of multimeric CD38 that correlates with its enzymatic activity.
Figure 6. VLRB MM3 binding correlates with CD38 enzymatic activity.

(A) VLRB MM3 binding to exogenous (BJAB, top panel) and endogenous (Daudi, bottom panel) CD38. BJAB cells were transiently transfected with CD38-EGFP-GyrB fusion constructs, followed by coumermycin treatment (1 μM) in the presence of the nonhydrolyzable inhibitor araF or NAD controls for 40 minutes prior to addition of Abs. VLRB MM3 binding to transfected BJAB cells was assessed for GFP-positive cells corresponding to gate II of Figure 5. Data represent the median fluorescence intensity (n = 4) ± SD, normalized to values obtained for cells without coumermycin treatment. *P < 0.05, by Student’s t test. (B) Assessment of CD38 enzymatic activity following VLRB MM3 treatment in BJAB (top panels) and Daudi (bottom panels) B cells. BJAB cells were transiently transfected with CD38-EGFP-GyrB fusion constructs, followed by isolation of transfected cells and coumermycin treatment. NAD hydrolase activity was assessed and is shown as the ratio of coumermycin-treated cells relative to untreated cells (top, left panel). Coumermycin-treated cells were preincubated with either VLR4 or VLRB MM3 prior to addition of the NAD hydrolase substrate. NAD activity of VLRB-treated cells is shown relative to that of VLR4-treated controls (top, right panel). Statistical significance was determined using a 1-sample Student’s t test (n = 6). Assessment of endogenous CD38 enzymatic activity in Daudi B cells was performed following treatment with monoclonal VLR Abs (left panel) or conventional mAbs (right panel). NAD activity of the treated cells is shown as the ratio relative to the control Ab–treated cells. Statistical significance was determined using a 1-sample Student’s t test (n = 6). araF, β-ara-2′-deoxy-2′-fluoro NAD; CM, coumermycin; MFI, mean fluorescence intensity; norm., normalized; rel., relative; VLRB, variable lymphocyte receptor B.
VLRB MM3 recognition of malignant PCs in multiple myeloma
Since the VLRB MM3 Ab was isolated following immunization of lampreys with multiple myeloma cells and also recognizes all of the PCs in healthy donors, it was anticipated that VLRB MM3 would recognize the malignant PCs in all patients with multiple myeloma. However, when we examined the reactivity of VLRB MM3 with BM aspirates from 60 individuals diagnosed with multiple myeloma, only 51 (85%) of the myelomas were positive. The majority of multiple myelomas also expressed CD38, a commonly used marker for both normal and malignant PCs. In individual samples in which a small population of apparently normal VLRB MM3-positive PCs were found to coexist with a much larger clone of MM3-negative myeloma cells, both populations of PCs demonstrated CD38 expression of comparable magnitude (Supplemental Figure 3).
Discussion
Characterization of the VLR-based immune system of the evolutionarily distant sea lamprey has revealed that the ability to mount adaptive immune responses preceded the divergence of jawless and jawed vertebrates (22). Nonconventional VLRB Abs that are made by the jawless vertebrates may also provide novel reagents for biomedical research and clinical applications. Using a strategy to generate monoclonal VLRB Abs (43), we have immunized lamprey larvae with multiple myeloma cells to generate the PC-specific VLRB MM3 Ab. The monoclonal VLRB MM3 Ab recognizes a cell-surface antigen that is expressed at all stages of PC differentiation in humans, from plasmablasts to the most mature PCs, and that is also expressed on PCs in rhesus macaque and sooty mangabey NHPs. Currently, PCs are identified by the use of panels of conventional Abs against antigens not exclusively expressed by PCs. As an example, the CD138 antigen, a commonly used PC marker, is absent on certain PC populations (12, 15) and is also present on cells of non–B cell lineages, including epithelial cells and monocytes and macrophages (13, 44). As a PC-specific reagent, the VLRB MM3 Ab can be used alone for precise identification of PCs.
