

Abstract
The majority of pancreatic cancer (PC) patients are clinically presented with obstructive jaundice with elevated levels of circulatory bilirubin and alkaline phosphatases. In the current study, we examined the implications of bile acids (BA), an important component of bile, on the pathophysiology of PC and investigated their mechanistic association in tumor-promoting functions. Integration of results from PC patient samples and autochthonous mouse models showed an elevated levels of BA (p<0.05) in serum samples compared to healthy controls. Similarly, an elevated BA levels was observed in pancreatic juice derived from PC patients (p<0.05) than non-pancreatic non-healthy (NPNH) controls, further establishing the clinical association of BA with the pathogenesis of PC. The tumor-promoting functions of BA were established by observed transcriptional upregulation of oncogenic MUC4 expression. Luciferase reporter assay revealed distal MUC4 promoter as the primary responsive site to BA. In silico analysis recognized two c-Jun binding sites at MUC4 distal promoter, which was biochemically established using ChIP assay. Interestingly, BA treatment led to an increased transcription and activation of c-Jun in a FAK-dependent manner. Additionally, BA receptor, namely FXR, which is also upregulated at transcriptional level in PC patient samples, was demonstrated as an upstream molecule in BA-mediated FAK activation, plausibly by regulating Src activation. Altogether, these results demonstrate that elevated levels of BA increase the tumorigenic potential of PC cells by inducing FXR/FAK/c-Jun axis to upregulate MUC4 expression, which is overexpressed in pancreatic tumors and is known to be associated with progression and metastasis of PC.
Keywords: Bile acids, Pancreatic cancer, MUC4, FAK, FXR, c-Jun
1. Introduction
In 2014, about 45,000 new cases of pancreatic cancer (PC) were diagnosed in the United States, of which pancreatic ductal adenocarcinoma (PDAC) represents the major histological type (Siegel et al., 2014). The majority of tumors (about 75%) arise at the head of the pancreas (Wisinski et al., 2007). Anatomically, the pancreatic duct is placed close to the common bile duct, and unite at the point known as the ampulla of Vater, and secrete their contents into the duodenum, the proximal site of the intestine (Bardeesy and DePinho, 2002). Approximately, 70% of PC patients develop extrahepatic cholestasis due to blockage of the common bile duct by increasing tumor size and results in multiple organ failure and early death (Nakamura et al., 2002). Due to bile duct obstruction, extrahepatic cholestasis exhibits obstructive jaundice, hyperbilirubinemia and elevated circulatory levels of bile acids (BA).
BA are amphiphilic molecules and are the main component of bile along with cholesterol, phospholipids, and bilirubin (Baptissart et al., 2013). By utilizing a series of enzymatic modifications, BA are synthesized in the liver using cholesterol as a precursor. BA are further modified by bacterial species present in the colon to form secondary BA (Baptissart et al., 2013). Dietary fat is a stimulus for BA secretion into the intestine, which is required for the proper digestion of fatty foods (Baptissart et al., 2013). Though bile-reflux has been associated with esophageal and gastric cancers, its association with PC pathogenesis has not been investigated (Sifrim, 2013; Tsoukali and Sifrim, 2013). A recently performed meta-analysis has revealed an increased risk of PDAC in patients with the cholecystectomy history (Lin et al., 2012). It has been proposed that the increased levels of cholecystokinin, which is known to stimulate the growth of human PC cell lines, promote pancreatic carcinogenesis in hamsters (Howatson and Carter, 1985).
BA have been shown to participate in tumor progression using multiple mechanisms including, alteration in the expression of oncogenic mucins (Mariette et al., 2004; Piessen et al., 2007). Interestingly, PC is characterized by aberrant mucins expression (Kaur et al., 2013c; Joshi et al., 2014; Joshi et al., 2015). Among various mucins expressed in PC, MUC4 is one of the top-differentially expressed protein compared to normal pancreas (Andrianifahanana et al., 2001; Iacobuzio-Donahue et al., 2003). We and others have established the oncogenic role of MUC4 in PC, and inhibition of MUC4 expression led to reduced PC cell proliferation, migration, and chemoresistance (Moniaux et al., 2007; Chaturvedi et al., 2007; Lakshmanan et al., 2015). In the present study, we have evaluated the role of BA in the regulation of MUC4 expression in PC. The findings from the current study, for the first time, have demonstrated that BA levels are significantly high in the serum and pancreatic juice samples obtained from PC patients. Using defined spontaneous mouse model of PC, we found that BA levels increased with the severity of PC, which we mechanistically linked with BA-mediated expression of oncogenic MUC4 through FAK-dependent activation of c-Jun. Further studies demonstrated the role of FXR as the upstream molecule in this FAK/c-Jun/MUC4 axis.
2. Materials and methods
2.1. Cell culture and reagents
All human PC cell lines were obtained from ATCC, except T3M4 and CD18/HPAF. CD18/HPAF is a metastatic clone derived from the HPAF cell line (Mullins et al., 1991), whereas T3M4 cell line is derived from lymph node metastasis of pancreatic adenocarcinoma (Okabe et al., 1983). Human ductal pancreatic epithelial (HDPE) cells were a generous gift of Dr. Thiru Arumugam (MD Anderson, Houston, Texas) and cultured in keratinocyte serum-free (KSF) medium supplemented with epidermal growth factor and bovine pituitary extract. All PC cell lines were cultured in DMEM (supplemented with 10% heat-inactivated FBS, penicillin, and streptomycin (100 μg/ml)) at 37°C with 5% CO 2 and were tested mycoplasma-free before conducting the experiments. Deoxycholic (DCA) and chenodeoxycholic acid (CDCA) were dissolved in sterile ethanol. For inhibition studies, wortamannin (phosphoinositide 3-kinase (PI3K) inhibitor, 1 μM, Cell Signaling Technology), SP100625 (JNK inhibitor; 35 μM, Merck Millipore), FAK inhibitor 14 (FAK inhibitor, 15 μM, Cayman’s chemical), U1026 (MAPK inhibitor, 10 μM, Promega) and actinomycin-D (2 μg/ml, Sigma-Aldrich) were given 1h prior to BA treatment. To transiently knockdown FXR, commercially available FXR siRNA (Santa Cruz biotechnologies (SCB), Dallas, TX, USA) were used. For transfection purposes, lipofectamine 2000 (Life Technologies; Carlsbad, CA, USA) was used, according to the manufacturer’s protocol.
2.2. Procurement of human and murine PDAC samples
All human PDAC samples used in the present study were de-identified and a written informed consent was received from all recruited patients before enrollment at respective institutions. The collection of secreted pancreatic juice upon secretin induction from PC patients was approved by the Mayo Clinic Institutional Review Board (IRB#07-0000099) and the detailed information has been provided in our previous publication (Kaur et al., 2013a). Plasma samples were collected using an approved protocol (IRB number PRO07030072) at University of Pittsburgh (Pittsburgh, PA) from PC patients (Kaur et al., 2013b). The samples from autochthonous murine model, KrasG12D/+; p53R172H/+; Pdx1-Cre (KPC), and their contemporary littermates, were collected at 5, 7, 10, 15, 20 and 25 weeks (wk) of age. The mouse model was housed at UNMC animals facility and generated by crossing LSL-KrasG12D with LSL-Trp53R172H/+ transgenic mice as described previously (Hingorani et al., 2005; Rachagani et al., 2012). The tissue specimen from Whipple procedure were obtained from UNMC tissue bank and used for immunohistoflorescence analysis. For mRNA expression profiling, frozen PC tissues were obtained from cooperative human tumor network (CHTN) and UNMC.
2.3. Total BA estimation method
To analyze total BA concentration in pancreatic juice and plasma samples, we used a highly sensitive bile acid estimation assay kit (Diazyme, NBT, DZO92A-k). To increase the precision of the test, each sample was analyzed in triplicates. We used deoxycholic acid for making the reference plot using serial dilution from 1.25 μ mol/L to 150 μ mol/L. After completing the BA estimation assay according to the manufacturer’s protocol, ELISA plates were read at 405 nm and the collected data was analyzed using SOFTMAX PRO software (Molecular Devices Corp., Sunnyvale, CA).