Mass spectrometric studies unambiguously indicated that the CD38 glycoprotein is the antigen recognized by the VLRB MM3 Ab. CD38 is expressed at high levels on PCs, and thus it was anticipated that MM3 would recognize all cells, which express sufficiently high CD38 levels. However, we demonstrate differential VLRB MM3 recognition of malignant and nonmalignant PCs with comparable CD38 expression levels, which suggests that VLRB MM3 may recognize a unique CD38 epitope expressed on PCs that is not solely dependent on sufficient levels of CD38 cell-surface expression. CD38 is a multifunctional ectoenzyme having ADP-ribosyl cyclase, NAD+ nucleosidase, and cyclic ADP-ribose hydrolase activities and a high degree of similarity to the adenosine diphosphate ribosyl cyclase of the mollusk Aplysia californica (45). Guided by earlier studies indicating that CD38 molecules exist in monomeric, dimeric, and tetrameric forms and that the tetramer is required for enzymatic activity, we found that VLRB MM3 binding could be enhanced by artificially inducing CD38 dimerization without changing the overall level of CD38 expression. This finding suggests that the VLRB MM3 Ab may recognize dimeric or potentially tetrameric CD38. Our further observations that a nonhydrolyzable analog of NAD interferes with VLRB MM3 binding to CD38 and that pretreatment of the cells with VLRB MM3 prior to addition of a fluorescent substrate of CD38 interfered with its enzymatic activity raise the possibility that VLRB MM3 may recognize an epitope present on active CD38. The apparent competition of VLRB MM3 with the nonhydrolyzable CD38 substrate suggests that the VLRB MM3 target epitope could be located in the enzymatic cleft of the protein. This mode of antigen recognition by a monoclonal VLR Ab would be comparable to that described for the lamprey VLRB.2D Ab, which recognizes hen egg lysozyme by inserting the variable loop that protrudes from the capping C-terminal LRR into the catalytic site of the enzyme, which is inaccessible to conventional Abs (27). Determination of the structure of VLRB MM3 in complex with its CD38 antigen will resolve this question.
The correlation of VLRB MM3 recognition of PCs with enzymatic activity of the CD38 molecule may have interesting implications. It is conceivable that the products of this enzymatic activity (cADPR, ADPR, and NAADP+), which regulate cellular calcium mobilization, may influence cells in the microenvironment that provide a survival niche for long-lived PCs. On the other hand, CD38 was also reported to colocalize with the antigen receptor complex in lipid rafts (46) and to interact with the nonsubstrate ligands CD31 and hyaluronic acid (47, 48). It will be informative to determine whether these interactions and subsequent cellular responses differ depending on whether CD38 can be recognized by VLRB MM3. In addition, activation status–dependent recognition of CD38 by VLRB MM3 may find application as an additional prognostic factor in the clinicopathological assessment of disorders in which the evaluation of cell-surface expression of CD38 is already established practice.
Our analysis of VLRB MM3 reactivity with healthy and malignant PCs indicated that approximately 15% of multiple myelomas are not recognized by this reagent. A pilot survey suggests that reduced or absent VLRB MM3 binding is correlated with the presence of the poor prognostic factors del17p, t(14;16), or t(4;14) defined by FISH chromosomal analysis (D.L. Jaye, unpublished observations), but this suggestion will require confirmation with larger patient cohorts. The expression of high CD38 levels in B cell chronic lymphocytic leukemia is associated with a more aggressive disease course (49). CD38 is also a promising therapeutic target for mAbs in the treatment of multiple myeloma (50, 51). Our study indicates that the epitope recognized by the lamprey VLRB MM3 Ab may present a novel target for PC-specific therapeutic intervention.
In conclusion, we describe the isolation of a lamprey mAb with PC specificity. The VLRB MM3 Ab recognizes an epitope on the CD38 ectoenzyme that is present on all normal PCs and in most multiple myelomas in humans. The binding of VLRB MM3 to CD38 correlates with CD38 NAD hydrolase activity and suppresses this enzymatic function. This Ab offers a unique reagent for evaluation, and possibly modification, of immune responses and PC disorders.
Methods
Cells and reagents
HEK293T cells were cultured in DMEM supplemented with glutamine, 100 U/ml penicillin-streptomycin, and 10% FBS. All human hemopoietic cell lines were provided by S. Trudel and G. Ehrhardt (University of Toronto, Toronto, Ontario, Canada) and M. Cooper (Emory University, Atlanta, Georgia, USA) and were cultured in RPMI 1640 supplemented with glutamine, 100 U/ml penicillin-streptomycin, 50 μM 2-mercaptoethanol, and 10% FBS. Cells were maintained in a humidified atmosphere at 37°C and 5% CO2. Abs against cell-surface antigens CD19 (clone HIB-19); CD20 (clone H1(FB1)); CD27 (clone M-T271); CD38 (clone HIT-2); CD138 (clone MI15); CD10 (clone HI10a); CD45 (clone HI30); CD24 (clone ML5); and IgD (clone IA6-2) were obtained from BD Biosciences. Abs against CD3 (clone HIT-3a) were purchased from BioLegend. Monoclonal anti-GFP Abs (clone B-2) were purchased from Santa Cruz Biotechnology Inc. Fluorescently labeled secondary Abs were purchased from SouthernBiotech. Monoclonal anti-VLRB Ab (clone 4C4) was described previously (52). Rabbit anti–β-actin Abs, coumermycin, NAD+, and nicotinamide 1,N6-ethenoadenine dinucleotide were obtained from Sigma-Aldrich; NHS-biotin, DTSSP, and BS3 from ThermoFisher; and araF from BioLog.
Tonsil samples were obtained from the Hospital for Sick Children (Toronto, Ontario, Canada). BM aspirates and spleen tissue were obtained from Emory University Hospital (Atlanta, Georgia, USA) and Princess Margaret Hospital (Toronto, Ontario, Canada). Peripheral blood was obtained from healthy donors and was heparinized.
Isolation of monoclonal VLRB Abs
Sea lamprey larvae (10–12 cm in length) were immunized with 5 × 106 BM aspirate cells from a patient with multiple myeloma in 0.66× PBS (25, 28). Prior to immunization, BM aspirate cells were subjected to depletion of CD45+ cells using magnetic separation. The animals were boosted twice in 2-week intervals before harvest of lymphocyte-like cells from the blood. Total RNA was extracted from the harvested lymphocytes using the RNeasy system (QIAGEN), and cDNA was generated using random oligo hexamers. The cDNA was used as a template for PCR amplification of VLRB sequence using the oligo sequences 5′-ATATGCTAGCCACCATGTGGATCAAGTGGATCGCCACGC-3′ and 5′-ATATACCGGTTCAACGTTTCCTGCAGAGGGCG-3′ as sense and antisense primers, respectively. The resulting VLRB library was cloned into the eukaryotic expression vector pIRESpuro2 and transformed into DH5α-competent bacteria. Plasmid DNA was isolated from individual bacterial colonies and used for transfection of HEK293T cells. The VLRB-containing culture supernatant was verified by Western blotting for the presence of monoclonal VLR Abs as described previously (28) and subsequently used for staining of BM aspirate cells. Plasmids encoding monoclonal VLRB Abs recognizing BM aspirate cells were selected for further analysis and their composition determined by DNA sequencing. Monomeric VLRB MM3 was generated by deleting sequences encoding the C-terminal 42 aa of the protein (28).
Generation of recombinant monoclonal VLRB Abs
VLRB sequences were subcloned into the eukaryotic expression vector p367HH, a modified version of the pIRESpuro2 vector that contains sequences encoding the invariant VLRB stalk region as well as the 6xHis and HA epitope tags. The plasmids were transfected into HEK293T cells using the polyethylenimine (PEI) method (53). Three days after transfection, the culture supernatants were analyzed by Western blotting for the presence of VLRB Abs and used for staining of primary cells and cell lines. Alternatively, the cells were drug selected with puromycin (1 μg/ml) and larger volumes of culture supernatant subjected to affinity chromatography using Ni-columns to obtain purified VLRB Abs as described previously (28). In some experiments, the LRR units encoding the VLRB antigen–binding region were fused to the Fc domain of rabbit IgG and transfected into HEK293T cells, and the resulting Fc-fusion proteins were purified using protein G Sepharose beads.
Staining of cell lines and primary cells with VLRB Abs
Cell lines were incubated with VLRB-containing culture supernatant or purified monoclonal VLRB Abs (for 20 minutes on ice at 500 ng/ml in PBS containing 1% BSA). Subsequently, the cells were incubated with anti-VLRB Ab 4C4, followed by detection of bound VLRB molecules by fluorescently labeled goat anti-mouse Abs. Alternatively, bound VLRB Abs were detected by incubation with fluorescently labeled anti-VLRB Ab 5-8A3 or with anti–HA-epitope tag Abs. Peripheral blood mononuclear cells (PBMCs) and tonsillar lymphocytes were isolated by density-gradient centrifugation over lymphocyte separation medium for 20 minutes at 750 g. Primary cells were incubated with monoclonal VLRB Abs as described, followed by addition of a cocktail of Abs containing directly labeled anti-VLRB Abs as well as mouse mAbs reactive with selective cell-surface antigens. In cases in which bound VLRB Abs were detected using mouse monoclonal anti-VLRB Ab 4C4 and goat anti-mouse fluorescently labeled secondary reagents, the cells were blocked extensively with 10% normal mouse serum for 15 minutes on ice prior to addition of a cocktail of lineage-specific, fluorescently labeled mouse mAbs. In some cases, cells were incubated with fusion proteins consisting of the antigen-binding unit of VLRB MM3 fused to the Fc domain of rabbit IgG at a concentration of 250 ng/ml. Bound VLRB MM3-Ig fusion proteins were in turn detected by incubation with rabbit Fc–specific fluorescently labeled Abs. Dead cells were excluded by inclusion of propidium iodide (1 μg/ml) or Aqua Dead Cell Staining Reagent (Life Technologies). Acquired flow cytometric data were analyzed using the FlowJo software package.