2.4. Immunoblotting
Briefly, cells were lysed with a radioimmunoprecipitation (RIPA) buffer (50 mM Tris-HCl pH-7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS), supplemented with protease inhibitor mixture, 2 mM Na3VO4, 10 mM NaF and 1 mM PMSF, for 15 minutes at 40C. Cell lysates were quantified and proteins were resolved on 2% agarose gel electrophoresis for high molecular weight MUC4 protein, whereas proteins less than 250 kDa were resolved by 10% SDS–PAGE under reducing conditions and blotted onto a PVDF membrane (Millipore). Following electrophoresis, the membrane were blocked in 5% non-fat dry milk in PBS for 1h and incubated overnight with primary antibodies at 40C. The primary antibodies used in the study are as follows: β-Actin (Sigma, 1:5000), c-Jun (SC-1694, Santa Cruz Biotechnologies (SCB), 1:500), p-c-Jun (9261, Cell Signaling, 1:1000), c-src (SC-18, SCB, 1:500), p-src (6943, Cell Signaling, 1:1000), FAK (SC-558, SCB, 1:500), p-FAK (8556, Cell Signaling, 1:1000), FXR (SC-134481, SCB, 1:500) and MUC4 (In house generated, 1:1000). Blots were washed and probed with respective secondary peroxidase-conjugated antibodies. Following, the protein bands were visualized using enhanced chemiluminescence (ECL) method (Thermoscientific) and quantified using image Studio Lite ver. 5.2 (LI-COR biosciences).
2.5. Immunofluorescence microscopy
PC cells were grown on sterilized cover slips for 24h, serum starved for 8h and treated with appropriate vehicle control (media, ethanol or DMSO), DCA, CDCA, and inhibitors for indicated time-points. After the treatment, immunofluorescence staining with specific primary antibodies was performed as described previously (Kumar et al., 2015b). For immunohistofluorescence, we deparaffinized tissue sections with xylene, rehydrated with decreasing concentrations of ethanol and incubated tissues for 30 min with 3% H2O2: methanol solution. Subsequently, antigen retrieval was performed and tissues sections were blocked in 2.5% horse serum for 2h and incubated with primary antibodies. Following, tissues were processed as described previously (Kumar et al., 2015b). The primary antibodies used are c-Jun (SC-1694, SCB, 1:500), FXR (SC-558, SCB, 1:200) and MUC4 (In-house generated, 1:400). All the images were taken by using LSM 510 microscope, a laser scanning confocal microscope (Carl Zeiss GmbH, Thornwood, NY) in the respective channels.
2.6. RNA isolation and RT-PCR
Quantitative real-time PCR amplifications, using gene-specific primers (Table-S1) were carried out using the standardized protocol established in our lab (Kumar et al., 2015b), using SYBR Green chemistry. The relative fold differences in gene expression were calculated using the ΔΔCt method with β-actin as a normalization control (Livak and Schmittgen, 2001). For RT-PCR, the amplification of both FXR and β-actin genes was done using the following steps: initial denaturation at 95°C for 5 min, followed by 35 prog rammed cycles at 95°C for 1 min., 58°C for 1.5 min. and 72°C for 1 min, with a final incubatio n at 72°C for 10 min. The amplified product was detected by electrophoresis on 2% agarose gels.
2.7. Chromatin immunoprecipitation (ChIP)
PC cells (CD18/HPAF) were starved of serum for 8h and treated with DCA or CDCA for 4h. Afterward, the ChIP experiment was performed as described previously (Kumar et al., 2015a; Pai et al., 2016) and have been repeated more than three times. In breif, 1% formaldehyde was used to cross-linked chromatin, which was isolated and sheared into 500–1000 bp fragments by sonication (Bioruptor UCD-200, Diagenode; New York, NY, USA). As an input, 1% of the sonicated DNA was used. The remaining sonicated DNA fraction was used for the pull down using indicated antibodies overnight at 40C. Antibodies used were: 5 μg of anti-c-Jun (SCB#1694X) and IgG (negative control). MUC4 promoter targeted primers were used to identify enriched fragment through real-time qPCR. The details of the primers are given in Table-S2.
2.8. Luciferase reporter assay
To perform this assay, previously designed and established pGL3-MUC4 deletion constructs were used (Andrianifahanana et al., 2005). PC cell lines were plated in six-wells in triplicates and repeated thrice. Transient transfection was performed with MUC4 deletion constructs using lipofectamine 2000 (Life Technologies; Carlsbad, CA, USA). Next day, the media was first changed to 10% FBS containing DMEM for 12h (to alleviate cellular stress of transfected cells) and then to serum free media for additional 8h. Subsequently, transfected cells were treated with BA for 4h in serum free condition. Following treatment, cells were lysed using reporter lysis buffer (Promega; Madison, WI) and subsequently, the activity of luciferase and beta-galactosidase activity was measured using Steady-Glo Luciferase assay system (Promega, E2510) and β-galactosidase assay kit (Promega, E2000). Fold activation of luciferase activity in BA-treated cells were calculated and compared with untreated cells. In silico analysis was performed on designated MUC4 promoter region using ALGGEN PROMO software (where similarity score of >0.85 was used to screen transcription factor binding sites) to predict the binding of putative transcription factors.
2.9. Cytoplasmic and nuclear fractionation
Briefly, after 4h of 50 μM of DCA or CDCA treatment, CD18/HPAF cells were washed with ice-cold PBS and incubated with a cytoplasmic extraction buffer (10 mM HEPES, pH 7.4, 10 mM KCl, 0.2% NP-40, 0.1 mM EDTA, 10% glycerol, 1.5 mM MgCl2, supplemented with protease inhibitor, 1 mM DTT, 1 mM PMSF, 5 mM Na3VO4, 5 mM NaF) for 1h at 40C. Cells were centrifuged at 800 × g for 10 min. and the supernatant was labeled as cytoplasmic extract, and the remaining pellet was washed with PBS and then incubated for 1h with the nuclear extraction buffer (20 mM HEPES (pH 7.6), 420 mM NaCl, 1 mM EDTA, 20% glycerol, 1.5 mM MgCl2, 1 mM DTT, 1 mM PMSF, 5 mM Na3VO4, 5 mM NaF). Following incubation, the pellet was sonicated for 10 s at 60% amplitude, and then subjected to centrifugation at 13,000xg for 10 min. The obtained supernatant was collected as a nuclear extract. The purity of the fractions were checked by analyzing the expression of nuclear SP1 (#9389S, Cell Signaling, 1:1000) and cytoplasmic GAPDH (#5174S, Cell Signaling, 1:3000) proteins in collected lysates.
2.10. . Statistical Analysis
All results are representative of at least three independent experiments. The in vitro data are expressed as the mean±standard deviation (S.D.), whereas the in vivo data are represented as mean±standard error (S.E.). Statistical comparisons of the two groups were made using a student’s t-test (two-tailed, unpaired) using Microsoft Excel 2010, where a p value of less than 0.05 was considered statistically significant. For correlation analysis, the coefficient of determination (R2) was determined between two groups.
3. Results
3.1 BA levels are elevated in serum and pancreatic juice during PC
According to our hypothesis, BA play important roles in PC development by regulating the expression of oncogenic proteins, including MUC4. Therefore, we first analyzed the levels of BA in serum and pancreatic juice obtained from PC patients. There was significantly higher circulatory levels of BA (p=0.0014) with a mean concentration of 68±4.39 μM as compared to 38±3.09 μM observed in the control group (Fig. 1A). Similarly, we noticed a significant increase in mean value of circulatory BA levels in 10-15 wk (19.96±4.15 μM, p=0.002) and 20-25wk-old (27±6.31 μM, p= 0.009) KPC mice compared to their littermate controls (1.17±0.97 μM) (Fig. 1B), strengthening the association of BA with the pathobiology of PC disease. We included controls from different age group for BA estimation and did not observe any noticeable change in their serum BA levels, which is also evident from the demonstrated standard errors (Fig. 1B). Additionally, earlier report by Uchida K et al. have demonstrated that circulatory BA levels when expressed in terms of units per rat did not ostensibly change, regardless of their age (Uchida et al., 1978). Consistently, pancreatic juice obtained from PC patients (n=18) had significantly high BA levels (65±12.26 μM, p=0.048), compared to the non-pancreatic non-healthy (NPNH, patients with symptoms mimicking pancreatic disease but found to be free of pancreatic pathology) subjects (n=5), where the mean concentration of BA was 13.63±12.55 μM (Fig. 1C). Taken together, high BA levels in PC condition suggest their possible involvement in the pathobiology of PC.
Figure 1. Upregulation of BA in PC condition.