Identification of CD38 as antigen recognized by VLRB MM3
Daudi B cells (1 × 108) were surface biotinylated and stained with VLRB MM3, followed by cross-linkage using the amine-reactive, membrane-nonpermeable, thiol-sensitive DTSSP cross-linker for 30 minutes at room temperature. The reaction was quenched by addition of 100 mM Tris, pH 7.5, prior to lysis of the cells in 1 ml Nonidet P-40 lysis buffer (1% Nonidet P-40, 150 mM NaCl, 5 mM EDTA, 50 mM Tris, pH 7.5) in the presence of protease inhibitors (5 μg/ml leupeptin, 1 μg/ml pepstatin, 5 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, 40 μg/ml PMSF) for 10 minutes on ice. The cell lysates were centrifuged at 20,000 g for 10 minutes at 4°C, and the supernatants were subjected to IP using 5 μg anti-HA Abs and 30 μl of a 50% slurry of Protein G Sepharose (GE Healthcare). Five percent of the resulting IPs were subjected to SDS-PAGE under reducing conditions and analyzed by Western blotting using HRP-labeled streptavidin and ECL reagent. The remaining 95% of the IPs were used for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis of protein antigens purified by VLRB MM3 as described before (28). Briefly, VLRB MM3 IPs and control precipitates consisting of Daudi IPs without initial VLRB MM3 incubation were washed and eluted in 0.15% trifluoroacetic acid (TFA), neutralized to l C18 stage 100 mM NH4HCO3, and digested with trypsin (54). The tryptic peptides were purified using 200 μ tips (Thermo Scientific) and analyzed using a Q-Exactive LC-MS/MS instrument (Thermo Fisher). Acquired MS raw files were analyzed using MaxQuant software (version 1.3.0.5) as described previously (55).
Co-IP of CD38 with VLRB MM3
BJAB 5–38 cells (1 × 107) were stained with VLR4, VLRB32, or VLRB MM3 for 20 minutes on ice, followed by addition of the DTSSP cross-linker and cell lysis as described above. The VLR Abs were IP using 2 μg anti-HA tag Abs (clone 12CA5; Roche), and the IPs and whole-cell lysates (WCLs) were resolved by SDS-PAGE under reducing conditions and transferred onto nitrocellulose membranes. The membranes were incubated with monoclonal anti-GFP Abs and HRP-labeled anti-mouse secondary Abs, followed by incubation with ECL reagent and detection using a MicroChemi 4.2 instrument (Gentaur).
CD38 ablation by siRNA and CRISPR/Cas9
Ablation of CD38 expression was performed by transiently transfecting Daudi B cells with 10 pmol CD38-specific siRNA or scrambled control siRNA (Silencer Select, Ambion) and 500 ng pcDNA3.1 vector expressing the GFP fluorescent marker using the Amaxa Nucleofection T system (Lonza) and the setting O-20. Cells were analyzed for VLR Ab binding and CD38 expression levels 48 hours after transfection using a FACSCalibur instrument (BD Biosciences) in the presence of 1 μg/ml propidium iodide for dead cell exclusion. In an alternative approach, CRISPR/Cas9 technology was used to disrupt the CD38 gene in Daudi B cells. Two guide RNAs (gRNAs), 5′-CCTCGTCGTG-GTGCTCGCGG-3′ (gRNA-1) and 5′-TCGCGGTGGTCGTCCCGAGG-3′ (gRNA-2), were designed to target exon 1 of human CD38 using the DNA2.0 CRISPR gRNA Design Tool (DNA 2.0). The gRNAs were synthesized by DNA2.0 and cloned into the pD1301-AD CRISPR/Cas9 expression vector, which encodes for the gRNA driven by the U6 promoter and Cas9-2A-GFP under the control of the CMV promoter. Daudi cells were transfected by electroporation using a Nucleofector device and Nucleofector Kit V (Lonza). Transfected Daudi cells were evaluated by flow cytometry for CD38 recognition by anti-CD38 and VLRB MM3 Abs 5 days after electroporation.