A. The representative box-plot is showing levels of BA in the serum samples obtained from the PC patients (n=36) and healthy (n=10) individuals using a commercially available total BA estimation kit. The difference in BA levels between normal and PC patients were found to be statistically significant. B. To understand the association of BA with PC progression, we measured BA levels in established KPC mice model at early (5-7 wk), medium (10-15 wk) and advanced stages (20-25 wk). The BA levels were found to increase with the severity of the disease. C. Box-plot representing the levels of BA in the pancreatic juice obtained from PC patients. We observed significant increase in BA concentration in the pancreatic juice obtained from PC patients (65 μmol/L) compared to NPNH controls (13 μmol/L). (All values are mean ±S.E, ns means non-significant)
3.2. BA up-regulate MUC4 expression in PC cells
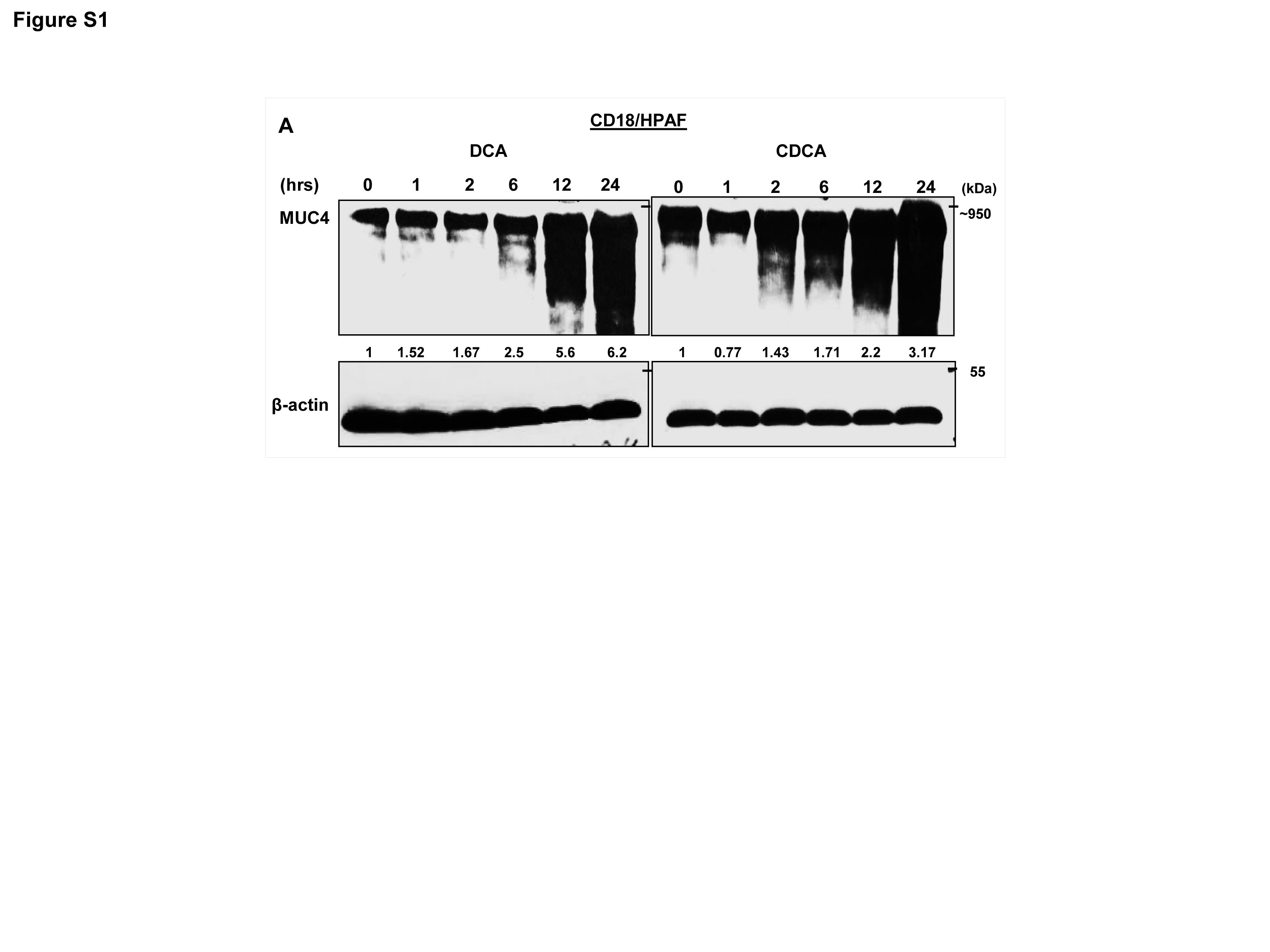
BA modulates the expression levels of mucins such as MUC1, MUC2, MUC4 and MUC5AC in oesophageal, gastric and colon cancers (Lee et al., 2010; Mariette et al., 2008; Song et al., 2011; Mariette et al., 2004; Piessen et al., 2007; Martin et al., 2011). Interestingly, PC is characterized by altered mucins expression. We along with others have established that mucins play important role in the pathogenesis of PC (Kaur et al., 2013c; Joshi et al., 2014; Joshi et al., 2015). In order to analyze the effect of BA on MUC4 expression, we treated PC cell lines with different concentrations of DCA and CDCA in a dose and time dependent manner. We observed a significant increase in MUC4 expression in CD18/HPAF cells at all concentrations, ranging from 5-100 μM with the maximal increase at 50 μM concentration for both DCA and CDCA in 24h (Fig. 2A. Fig. S1). BA-mediated increase in MUC4 expression was further confirmed in T3M4 (Fig. 2B) and CAPAN-1 cell lines (Fig. 2C). Furthermore, immunofluorescence experiment revealed significant increase in MUC4 expression in DCA or CDCA treated CD18/HPAF cell line (Fig. 2D). Altogether, the results suggest that BA may play important role in the pathogenesis of PC by upregulating MUC4 expression.
Figure 2. BA-mediated positive regulation of MUC4 protein.

A. CD18/HPAF cells were serum starved for 8h prior to BA treatment. Following 24h of BA treatment, cell lysates were collected, quantified and resolved using gel electrophoresis. Immunoblot showing increase in MUC4 expression upon DCA and CDCA treatment of CD18/HPAF cells at indicated concentrations. B. Immunoblot showing increase expression of MUC4 in DCA and CDCA treated T3M4 PC cells cells at indicated concentrations. C. Immunoblots confirming MUC4 upregulation by BA treatment in CAPAN1 cells. D. Representative confocal images showing the positive effect of BA on MUC4 expression in CD18/HPAF cells. (scale bar = 20 μM)
3.3. BA transcriptionally upregulates MUC4 expression in PC
In order to mechanistically evaluate BA-mediated upregulation of MUC4, PC cells were treated with DCA or CDCA in conjunction with actinomycin-D, which inhibits the process of transcription. There was 4.09- and 4.49-fold increase in MUC4 expression after DCA and CDCA treatements in CD18/HPAF cells, respectively, which was attenuated to 0.18- and 0.16-fold when treated in combination with actinomycin-D (Fig. 3A). Similarly, in T3M4 cells, we observed a 2.40-fold increase in MUC4 upon DCA treatment, was attenuated to 0.38-fold, when given in the presence of actinomycin-D, whereas a 2-fold MUC4 upregulation in CDCA treated T3M4 cells was reduced to 0.54-fold in the presence of CDCA and actinomycin-D treatment (Fig. 3A).
Figure 3. BA-mediated transcriptional regulation of MUC4.

A. After 8h of serum starvation, both CD18/HPAF and T3M4 cell lines were treated for 12h with 50 μM of DCA, CDCA or vehicle control (ethanol) in the presence or absence of actinomycin-D (2 μg/ml). Following treatment, cDNA was prepared from isolated RNA and used for real-time PCR to analyze the quantitative expression of MUC4 gene. The represented graph is demonstrating that inhibition of transcription attenuates DCA- and CDCA-mediated increase in MUC4 expression in both CD18/HPAF and T3M4 cell lines. B. Luciferase assay was performed in CD18/HPAF cell line transfected with MUC4 promoter-truncated constructs, followed by 4h treatment of 50 μM of DCA and CDCA. A significantly elevated luminescence was detected upon BA treatment, primarily at the distal promoter region. C. Similar to CD18/HPAF cells, T3M4 cells also showed significantly elevated luminescence at the distal promoter region upon BA (50 μM) treatment. (*p<0.05, ** p<0.01, *** p<0.001, ns means non-significant)
To highlight the DCA and CDCA responsive regions on the MUC4 promoter, Luciferase reporter assay was performed (Andrianifahanana et al., 2005). Our results demonstrated that both distal (P-1641) and proximal (P-1809 and P-2150) constructs were responsive to BA in CD18/HPAF cells (Fig. 3B). Of particular interest was the deletion construct P-1641, which evidenced a statistically significant 2.95- and 3.24-fold upregulation of the reporter gene in response to DCA and CDCA treatment, respectively (Fig. 3B). A similarly enhanced transcriptional activity by 1.93-fold was also noticed in DCA and CDCA treated CD18/HPAF cells transfected with P-2150 construct, however, these changes were insignificant. P-1809 construct demonstrated increase in luciferase activity by 1.21- and 1.91- fold upon DCA and CDCA treatment, respectively (Fig. 3B), nevertheless, these changes were significant only for CDCA treatment. Correspondingly, compared to untreated controls, T3M4 cells transfected with P-1641 fragments showed 3.04- and 2.55-fold increase (p<0.05), in luciferase activity upon DCA and CDCA treatment, respectively (Fig. 3C). P-1809 deletion construct demonstrated 1.53- and 1.78-fold increase in luciferase activity upon DCA and CDCA, respectively. Similarly, P-2150 construct exhibited 1.4- and 2-fold increase in luciferase activity in the presence of DCA and CDCA, respecitively. However, the increase in luciferase activity at proximal promoter regions upon BA treament were statistically insignificant in T3M4 cell lines. Taken together, our data suggests that the distal promoter region of MUC4 gene is mainly responsible for BA-mediated transcriptional upregulation of MUC4 in both CD18/HPAF and T3M4 cell lines.