Generation of GFP-fusion constructs
Generation of CD5-GFP constructs was described previously (28). cDNA encoding human CD38 was obtained from the plasmid repository of Harvard Medical School. The cDNA sequence was PCR amplified and cloned into the pEGFP-C1 vector (Clontech) to generate CD38-GFP fusion constructs. To generate CD38-GFP-GyrB fusion constructs with the linker sequence GSEGKSSGSGSESKVDTS separating the GFP and GyrB domains, sequences encoding the 226 N-terminal aa of bacterial gyrase B were amplified with oligonucleotides “740” (TATGCTAGCCACCATGTC-GAATTCTTATGACTCCTCC) and “742” (CTTGCTCTCGCTTCCGGAGCCGCTGCTCTTTCCCTC-GGATCCGCCGCCTTCATAGTGGAAGTGGTC), whereas the CD38-GFP fusion sequences were amplified with oligonucleotides “741” (AAGAGCAGCGGCTCCGGAAGCGAGAGCAAGGTGGA-CACATCTATGGTGAGCAAGGGCGAGGAGCTG) and “743” (ATAGCGGCCGCTCAGATCTCA-GATGTGCAAGATGAATC). The 30 nt overlap in the amplified DNA fragments allowed for PCR-mediated fusion in a subsequent round of PCR amplification with oligonucleotides 740 and 743, using the 2 primary PCR products as templates. The resulting PCR product was cloned into the eukaryotic expression vector pIRESpuro2 (Invitrogen) using the NheI and NotI restriction enzymes, and sequence fidelity was verified by DNA sequencing.
Overexpression of CD38 and CD5 fusion constructs in BJAB B cells
BJAB cells (3 × 106 per transfection) were washed twice with PBS prior to resuspension in 800 μl electroporation buffer (25 mM HEPES, pH 7.2, 0.75 mM Na2HPO4, 140 mM KCl, 5 mM NaCl, 2 mM MgCl2, 0.5% Ficoll 400) and electroporation using a Bio-Rad Gene Pulser Xcell instrument (275 V, 975 μF). The cells were incubated for 48 hours, followed by analysis for VLRB Ab binding. In experiments using CD38-GFP-GyrB fusion constructs, the cells were washed twice prior to addition of the indicated amounts of coumermycin and an additional incubation for 25 minutes at 37°C. Coumermycin was prepared as a 4.5-mM stock solution in DMSO. In experiments involving the CD38 substrate NAD+ or the nonhydrolyzable CD38 inhibitor araF, this incubation period was extended to 40 minutes to allow for enzymatic processing of NAD+. Subsequently, the cells were stained with the indicated Abs as well as propidium iodide for dead cell exclusion and and analyzed by flow cytometry using a Guava EasyCyte HT instrument (EMD Millipore). Binding of VLRB MM3 or conventional anti-CD38 Abs against endogenous CD38 in Daudi cells was determined accordingly, without addition of coumermycin. Acquired data were analyzed using FlowJo software.
Dimerization of CD38-GFP-GyrB following coumermycin treatment was verified by Western blotting using BJAB cells stably expressing the fusion protein. Following addition of coumermycin (1 μM) for 20 minutes, the cells were incubated with the amine-reactive, membrane nonpermeable, thiol-insensitive BS3 cross-linker for 30 minutes at room temperature prior to quenching of the reaction by addition of 100 mM Tris, pH 7.5, and lysis of the cells in 1 ml Nonidet P-40 lysis buffer. The samples were resolved by SDS-PAGE under reducing conditions and analyzed by Western blotting for the presence of covalently linked CD38-GFP-GyrB protein complexes using anti-GFP Abs as described above.
Detection of CD38 enzymatic activity
For detection of CD38 enzymatic activity in transiently transfected BJAB cells, the CD38-GFP-GyrB fusion protein was cloned into an expression vector coexpressing the extracellular and transmembrane domains of murine CD4 downstream of an internal ribosome entry site (IRES) element. Forty-eight hours after transfection, the transfected cells were purified using anti-mCD4 magnetic beads (STEMCELL Technologies). Subsequently, the cells were left untreated or were treated with coumermycin in the presence or absence of the VLR Abs VLRB MM3 and VLR4 for 15 minutes. Monoclonal VLR Abs or conventional Abs were used at concentrations of 100 ng/ml and 250 ng/ml, respectively. The cells were then plated at a density of 1 × 105 cells per well in 96-well plates prior to addition of 40 μM nicotinamide 1,N6-ethenoadenine dinucleotide. Enzymatic activity was determined following a 15-minute incubation using a Spectramax i3 instrument (Molecular Devices) at 310 nm/410 nm excitation/emission settings and normalized to the GFP signal obtained from the input material assessed at at 488 nm/510 nm excitation/emission settings. Enzymatic activity of endogenous CD38 in Daudi cells was determined accordingly, without addition of coumermycin or GFP normalization.
Statistics
Statistical significance was determined by a 2-tailed paired Student’s t test and a 1-sample t test. A P value of less than 0.05 was considered statistically significant.
Study approval
This study was approved by the ethics review boards of Emory University and the University of Toronto, in accordance with Declaration of Helsinki principles. All participants provided written informed consent. All animal work was approved by the IACUCs of Emory University and the University of Toronto.
Supplementary Material
Acknowledgments
We thank Hong Yi (Emory University, Atlanta, Georgia, USA) for help with electron microscopy. We are are grateful for the murine PC preparations provided by Yee Leung and J. Gommerman (University of Toronto, Toronto, Ontario, Canada). This study was supported by Canadian Cancer Society grant 2012-701054 (to G.R.A. Ehrhardt); NIH grant U19 AI096187 (to G.R.A. Ehrhardt and M.D. Cooper); and NIH grant R01 AI072435 (to M.D. Cooper). D.L. Jaye is supported by the Winship Cancer Institute of Emory University Cancer Center Support Grant P30CA138292, and the Electron Microscopy Core (Emory University, Atlanta, Georgia, USA) is supported by by NIH grant S10RR025679.
Footnotes
Author contributions
CY conducted and analyzed experiments. YL designed, conducted, and analyzed experiments. JTHC conducted and analyzed experiments. MS conducted and analyzed experiments. MP conducted and analyzed experiments. SK analyzed experiments. SK conducted experiments. JT conducted and analyzed experiments. WZ conducted and analyzed experiments. ZL conducted experiments. DD conducted experiments. EG provided specimens and critically appraised the manuscript. PC provided specimens and critically appraised the manuscript. EJP provided specimens and critically appraised the manuscript. DLJ provided reagents and analyzed experiments. ST provided reagents and analyzed experiments. MFM designed and analyzed experiments. MO designed and analyzed experiments. IS designed and analyzed experiments. BRH designed and analyzed experiments. FEYL designed and analyzed experiments. MDC designed and analyzed experiments and wrote the manuscript. GRAE designed, conducted, and analyzed experiments and wrote the manuscript.
Conflict of interest: The monoclonal VLRB MM3 Ab has been disclosed to the Office of Technology Transfer of Emory University. Authors listed on the disclosure are B.R. Herrin, M.D. Cooper, and G.R.A. Ehrhardt. F.E. Lee is the founder of MicroB-plex.
References
- 1.Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5(3):230–242. doi: 10.1038/nri1572. [DOI] [PubMed] [Google Scholar]
- 2.Hoyer BF, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. 2004;199(11):1577–1584. doi: 10.1084/jem.20040168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tarlinton DM, Hodgkin PD. Targeting plasma cells in autoimmune diseases. J Exp Med. 2004;199(11):1451–1454. doi: 10.1084/jem.20040719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 5.Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122(10):3456–3463. doi: 10.1172/JCI61188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuxa M, Busslinger M. Reporter gene insertions reveal a strictly B lymphoid-specific expression pattern of Pax5 in support of its B cell identity function. J Immunol. 2007;178(5):3031–3037. doi: 10.4049/jimmunol.178.5.3031. [DOI] [PubMed] [Google Scholar]
- 7.Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. Semin Immunol. 2011;23(5):341–349. doi: 10.1016/j.smim.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Zinkernagel RM, Bachmann MF, Kundig TM, Oehen S, Pirchet H, Hengartner H. On immunological memory. Annu Rev Immunol. 1996;14:333–367. doi: 10.1146/annurev.immunol.14.1.333. [DOI] [PubMed] [Google Scholar]
- 9.Manz RA, Thiel A, Radbruch A. Lifetime of plasma cells in the bone marrow. Nature. 1997;388(6638):133–134. doi: 10.1038/40540. [DOI] [PubMed] [Google Scholar]
- 10.Manz RA, Lohning M, Cassese G, Thiel A, Radbruch A. Survival of long-lived plasma cells is independent of antigen. Int Immunol. 1998;10(11):1703–1711. doi: 10.1093/intimm/10.11.1703. [DOI] [PubMed] [Google Scholar]