3.4. BA increase the expression and nuclear localization of c-Jun
Due to an observed maximal increase in the region −2572 to −3135 (present in P-1641) to BA treatment in PC cell lines, we performed in silico analysis to delineate putative transcription factors binding sites for transcription factors on this promoter region (Fig. S2). Two c-Jun binding sites were identified on MUC4 distal promoter (P-1641), which were absent on the proximal promoter fragment (P-1809 sequence) (Singh et al., 2007), and therefore, were suggestive of making distal promoter (P-1641) more responsive to BA treatment (Fig. 4A). It incited us to propose that BA-facilitated upregulation in MUC4 expression in PC cell lines is c-Jun dependent. Firstly, we were interested to know whether BA itself has any effect on c-Jun expression levels. Intriguingly, in CD18/HPAF cells, we observed 1.95-, 2.9-, and 3.46-fold increase (p<0.05) in c-Jun expression at 10, 50, and 100 μM of DCA treatment over untreated cells. On the other hand, 1.78-, 2.16-, and 3.87-fold increase (p<0.05) in c-Jun expression was noticed at 10, 50 and 100 μM concentration of CDCA treatment, respectively (Fig. 4B). The increased expression of c-Jun in response to both DCA and CDCA treatments was also confirmed by immunoblot analysis in PC cell lines (Fig. 4C). Immunofluorescence experiments also revealed a significant increase in c-Jun expression and nuclear localization in both DCA- and CDCA-treated CD18/HPAF cells (Fig. 4D). Further, nuclear and cytoplasm fractionation after BA treatment in CD18/HPAF cells, revealed significant increase in c-Jun expression in the nuclear extracts than untreated cells (Fig. 4E).
Figure 4. The effects of BA on MUC4 expression via activation and nuclear translocation of c-Jun.

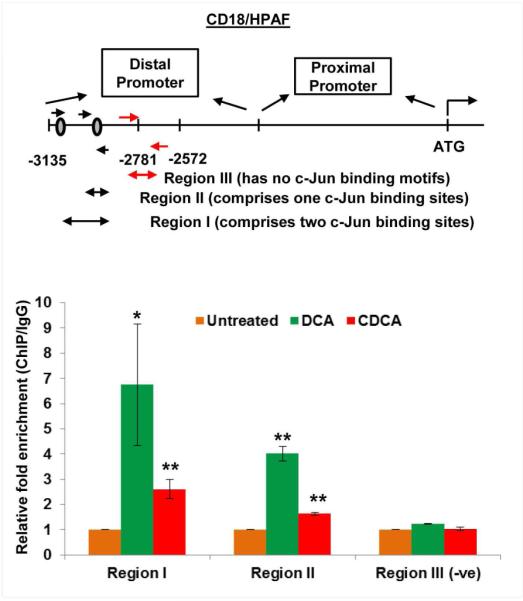
A. Sequence of the MUC4 distal promoter (P-1641) which has two binding sites for c-Jun protein (marked red). B. Graph showing increase in c-Jun mRNA expression in a dose-dependent manner in CD18/HPAF cell line, treated for 2 h with DCA and CDCA. C. CD18/HPAF and T3M4 cell lines were treated with BA (50 μM) for 4 h and cell lysates were collected. Immunoblot was performed to observe change in c-Jun expression in DCA- and CDCA-treated CD18/HPAF and T3M4 cell lines, compared to their respective untreated controls. D. Confocal images showing significant increase in c-Jun and MUC4 protein expression in CD18/HPAF cells treated with DCA or CDCA. Graph showing the quantification of the c-Jun positive nuclei in DCA and CDCA treated CD18/HPAF cells. E. Immunoblot showing significant increase in the expression levels of c-Jun in the nuclear fraction obtained from BA (25 μM)-treated CD18/HPAF cells, whereas cytoplasmic fraction did not demonstrate any noticeable alteration in c-Jun expression. F. ChIP experiment was performed to observe the effect on enrichment for c-Jun binding on MUC4 distal promoter in the presence or absence of DCA (50 μM) and CDCA (50 μM). We observed a significant increase in fold-enrichment at both region-I (containing two c-Jun binding sites) and region-II (containing one c-Jun binding sites). (*p<0.05, ** p<0.01, *** p<0.001, scale bar = 20 μM)
To investigate the direct involvement of c-Jun in BA-induced MUC4 expression, we performed ChIP assay to analyze c-Jun binding on MUC4 distal promoter (Fig. 4F). Using a primer set covering only one c-Jun binding site (or region-II), we observed 4.01- and 1.64-fold enrichment upon DCA and CDCA treatment of CD18/HPAF cells, respectively. However, primers encompassing both c-Jun binding sites (region-I), showed a significant (<0.05) enrichment of 6.74- and 2.61-folds, compared to untreated cells after DCA and CDCA treatments in CD18/HPAF cells (Fig. 4F), suggestive of the cumulative effects of both c-Jun binding sites in inducing the transcription of MUC4 gene. As a negative control, we synthesized primers against the non-c-Jun binding MUC4 promoter fragment and found no difference. Taken together, BA increase the expression and nuclear localization of c-Jun, which then occupy MUC4 promoter to increase its transcription.
3.5. BA mediated increase in FAK activation induced c-Jun expression
To elucidate the signaling pathways responsible for increased MUC4 transcription to BA treatment, CD18/HPAF cells were treated with a panel of inhibitors targeting different signaling pathways prior to BA treatment. Interestingly, pharmacological inhibition of both FAK and MAPK pathway showed attenuation of DCA- and CDCA-mediated MUC4 upregulation (Fig. 5A). Inhibition of PI3K pathway did not have perceptible effect on MUC4 expression, whereas inhibition of JNK suppressed CDCA-mediated upregulation of MUC4 (Fig. 5A). The involvement of FAK pathway in BA-mediated upregulation of MUC4 was further confirmed using an immunofluorescence analysis (Fig. 5B). Earlier, JNK and MAPK pathways have been associated with BA (Gupta et al., 2001; Qiao et al., 2001); however, effects of BA on FAK has not been studied so far, particularly, in terms of MUC4 regulation. Due to observed maximal effect of FAK pathway on BA-facilitated MUC4 expression, we decided to focus on FAK pathway and analyzed the activation status of FAK in BA-treated PC cells. As anticipated, we observed a high expression of activated FAK or pFAK (Y397) FAK in BA-treated PC cells (Fig. 5C), whereas expression of total FAK remains constant. Further, the selective pharmacological inhibition of FAK, led to significant decline in the expression levels of c-Jun and MUC4 in PC cell lines, both at transcript and protein levels, suggesting that c-JUN activation is mediated through FAK pathway (Fig. 5D and E). To further substantiate our results, we performed ChIP experiment and observed significant reduction in enrichment for c-Jun binding on MUC4 promoter when BA treatment was concomitantly given with FAK inhibitor, as compared to BA alone (Fig. 5F), suggesting that FAK activation is a prerequisite for DCA- and CDCA-mediated MUC4 upregulation in PC cells due to its direct involvement in the induction of c-Jun expression.
Figure 5. BA-mediated upregulation of MUC4 is dependent on FAK activation.