- 11.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8(3):363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 12.Halliley JL, et al. Long-lived plasma cells are contained within the CD19(−)CD38(hi)CD138(+) subset in human bone marrow. Immunity. 2015;43(1):132–145. doi: 10.1016/j.immuni.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeaman C, Rapraeger AC. Post-transcriptional regulation of syndecan-1 expression by cAMP in peritoneal macrophages. J Cell Biol. 1993;122(4):941–950. doi: 10.1083/jcb.122.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wrammert J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453(7195):667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, Brieva JA. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. 2002;99(6):2154–2161. doi: 10.1182/blood.v99.6.2154. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez-Garcia I, Ocana E, Jimenez-Gomez G, Campos-Caro A, Brieva JA. Immunization-induced perturbation of human blood plasma cell pool: progressive maturation, IL-6 responsiveness, and high PRDI-BF1/BLIMP1 expression are critical distinctions between antigen-specific and nonspecific plasma cells. J Immunol. 2006;176(7):4042–4050. doi: 10.4049/jimmunol.176.7.4042. [DOI] [PubMed] [Google Scholar]
- 17.Qian Y, et al. Elucidation of seventeen human peripheral blood B-cell subsets and quantification of the tetanus response using a density-based method for the automated identification of cell populations in multidimensional flow cytometry data. Cytometry B Clin Cytom. 2010;78(suppl 1):S69–S82. doi: 10.1002/cyto.b.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu D, et al. Immunophenotypic heterogeneity of normal plasma cells: comparison with minimal residual plasma cell myeloma. J Clin Pathol. 2012;65(9):823–829. doi: 10.1136/jclinpath-2012-200881. [DOI] [PubMed] [Google Scholar]
- 19.Finstad J, Good RA. The evolution of the immune response. 3. Immunologic responses in the lamprey. J Exp Med. 1964;120:1151–1168. doi: 10.1084/jem.120.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linthicum DS, Hildemann WH. Immunologic responses of Pacific hagfish. 3. Serum antibodies to cellular antigens. J Immunol. 1970;105(4):912–918. [PubMed] [Google Scholar]
- 21.Pancer Z, Amemiya CT, Ehrhardt GR, Ceitlin J, Gartland GL, Cooper MD. Somatic diversification of variable lymphocyte receptors in the agnathan sea lamprey. Nature. 2004;430(6996):174–180. doi: 10.1038/nature02740. [DOI] [PubMed] [Google Scholar]
- 22.Boehm T, McCurley N, Sutoh Y, Schorpp M, Kasahara M, Cooper MD. VLR-based adaptive immunity. Annu Rev Immunol. 2012;30:203–220. doi: 10.1146/annurev-immunol-020711-075038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo P, et al. Dual nature of the adaptive immune system in lampreys. Nature. 2009;459(7248):796–801. doi: 10.1038/nature08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirano M, et al. Evolutionary implications of a third lymphocyte lineage in lampreys. Nature. 2013;501(7467):435–438. doi: 10.1038/nature12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alder MN, et al. Antibody responses of variable lymphocyte receptors in the lamprey. Nat Immunol. 2008;9(3):319–327. doi: 10.1038/ni1562. [DOI] [PubMed] [Google Scholar]
- 26.Han BW, Herrin BR, Cooper MD, Wilson IA. Antigen recognition by variable lymphocyte receptors. Science. 2008;321(5897):1834–1837. doi: 10.1126/science.1162484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velikovsky CA, et al. Structure of a lamprey variable lymphocyte receptor in complex with a protein antigen. Nat Struct Mol Biol. 2009;16(7):725–730. doi: 10.1038/nsmb.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu C, et al. Purification and identification of cell surface antigens using lamprey monoclonal antibodies. J Immunol Methods. 2012;386(1–2):43–49. doi: 10.1016/j.jim.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silveira EL, et al. Vaccine-induced plasmablast responses in rhesus macaques: phenotypic characterization and a source for generating antigen-specific monoclonal antibodies. J Immunol Methods. 2015;416:69–83. doi: 10.1016/j.jim.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaminski DA, Wei C, Rosenberg AF, Lee FE, Sanz I. Multiparameter flow cytometry and bioanalytics for B cell profiling in systemic lupus erythematosus. Methods Mol Biol. 2012;900:109–134. doi: 10.1007/978-1-60761-720-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tipton CM, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol. 2015;16(7):755–765. doi: 10.1038/ni.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuber F, Lund FE. Structure and enzymology of ADP-ribosyl cyclases: conserved enzymes that produce multiple calcium mobilizing metabolites. Curr Mol Med. 2004;4(3):249–261. doi: 10.2174/1566524043360708. [DOI] [PubMed] [Google Scholar]
- 33.Hara-Yokoyama M, et al. Tetrameric interaction of the ectoenzyme CD38 on the cell surface enables its catalytic and raft-association activities. Structure. 2012;20(9):1585–1595. doi: 10.1016/j.str.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 34.Bruzzone S, Guida L, Franco L, Zocchi E, Corte G, De Flora A. Dimeric and tetrameric forms of catalytically active transmembrane CD38 in transfected HeLa cells. FEBS Lett. 1998;433(3):275–278. doi: 10.1016/s0014-5793(98)00929-6. [DOI] [PubMed] [Google Scholar]
- 35.Farrar MA, Olson SH, Perlmutter RM. Coumermycin-induced dimerization of GyrB-containing fusion proteins. Methods Enzymol. 2000;327:421–429. doi: 10.1016/s0076-6879(00)27293-5. [DOI] [PubMed] [Google Scholar]
- 36.Funaro A, et al. Involvement of the multilineage CD38 molecule in a unique pathway of cell activation and proliferation. J Immunol. 1990;145(8):2390–2396. [PubMed] [Google Scholar]
- 37.Zupo S, Rugari E, Dono M, Taborelli G, Malavasi F, Ferrarini M. CD38 signaling by agonistic monoclonal antibody prevents apoptosis of human germinal center B cells. Eur J Immunol. 1994;24(5):1218–1222. doi: 10.1002/eji.1830240532. [DOI] [PubMed] [Google Scholar]
- 38.Kitanaka A, Ito C, Coustan-Smith E, Campana D. CD38 ligation in human B cell progenitors triggers tyrosine phosphorylation of CD19 and association of CD19 with lyn and phosphatidylinositol 3-kinase. J Immunol. 1997;159(1):184–192. [PubMed] [Google Scholar]
- 39.Lund FE, et al. CD38 induces apoptosis of a murine pro-B leukemic cell line by a tyrosine kinase-dependent but ADP-ribosyl cyclase- and NAD glycohydrolase-independent mechanism. Int Immunol. 2006;18(7):1029–1042. doi: 10.1093/intimm/dxl037. [DOI] [PubMed] [Google Scholar]
- 40.Hara-Yokoyama M, et al. Alteration of enzymatic properties of cell-surface antigen CD38 by agonistic anti-CD38 antibodies that prolong B cell survival and induce activation. Int Immunopharmacol. 2008;8(1):59–70. doi: 10.1016/j.intimp.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 41.Vaisitti T, et al. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia. 2010;24(5):958–969. doi: 10.1038/leu.2010.36. [DOI] [PubMed] [Google Scholar]
- 42.Kumagai M, et al. Ligation of CD38 suppresses human B lymphopoiesis. J Exp Med. 1995;181(3):1101–1110. doi: 10.1084/jem.181.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herrin BR, et al. Structure and specificity of lamprey monoclonal antibodies. Proc Natl Acad Sci U S A. 2008;105(6):2040–2045. doi: 10.1073/pnas.0711619105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong ZM, Choo B, Li M, Carey DJ, Cano-Gauci DF, Buick RN. Syndecan-1 is up-regulated in ras-transformed intestinal epithelial cells. Br J Cancer. 1998;77(6):890–896. doi: 10.1038/bjc.1998.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.States DJ, Walseth TF, Lee HC. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem Sci. 1992;17(12):495. doi: 10.1016/0968-0004(92)90337-9. [DOI] [PubMed] [Google Scholar]
- 46.Deaglio S, et al. CD38/CD19: a lipid raft-dependent signaling complex in human B cells. Blood. 2007;109(12):5390–5398. doi: 10.1182/blood-2006-12-061812. [DOI] [PubMed] [Google Scholar]
- 47.Nishina H, Inageda K, Takahashi K, Hoshino S, Ikeda K, Katada T. Cell surface antigen CD38 identified as ectoenzyme of NAD glycohydrolase has hyaluronate-binding activity. Biochem Biophys Res Commun. 1994;203(2):1318–1323. doi: 10.1006/bbrc.1994.2326. [DOI] [PubMed] [Google Scholar]
- 48.Deaglio S, et al. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol. 1998;160(1):395–402. [PubMed] [Google Scholar]
- 49.Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood. 2011;118(13):3470–3478. doi: 10.1182/blood-2011-06-275610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flemming A. Deal watch: J&J and Genmab deal to push forward CD38 as a blood cancer target. Nat Rev Drug Discov. 2012;11(11):822. doi: 10.1038/nrd3890. [DOI] [PubMed] [Google Scholar]
- 51.de Weers M, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 52.Alder MN, Rogozin IB, Iyer LM, Glazko GV, Cooper MD, Pancer Z. Diversity and function of adaptive immune receptors in a jawless vertebrate. Science. 2005;310(5756):1970–1973. doi: 10.1126/science.1119420. [DOI] [PubMed] [Google Scholar]
- 53.Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Release. 1999;60(2–3):149–160. doi: 10.1016/s0168-3659(99)00090-5. [DOI] [PubMed] [Google Scholar]
- 54.Tong J, Taylor P, Moran MF. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol Cell Proteomics. 2014;13(7):1644–1658. doi: 10.1074/mcp.M114.038596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.