A. Concomitant treatment of 25 μM of DCA or CDCA in the presence or absence of selective pharmacological signaling inhibitors for 12h led us to know that the FAK pathway is mainly responsible for MUC4 upregulation upon BA exposure, as attenuation of this pathway maximally suppresses the BA-mediated upregulation of MUC4, compared to the other signaling inhibitors. Besides FAK, inhibiton of MAPK pathway also led to reduced MUC4 expression. B. Images obtained from immunofluorescence experiment showing MUC4 upregulation in DCA (25 μM) and CDCA (25 μM) treated CD18/HPAF cells, which is attenuated upon inhibiting FAK activity (or phosphorylation). C. Increase in FAK activity was confirmed by analyzing pFAK (Tyr397) expression upon BA (25 μM) treatment of CD18/HPAF and CAPAN1 cell lines for 4h. D. Graphical representation of relative mRNA expression for MUC4 and c-Jun gene altered upon inhibition of FAK pathway in both CD18/HPAF and T3M4 cell lines. E. Immunoblot showng that inhibition of FAK pathway, using 15 μM of FAK Inhibitor 14, leads to downregulation of MUC4, pFAK and c-Jun in CD18/HPAF cells. F. Graph representing the relative fold enrichment for c-Jun on AP-1 sequence motifs present on MUC4 distal promoter when CD18/HPAF cells were concomitantly treated with DCA and CDCA in the presence and absence of FAK inhibitor for 4 hours. (*p<0.05, scale bar = 20 μM)
3.6. FXR activation is a essential for BA-mediated MUC4 upregulation via src/FAK/c Jun axis
Farenosoid-X-receptor (FXR) is known to be activated by BA. Upon its activation, FXR gets translocated to the nucleus, where it alters the trancriptional expression of multiple genes (Fig. 6A). Interestingly, the overall expression of FXR did not get influence by BA treatment, as FXR levels were high in the cytoplasmic fraction of untreated cells than DCA and CDCA treated cells. The insignificant changes on FXR levels upon BA exposure were also noticed at mRNA levels (Fig. S3A). Expression profiling of FXR receptor in PC cell lines showed its significant overexpression in HPAC, CD18/HPAF, CAPAN1, Panc10.05 and Panc1 cell lines (Fig. 6B, Fig. S3B), compared to normal pancreatic cells (HDPE). Interestingly, significantly high FXR levels in CD18/HPAF cells explains drastic increase in MUC4 expression even at very low concentration of BA treatment compared to T3M4 and CAPAN1 cell lines (Fig. 2A-C). Due to observed downregulation of activated FAK expression levels along with c-Jun levels upon transient knockdown of FXR in CD18/HPAF and T3M4 PC cell lines, it is likely that FXR is acting upstream in this FAK/c-Jun/MUC4 axis (Fig. 6C).The key question which arises is that how FXR expression regulates the activity of FAK. It is well-known in the literature that src kinase is one of the critical regulator of FAK activity (Westhoff et al., 2004). As we have observed thar BA treatment do affect the phosphorylation of src (Fig. S3C), we assumed that FXR-mediated phosphorylation of FAK is p-src-dependent, and FXR knocked down PC cells indeed showed significant reduction in p-src levels compared to si control (Fig. 6C). To further substantiate our results, we gave BA treatment to FXR knockdown CD18/HPAF cells and found significant abrogation of BA-mediated MUC4 upregulation (Fig. 6D). A 2.1-fold increase in MUC4 expression due to DCA treatment was reduced to 1.32-fold in FXR silenced CD18/HPAF cells (Fig. 6D). Similarly, a 1.92-fold increase in MUC4 expression upon CDCA treatment was reduced to 1.13-fold when CDCA treatment was given to FXR knockdown cells (Fig. 6D). Altogether, the results suggest that FXR activation due to BA exposure is responsible for the initiation of FAK/c-Jun/MUC4 axis in PC cells, by plausibly regulating the activity of src kinase.
Figure 6. Activation of FXR is required for BA-mediated MUC4 upregulation via Src/FAK/c-Jun axis.

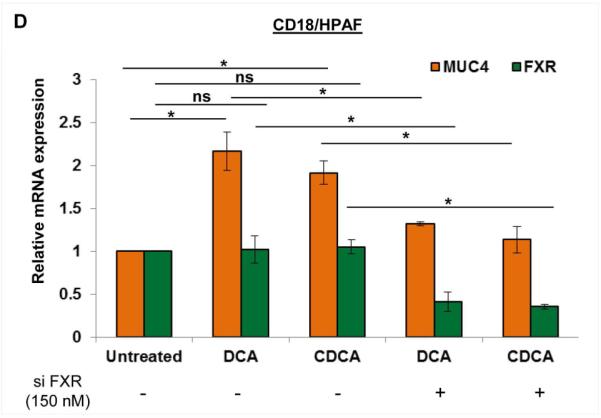
A. Nuclear fraction obtained from 50 μM of DCA and CDCA treated CD18/HPAF cells, demonstrated increased levels of FXR compared to the untreated control. On the other hand, FXR expression was more on the cytoplasmic fraction in untreated cells than DCA and CDCA treated cells. B. FXR expression was found to be significantly high in PC cell lines than normal pancreatic cells (HDPE). C. FXR was transiently knockdown in CD18/HPAF and T3M4 cell lines using 150 nM of siRNA oligos and confirmed using immunoblotting. Interestingly, FXR knockdown cells exhibited significant decline in FAK, pFAK (Tyr397), src, p-src (Tyr416), c-Jun, p-c-Jun (Ser63) and MUC4, suggestive of FXR involvement as the most upstream molecule in this BA-mediated FAK/c-Jun/MUC4 axis. D. The graphical representation of the result obtained from real-time PCR showing that knockdown of FXR in CD18/HPAF cell line leads to significant attenuation of both DCA (25 μM) or CDCA (25 μM)-mediated MUC4 upregulation. (*p<0.05, **p<0.01, ns means non-significant)
3.7. Clinical association between MUC4 and BA receptor FXR
In PC patient samples, we clearly observed that similar to PC cell lines, mRNA expression for FXR was higher in 47% of PC tissues (n=15), as compared to the tumor adjacent normal pancreatic tissues (n=4) (Fig. 7A). Though the upregulation of FXR in PC patients was not statistically significant (p>0.05), but considering significant increase in the levels of BA, which are activators of FXR receptor, both in the circulation and pancreatic juice of PC patients, we can speculate that pancreatic tumors have increased activity of FXR receptor, which is sufficient to initiate FAK/c-Jun/MUC4 signaling cascade. In order to confirm an in vivo association between MUC4 and FXR, we measured the transcript levels of MUC4 in same clinical samples and performed regression analysis (Fig. 7B). A fairly positive correlation (R2=0.60) between MUC4 and FXR, further substantiated our in vitro findings. Moreover, using confocal microscopy, we observed co-expression of both FXR and MUC4 at the same PC tissue spots (Fig. 7C).
Figure 7. Clinical association between MUC4 and FXR in PC tissues.

A. Expression profiling of FXR was performed in cDNA samples prepared from pancreatic tumor tissues (n=15) and tumor adjacent normal (n=4). Similar to its agonists, the levels of FXR was upregulated in tissues obtained from PC patients than tumor adjacent tissues. B. Data showing regression analysis which was performed to correlate MUC4 and FXR in clinical samples at transcriptional levels. C. PC tissues (obtained from Whipple procedure) showed the co-expression of both MUC4 and FXR at same tissue spots, suggestive of their direct association. (scale bar = 20 μM). D. Diagrammatic representation of the overall summary of the paper: Treatment with BA leads to the activation of FXR receptor, which gets engage in the activation of FAK pathway, possibly by activating src kinase. Increase in FAK-mediated signaling leads to an increased transcription of c-Jun gene. Increased expression and activation of c-Jun is followed by its increased nuclear translocation, leading to increased MUC4 transcription, which plays an important role in the proliferation, survival, metastasis and chemoresistance of pancreatic tumors.
4. Discussion
Anatomically, the common bile duct and the pancreatic duct are close in proximity, and unite at the ampulla of Vater. This led us to believe that BA can reflux to the pancreatic duct under pathological conditions. Growing pancreatic tumor often obstruct the bile ducts, preventing the flow of bile to the duodenum, leading to jaundice, a frequently occuring clinical manifestation in PC patients (Chen et al., 2015). Studies have established the tumor-promoting effects of BA in multiple cancers, including Barrett's metaplasia and colorectal, biliary, and hepatocellular cancers (Changbumrung et al., 1990; Degirolamo et al., 2011; Piessen et al., 2007; Souza et al., 2011). However, the role of BA in PC has not been clearly understood, which prompted us to investigate its tumorigenic properties in PC. In order to establish our hypothesis, BA levels were measured in the serum and pancreatic juice obtained from PC patients and NPNH individuals. Encouragingly, we observed a significant increase in BA concentration in those PC patients compared to controls. We also observed increased mRNA expression of BA receptor, FXR, in PC tumors compared to a normal pancreas. Due to increased BA levels, which act as FXR agonist, it can be speculated that not only its expression, activity of FXR also get increased under PC condition, which we have confirmed as well due to increased nuclear expression levels of FXR upon BA treatment. Similar to our observation, Lee et al. have also observed increased expression of FXR in the PC tissues and established its protumorigenic role in PC disease condition (Lee et al., 2011). Altogether, this is a first experimental evidence establishing that BA do enter the pancreatic duct and increases the tumorigenic potential of PC cells by altering the expression of oncogenic MUC4 mucin.
Our luciferase reporter assay established MUC4 distal promoter as the major BA responsive site. Further, in silico analysis demonstrated the presence of two activator protein 1 (AP-1) motifs in this region, which has also been reported in our earlier study (Singh et al., 2007). Consistent with the previous findings observed in gastric cells (Park et al., 2008), we noticed that BA treatment increase c-Jun expression, one of the members of the AP-1 family. Furthermore, ChIP experiments confirmed an increase in c-Jun binding onto MUC4 distal promoter when treated with BA. Interestingly, by utilizing the same c-Jun transcription factor, BA are known to increase the transcription of cyclooxygenase gene, by facilitating increased c-Jun binding on COX promoter in esophageal adenocarcinoma cells (Zhang et al., 1998). Importantly, c-Jun overexpression has already been associated with carcinogenesis and cancer progression in multiple cancers (Okutomi et al., 2003; Tessari et al., 1999). Although BA responsiveness was primarily observed at MUC4 distal promoter (P-1641), we also observed increased luciferase activity in proximal promoter region, P-1809 transfected CD18/HPAF cells, upon CDCA treatment, implying the involvement of other transcription factors in CDCA-mediated upregulation of CD18/HPAF cells, and requires further investigation (Singh et al., 2007). Inspite of the presence of c-Jun bindings sites on MUC4 proximal promoter (P-2150), we observed an insignificant increase in luciferase activity upon BA treatment, suggesting that BA might be affecting the expression and binding of transcription factors having inhibitory effects on proximal promoter region, and therefore, neutralizing the positive effects of c-Jun.
Multiple forms of BA have been previously identified as potent inducers of MUC4 expression in esophageal carcinogenesis associated with bile reflux (Degirolamo et al., 2011; Mariette et al., 2004). Mechanistically, PI3K signaling, protein kinase C and hepatocyte nuclear factor-1α were attributed to BA-facilitated increase in MUC4 expression (Mariette et al., 2004; Piessen et al., 2007). However, in the current study, we have established the role of FAK in MUC4 regulation in PC cells upon BA treatment. Moreover, downregulation of c-Jun expression upon FAK inhibition, suggesting that c-Jun activation is a downstream event occurring after FAK activation. Nadruz et al. have also established the link between c-Jun and FAK molecules in ventricular myocytes (Nadruz, Jr. et al., 2005). Unlike CDCA, the inhibition of the JNK pathway had no remarkable effect on DCA-induced MUC4 expression, suggesting that different BA transduce differential signaling to modulate the expression of the target molecules. Moreover, the data also implies differential mode of c-Jun activation in the presence of DCA and CDCA. Earlier studies have shown that c-Jun can get activated in JNK-independent manner (Besirli and Johnson, Jr., 2003; Deng et al., 2012). For instance, in neuronal cells, DNA damage causing induction of neuronal c-Jun kinase has been shown to increase c-Jun phosphorylation (Besirli and Johnson, Jr., 2003). Upon injury, c-Jun is found to be activated in Schwann cells by MAP kinases, which is again occurring independent of JNK (Deng et al., 2012). In addition to FAK pathway, inhibition of MAPK pathway also led to attenuation of BA-mediated MUC4 upregulation, which further strengthened our notion that MAPK pathway could be involved in c-Jun activation. Future studies will be focused to understand the in-depth involvement of different signaling pathways in MUC4 regulation after BA treatment.
BA are known to interact with nuclear receptors family including; FXR and pregnane X receptor (PXR) in order to modulate the transcription of their target genes. In the current study, for the first time, we have established the direct involvement of FXR protein in MUC4 regulation. In the clinical samples, we observed a positive correlation between FXR and MUC4 mRNA expression levels. Upon FXR knocked down, BA-mediated upregulation of MUC4 and activation of FAK and c-Jun were abrogated, placing FXR as an upstream molecule in this FAK/c-Jun/MUC4 axis in PC. Das A et al have shown that FXR promotes the migration of endothelial cells by regulating the expression of FAK and MMP9 (Das et al., 2009). However, the molecular mechanism of FXR-facilitated FAK activation is still unexplored. Due to observed increase in src kinase activity upon BA treatment, we assumed its role in this FXR-mediated increase in FAK activation, as FXR silencing led to reduced p-Src levels in PC cell lines. Interestingly, previous study in our lab has also shown that Guggulsterone, a selective pharmacological FXR inhibitor, also leads to a MUC4 downregulation at transcriptional level in PC cells by utilizing src/FAK pathway (Macha et al., 2013). In addition to FXR, other BA receptors could also be implication in BA-facilitated MUC4 upregulation. Interestingly, TGR5 has found to be upregulated in 67% of PC patients (data not shown) and recent report has shown its tumorigenic role in gastrointestinal cancers, including PC (Nagathihalli et al., 2014). Further studies will be helpful and required to mechanistically delineate the association between TGR5 and PC disease condition.
Future studies will be directed to get the better insight of BA on the pathobiology of PC by bile duct ligation or cholecystectomy using autochthonous murine models , which will delineate the role of BA on pancreatic tumor growth and metastasis. Moreover, the significantly induced levels of BA indicates their possible usefulness for diagnostic purposes, and needs to be validated in more number of patient samples to assess and establish its clinical utility.
5. Conclusions
Altogether, the current study, for the first time, has established that BA levels rises both in the circulation and pancreatic juice in PC, and they exert their protumorigenic functions by upregulating oncogenic MUC4 expression. Mechanistically, we have demonstrated that BA binding to FXR receptor leads to FAK activation, followed by increased c-Jun expression and its nuclear translocation, which in turn causes increased transcription of the MUC4 gene (Fig. 7D). The current study also supports emerging epidemiological data that, similar to colorectal cancer, fat-rich diet could be one of the risk factors for PC development and progression. Therefore, targeting BA receptors an administration of BA antagonists can significantly impact the outcome of PC patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
BA levels are elevated in the serum and pancreatic juice of PC patients.
BA induces MUC4 expression, primarily at transcriptional level, in PC cell lines.
BA-mediated increase in MUC4 is occurring via FAK-induced c-Jun expression.
BA receptor, namely FXR, is acting upstream in BA-mediated FAK/c-Jun/MUC4 axis.
FXR is overexpressed in PC tissues and showed a positive association with MUC4
Acknowledgments
The work was supported, in part, by grants from the National Insititutes of Health (UO1 CA111294, P50 CA127297, U54 CA163120 and RO1 CA183459). The authors are indebted to Kavita Mallya, James Talaska, Janice Taylor, and Phillip Hexley for technical support and Drs. Angie Rizzino and Shilpa Buch for letting us use their facilities. Lastly, we would like to thank UNMC Graduate Studies for the graduate fellowship support to Suhasini Joshi and Eric Cruz.
Abbreviations
- PC
pancreatic cancer
- PDAC
pancreatic ductal adenocarcinoma
- BA
bile acids
- DCA
deoxycholic acid
- CDCA
chenodeoxycholic acid
- FXR
farnesoid-x-receptor
- FAK
focal adhesion kinase
- MAPK
mitogen activated protein kinase
- JNK
c-Jun N-terminal kinase
- PI3K
Phosphoinositide 3-kinase
- ChIP
Chromatin immunoprecipitation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflicts of interest
References
- Andrianifahanana M, Agrawal A, Singh AP, Moniaux N, van S,I, Aubert JP, Meza J, Batra SK. Synergistic induction of the MUC4 mucin gene by interferon-gamma and retinoic acid in human pancreatic tumour cells involves a reprogramming of signalling pathways. Oncogene. 2005;40:6143–6154. doi: 10.1038/sj.onc.1208756. 1208756 [pii];10.1038/sj.onc.1208756 [doi] [DOI] [PubMed] [Google Scholar]
- Andrianifahanana M, Moniaux N, Schmied BM, Ringel J, Friess H, Hollingsworth MA, Buchler MW, Aubert JP, Batra SK. Mucin (MUC) gene expression in human pancreatic adenocarcinoma and chronic pancreatitis: a potential role of MUC4 as a tumor marker of diagnostic significance. Clin. Cancer Res. 2001;12:4033–4040. [PubMed] [Google Scholar]
- Baptissart M, Vega A, Maqdasy S, Caira F, Baron S, Lobaccaro JM, Volle DH. Bile acids: from digestion to cancers. Biochimie. 2013;3:504–517. doi: 10.1016/j.biochi.2012.06.022. S0300-9084(12)00257-X [pii];10.1016/j.biochi.2012.06.022 [doi] [DOI] [PubMed] [Google Scholar]
- Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat. Rev. Cancer. 2002;12:897–909. doi: 10.1038/nrc949. 10.1038/nrc949 [doi];nrc949 [pii] [DOI] [PubMed] [Google Scholar]
- Besirli CG, Johnson EM., Jr. JNK-independent activation of c-Jun during neuronal apoptosis induced by multiple DNA-damaging agents. J Biol. Chem. 2003;25:22357–22366. doi: 10.1074/jbc.M300742200. 10.1074/jbc.M300742200 [doi];M300742200 [pii] [DOI] [PubMed] [Google Scholar]
- Changbumrung S, Tungtrongchitr R, Migasena P, Chamroenngan S. Serum unconjugated primary and secondary bile acids in patients with cholangiocarcinoma and hepatocellular carcinoma. J Med Assoc. Thai. 1990;2:81–90. [PubMed] [Google Scholar]
- Chaturvedi P, Singh AP, Moniaux N, Senapati S, Chakraborty S, Meza JL, Batra SK. MUC4 mucin potentiates pancreatic tumor cell proliferation, survival, and invasive properties and interferes with its interaction to extracellular matrix proteins. Mol. Cancer Res. 2007;4:309–320. doi: 10.1158/1541-7786.MCR-06-0353. 1541-7786.MCR-06-0353 [pii];10.1158/1541-7786.MCR-06-0353 [doi] [DOI] [PubMed] [Google Scholar]
- Chen YG, Pan HH, Dai MS, Lin C, Lu CS, Su SL, Chang PY, Huang TC, Chen JH, Wu YY, Chen YC, Ho CL. Impact of Comorbidity and Age on Determinants Therapeutic Strategies in Advanced Pancreatic Head Cancer Patients With Obstructive Jaundices. Medicine (Baltimore) 2015;31:e1298. doi: 10.1097/MD.0000000000001298. 10.1097/MD.0000000000001298 [doi];00005792-201508010-00042 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Yaqoob U, Mehta D, Shah VH. FXR promotes endothelial cell motility through coordinated regulation of FAK and MMP-9. Arterioscler. Thromb. Vasc. Biol. 2009;4:562–570. doi: 10.1161/ATVBAHA.108.182725. ATVBAHA.108.182725 [pii];10.1161/ATVBAHA.108.182725 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degirolamo C, Modica S, Palasciano G, Moschetta A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011;10:564–572. doi: 10.1016/j.molmed.2011.05.010. S1471-4914(11)00097-9 [pii];10.1016/j.molmed.2011.05.010 [doi] [DOI] [PubMed] [Google Scholar]
- Deng Z, Sui G, Rosa PM, Zhao W. Radiation-induced c-Jun activation depends on MEK1-ERK1/2 signaling pathway in microglial cells. PLoS. One. 2012;5:e36739. doi: 10.1371/journal.pone.0036739. 10.1371/journal.pone.0036739 [doi];PONE-D-11-15222 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stravitz RT, Dent P, Hylemon PB. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2001;19:15816–15822. doi: 10.1074/jbc.M010878200. 10.1074/jbc.M010878200 [doi];M010878200 [pii] [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;5:469–483. doi: 10.1016/j.ccr.2005.04.023. S1535-6108(05)00128-5 [pii];10.1016/j.ccr.2005.04.023 [doi] [DOI] [PubMed] [Google Scholar]
- Howatson AG, Carter DC. Pancreatic carcinogenesis-enhancement by cholecystokinin in the hamster-nitrosamine model. Br. J Cancer. 1985;1:107–114. doi: 10.1038/bjc.1985.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K, Hollingsworth MA, Cameron JL, Yeo CJ, Kern SE, Goggins M, Hruban RH. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;24:8614–8622. [PubMed] [Google Scholar]
- Joshi S, Kumar S, Bafna S, Rachagani S, Wagner KU, Jain M, Batra SK. Genetically engineered mucin mouse models for inflammation and cancer. Cancer Metastasis Rev. 2015 doi: 10.1007/s10555-015-9549-1. 10.1007/s10555-015-9549-1 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Kumar S, Choudhury A, Ponnusamy MP, Batra SK. Altered Mucins (MUC) trafficking in benign and malignant conditions. Oncotarget. 2014;17:7272–7284. doi: 10.18632/oncotarget.2370. 2370 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Baine MJ, Guha S, Ochi N, Chakraborty S, Mallya K, Thomas C, Crook J, Wallace MB, Woodward TA, Jain M, Singh S, Sasson AR, Skinner V, Raimondo M, Batra SK. Neutrophil gelatinase-associated lipocalin, macrophage inhibitory cytokine 1, and carbohydrate antigen 19-9 in pancreatic juice: pathobiologic implications in diagnosing benign and malignant disease of the pancreas. Pancreas. 2013a;3:494–501. doi: 10.1097/MPA.0b013e31826a8597. 10.1097/MPA.0b013e31826a8597 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Chakraborty S, Baine MJ, Mallya K, Smith LM, Sasson A, Brand R, Guha S, Jain M, Wittel U, Singh SK, Batra SK. Potentials of plasma NGAL and MIC-1 as biomarker(s) in the diagnosis of lethal pancreatic cancer. PLoS. One. 2013b;2:e55171. doi: 10.1371/journal.pone.0055171. 10.1371/journal.pone.0055171 [doi];PONE-D-12-23319 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Kumar S, Momi N, Sasson AR, Batra SK. Mucins in pancreatic cancer and its microenvironment. Nat. Rev. Gastroenterol. Hepatol. 2013c;10:607–620. doi: 10.1038/nrgastro.2013.120. nrgastro.2013.120 [pii];10.1038/nrgastro.2013.120 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Das S, Rachagani S, Kaur S, Joshi S, Johansson SL, Ponnusamy MP, Jain M, Batra SK. NCOA3-mediated upregulation of mucin expression via transcriptional and post-translational changes during the development of pancreatic cancer. Oncogene. 2015a;37:4879–4889. doi: 10.1038/onc.2014.409. onc2014409 [pii];10.1038/onc.2014.409 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Torres MP, Kaur S, Rachagani S, Joshi S, Johansson SL, Momi N, Baine MJ, Gilling CE, Smith LM, Wyatt TA, Jain M, Joshi SS, Batra SK. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene. 2015b;16:2052–2060. doi: 10.1038/onc.2014.154. onc2014154 [pii];10.1038/onc.2014.154 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan I, Seshacharyulu P, Haridas D, Rachagani S, Gupta S, Joshi S, Guda C, Yan Y, Jain M, Ganti AK, Ponnusamy MP, Batra SK. Novel HER3/MUC4 oncogenic signaling aggravates the tumorigenic phenotypes of pancreatic cancer cells. Oncotarget. 2015:3912. doi: 10.18632/oncotarget.3912. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Crawley S, Hokari R, Kwon S, Kim YS. Bile acid regulates MUC2 transcription in colon cancer cells via positive EGFR/PKC/Ras/ERK/CREB, PI3K/Akt/IkappaB/NF-kappaB and p38/MSK1/CREB pathways and negative JNK/c-Jun/AP-1 pathway. Int. J. Oncol. 2010;4:941–953. doi: 10.3892/ijo_00000573. [DOI] [PubMed] [Google Scholar]
- Lee JY, Lee KT, Lee JK, Lee KH, Jang KT, Heo JS, Choi SH, Kim Y, Rhee JC. Farnesoid X receptor, overexpressed in pancreatic cancer with lymph node metastasis promotes cell migration and invasion. Br. J. Cancer. 2011;6:1027–1037. doi: 10.1038/bjc.2011.37. bjc201137 [pii];10.1038/bjc.2011.37 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Zeng Z, Wang X, Wu Z, Wang J, Wang C, Sun Q, Chen Y, Quan H. Cholecystectomy and risk of pancreatic cancer: a meta-analysis of observational studies. Cancer Causes Control. 2012;1:59–67. doi: 10.1007/s10552-011-9856-y. 10.1007/s10552-011-9856-y [doi] [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;4:402–408. doi: 10.1006/meth.2001.1262. 10.1006/meth.2001.1262 [doi];S1046-2023(01)91262-9 [pii] [DOI] [PubMed] [Google Scholar]
- Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, Jain M. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013;2:166–177. doi: 10.1016/j.canlet.2013.07.037. S0304-3835(13)00557-0 [pii];10.1016/j.canlet.2013.07.037 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariette C, Perrais M, Leteurtre E, Jonckheere N, Hemon B, Pigny P, Batra S, Aubert JP, Triboulet JP, Van S,I. Transcriptional regulation of human mucin MUC4 by bile acids in oesophageal cancer cells is promoter-dependent and involves activation of the phosphatidylinositol 3-kinase signalling pathway. Biochem. J. 2004:701–708. doi: 10.1042/BJ20031132. Pt 3. 10.1042/BJ20031132 [doi];BJ20031132 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariette C, Piessen G, Leteurtre E, Hemon B, Triboulet JP, Van S,I. Activation of MUC1 mucin expression by bile acids in human esophageal adenocarcinomatous cells and tissues is mediated by the phosphatidylinositol 3-kinase. Surgery. 2008;1:58–71. doi: 10.1016/j.surg.2007.07.043. S0039-6060(07)00638-1 [pii];10.1016/j.surg.2007.07.043 [doi] [DOI] [PubMed] [Google Scholar]
- Martin NA, Mount Patrick SK, Estrada TE, Frisk HA, Rogan DT, Dvorak B, Halpern MD. Active transport of bile acids decreases mucin 2 in neonatal ileum: implications for development of necrotizing enterocolitis. PLoS. One. 2011;12:e27191. doi: 10.1371/journal.pone.0027191. 10.1371/journal.pone.0027191 [doi];PONE-D-11-13529 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniaux N, Chaturvedi P, Varshney GC, Meza JL, Rodriguez-Sierra JF, Aubert JP, Batra SK. Human MUC4 mucin induces ultra-structural changes and tumorigenicity in pancreatic cancer cells. Br. J Cancer. 2007;3:345–357. doi: 10.1038/sj.bjc.6603868. 6603868 [pii];10.1038/sj.bjc.6603868 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins TD, Kern HF, Metzgar RS. Ultrastructural differentiation of sodium butyrate-treated human pancreatic adenocarcinoma cell lines. Pancreas. 1991;5:578–587. doi: 10.1097/00006676-199109000-00012. [DOI] [PubMed] [Google Scholar]
- Nadruz W, Jr., Corat MA, Marin TM, Guimaraes Pereira GA, Franchini KG. Focal adhesion kinase mediates MEF2 and c-Jun activation by stretch: role in the activation of the cardiac hypertrophic genetic program. Cardiovasc. Res. 2005;1:87–97. doi: 10.1016/j.cardiores.2005.05.011. S0008-6363(05)00253-1 [pii];10.1016/j.cardiores.2005.05.011 [doi] [DOI] [PubMed] [Google Scholar]
- Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, Merchant NB. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-alpha: impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res. 2014;7:2062–2072. doi: 10.1158/0008-5472.CAN-13-2329. 0008-5472.CAN-13-2329 [pii];10.1158/0008-5472.CAN-13-2329 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Hirai R, Kitagawa M, Takehira Y, Yamada M, Tamakoshi K, Kobayashi Y, Nakamura H, Kanamori M. Treatment of common bile duct obstruction by pancreatic cancer using various stents: single-center experience. Cardiovasc. Intervent. Radiol. 2002;5:373–380. doi: 10.1007/s00270-002-0426-2. 10.1007/s00270-002-0426-2 [doi] [DOI] [PubMed] [Google Scholar]
- Okabe T, Yamaguchi N, Ohsawa N. Establishment and characterization of a carcinoembryonic antigen (CEA)-producing cell line from a human carcinoma of the exocrine pancreas. Cancer. 1983;4:662–668. doi: 10.1002/1097-0142(19830215)51:4<662::aid-cncr2820510419>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Okutomi Y, Shino Y, Komoda F, Hirano T, Ishihara T, Yamaguchi T, Saisho H, Shirasawa H. Survival regulation in pancreatic cancer cells by c-Jun. Int J Oncol. 2003;4:1127–1134. [PubMed] [Google Scholar]
- Pai P, Rachagani S, Lakshmanan I, Macha MA, Sheinin Y, Smith LM, Ponnusamy MP, Batra SK. The canonical Wnt pathway regulates the metastasis-promoting mucin MUC4 in pancreatic ductal adenocarcinoma. Mol. Oncol. 2016;2:224–239. doi: 10.1016/j.molonc.2015.10.005. S1574-7891(15)00183-0 [pii];10.1016/j.molonc.2015.10.005 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WI, Park MJ, An JK, Choi YH, Kim HY, Cheong J, Yang US. Bile acid regulates c-Jun expression through the orphan nuclear receptor SHP induction in gastric cells. Biochem. Biophys. Res. Commun. 2008;2:437–443. doi: 10.1016/j.bbrc.2008.02.065. S0006-291X(08)00294-5 [pii];10.1016/j.bbrc.2008.02.065 [doi] [DOI] [PubMed] [Google Scholar]
- Piessen G, Jonckheere N, Vincent A, Hemon B, Ducourouble MP, Copin MC, Mariette C, van S,I. Regulation of the human mucin MUC4 by taurodeoxycholic and taurochenodeoxycholic bile acids in oesophageal cancer cells is mediated by hepatocyte nuclear factor 1alpha. Biochem. J. 2007;1:81–91. doi: 10.1042/BJ20061461. BJ20061461 [pii];10.1042/BJ20061461 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L, Studer E, Leach K, McKinstry R, Gupta S, Decker R, Kukreja R, Valerie K, Nagarkatti P, El DW, Molkentin J, Schmidt-Ullrich R, Fisher PB, Grant S, Hylemon PB, Dent P. Deoxycholic acid (DCA) causes ligand-independent activation of epidermal growth factor receptor (EGFR) and FAS receptor in primary hepatocytes: inhibition of EGFR/mitogen-activated protein kinase-signaling module enhances DCA-induced apoptosis. Mol. Biol. Cell. 2001;9:2629–2645. doi: 10.1091/mbc.12.9.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachagani S, Torres MP, Kumar S, Haridas D, Baine M, Macha MA, Kaur S, Ponnusamy MP, Dey P, Seshacharyulu P, Johansson SL, Jain M, Wagner KU, Batra SK. Mucin (Muc) expression during pancreatic cancer progression in spontaneous mouse model: potential implications for diagnosis and therapy. J. Hematol. Oncol. 2012:68. doi: 10.1186/1756-8722-5-68. 1756-8722-5-68 [pii];10.1186/1756-8722-5-68 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;1:9–29. doi: 10.3322/caac.21208. 10.3322/caac.21208 [doi] [DOI] [PubMed] [Google Scholar]
- Sifrim D. Management of bile reflux. Gastroenterol. Hepatol. (N. Y. ) 2013;3:179–180. [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Chauhan SC, Andrianifahanana M, Moniaux N, Meza JL, Copin MC, Van S,I, Hollingsworth MA, Aubert JP, Batra SK. MUC4 expression is regulated by cystic fibrosis transmembrane conductance regulator in pancreatic adenocarcinoma cells via transcriptional and post-translational mechanisms. Oncogene. 2007;1:30–41. doi: 10.1038/sj.onc.1209764. 1209764 [pii];10.1038/sj.onc.1209764 [doi] [DOI] [PubMed] [Google Scholar]
- Song S, Byrd JC, Guha S, Liu KF, Koul D, Bresalier RS. Induction of MUC5AC mucin by conjugated bile acids in the esophagus involves the phosphatidylinositol 3-kinase/protein kinase C/activator protein-1 pathway. Cancer. 2011;11:2386–2397. doi: 10.1002/cncr.25796. 10.1002/cncr.25796 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza RF, Freschi G, Taddei A, Ringressi MN, Bechi P, Castiglione F, Rossi DD, Triadafilopoulos G, Wang JS, Chang AC, Barr H, Bajpai M, Das KM, Schneider PM, Krishnadath KK, Malhotra U, Lynch JP. Barrett's esophagus: genetic and cell changes. Ann. N. Y. Acad. Sci. 2011:18–35. doi: 10.1111/j.1749-6632.2011.06043.x. 10.1111/j.1749-6632.2011.06043.x [doi] [DOI] [PubMed] [Google Scholar]
- Tessari G, Ferrara C, Poletti A, Dubrovich A, Corsini A, Del FG, Naccarato R. The expression of proto-oncogene c-jun in human pancreatic cancer. Anticancer Res. 1999;1B:863–867. [PubMed] [Google Scholar]
- Tsoukali E, Sifrim D. Investigation of extraesophageal gastroesophageal reflux disease. Ann. Gastroenterol. 2013;4:290–295. [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Nomura Y, Kadowaki M, Takase H, Takano K, Takeuchi N. Age-related changes in cholesterol and bile acid metabolism in rats. J. Lipid Res. 1978;5:544–552. [PubMed] [Google Scholar]
- Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell Biol. 2004;18:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. 10.1128/MCB.24.18.8113-8133.2004 [doi];24/18/8113 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisinski KB, Wahl AO, Small W, Jr., Benson AB., III Inoperable pancreatic cancer: standard of care. Oncology (Williston. Park) 2007;13:1558–1564. 169734 [pii] [PubMed] [Google Scholar]
- Zhang F, Subbaramaiah K, Altorki N, Dannenberg AJ. Dihydroxy bile acids activate the transcription of cyclooxygenase-2. J. Biol. Chem. 1998;4:2424–2428. doi: 10.1074/jbc.273.4.2424. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.