

Abstract
Vascular permeability regulated by the vascular endothelial growth factor (VEGF) through endothelial-barrier junctions is essential for inflammation. Mechanisms regulating vascular permeability remain elusive. Although ‘Akt’ and ‘Src’ have been implicated in the endothelial-barrier regulation, it is puzzling how both agents that protect and disrupt the endothelial-barrier activate these kinases to reciprocally regulate vascular permeability. To delineate the role of Akt1 in endothelial-barrier regulation, we created endothelial-specific, tamoxifen-inducible Akt1 knockout mice and stable ShRNA-mediated Akt1 knockdown in human microvascular endothelial cells. Akt1 loss leads to decreased basal and angiopoietin1-induced endothelial-barrier resistance, and enhanced VEGF-induced endothelial-barrier breakdown. Endothelial Akt1 deficiency resulted in enhanced VEGF-induced vascular leakage in mice ears, which was rescued upon re-expression with Adeno-myrAkt1. Furthermore, co-treatment with angiopoietin1 reversed VEGF-induced vascular leakage in an Akt1-dependent manner. Mechanistically, our study revealed that while VEGF-induced short-term vascular permeability is independent of Akt1, its recovery is reliant on Akt1 and FoxO-mediated claudin expression. Pharmacological inhibition of FoxO transcription factors rescued the defective endothelial-barrier due to Akt1 deficiency. Here we provide novel insights on the endothelial-barrier protective role of VEGF in the long-term and the importance of Akt1-FoxO signaling on tight-junction stabilization and prevention of vascular leakage through claudin expression.
Keywords: VEGF, Angiopoietin-1, Akt, VE-cadherin, Claudin, vascular permeability
Introduction
Vascular permeability, a highly complex and coordinated event is important in cardiovascular diseases, cancer and inflammation [1,2]. Many pro- and anti-vascular permeability agents such as vascular endothelial growth factor (VEGF) [3,4] and angiopoietin1 (Ang-1) [5,6], trigger common pathways such as ‘Src’ and ‘Akt’ in the differential regulation of endothelial-barrier [7]. In contrast to VEGF, Ang-1 enhances endothelial-barrier function, and blocking Ang-1 signaling disrupts endothelial-barrier [8–11]. Whereas, VEGF utilizes cSrc for the endothelial-barrier breakdown, Ang-1 antagonizes VEGF action through mDia-mediated sequestration of cSrc, thus putting a break on VEGF-mediated VE-cadherin internalization [6]. In contrast, Robo-4 opposes VEGF-induced vascular permeability via cSrc inhibition [12]. VEGF and Ang-1 have also been shown to exert opposing effects on endothelial junctions via differential regulation of cSrc involving Syx [13]. A similar competition between pro- and anti-vascular permeability agents for Akt pathway in the differential regulation of endothelial-barrier has not yet been determined.
Interestingly, both vascular permeability inducing [VEGF, tumor necrosis factor-α (TNFα)] and inhibiting [Ang-1, sphingosine-1-phosphate (S1P), Robo4] agents activate Akt [5,12,14–16]. Previous studies conflicted between vascular permeability promoting role of Akt1-endothelial nitric oxide synthase (eNOS) pathway [17] versus vascular protective role of Akt-mTOR signaling [18]. Our previous studies demonstrated increased vascular permeability in response to Adeno-VEGF in Akt1−/− mice [3,19]. Although subsequent studies [20–22],[23] supported our findings, until today, how Akt1 activation by VEGF and Ang-1 results in reciprocal regulation of the endothelial-barrier in the short-term is never addressed. It is also not clear what pathway is responsible for the restoration of endothelial-barrier integrity following VEGF-induced endothelial-barrier breakdown.
Based on our previous studies and the existing literature, we hypothesized that although VEGF induces short-term vascular permeability utilizing cSrc, recovery from VEGF-induced vascular permeability and endothelial-barrier enhancement by Ang-1 is reliant on sustained activation of Akt1. To test this, we generated Akt1-depleted human microvascular endothelial cells (HMEC) through ShRNA and tamoxifen-inducible, VE-cadherin promoter driven, endothelial-specific Akt1 knockout (VECad-Cre-Ak1) mice. Our studies revealed that Akt1 depletion prevents restoration of basal endothelial-barrier resistance following VEGF-induced permeability and impairs Ang-1-mediated endothelial-barrier enhancement in vitro and vascular permeability in vivo. Although VEGF-induced acute endothelial-barrier disruption was independent of Akt1, long-term effects of VEGF and Ang-1 on barrier protection was dependent on Akt1 activity through nuclear localization of FoxO, and expression of various TJ proteins, mainly claudin-5. Together, we demonstrate a specific role for the Akt1-FoxO pathway in endothelial-barrier protection and provide novel insights into its therapeutic potential for the treatment of vascular permeability-related clinical conditions.
Materials and methods
Generation of ‘VECad-Cre-Akt1’ transgenic mouse model
All experiments were performed with approval by the Charlie Norwood VAMC Institutional Animal Care and Use Committee (Approval reference #13-09-062). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals. Carbon dioxide asphyxiation followed by cervical dislocation was performed for Euthanesia. Isoflurane inhalation was used as anesthesia. For our study, we generated an endothelial-specific, tamoxifen-inducible Akt1 knockout mouse model (VECad-Cre-Akt1) by crossing Akt1 LoxP mice with VE-Cadhein Cre mice [24] in pure C57BL6 background. The mice were back-crossed 6 times prior to use in experiments. Age and sex matched 8–12 week-old tamoxifen untreated littermates and VECad-Cre-Akt1 mice on a C57BL/6 background were used in the study. The left ear in each of these mice was used as a control for the treatment in the right ear in the same mice. For a direct comparison of control and VECad-Cre-Akt1 mice on basal vascular permeability, controls used were wild-type (WT) mice treated with tamoxifen. Genotyping was performed using specific primers for A1-3Loxp: TCACAGAGATCCACCTGTGC, and A1-4113R: GCAGCGGATGATAAAGGTGT. Tamoxifen (Sigma, St. Louis, MO) stock solution (100mg/ml) was prepared dissolving in absolute ethanol and stored in aluminum foil covered plastic tubes at −20 °C. Right before injection, sterile corn oil was added to dilute tamoxifen to a final concentration of 10mg/ml. Tamoxifen (1mg/10g dose) was administered to the mice using a 27G needle via intraperitoneal (i.p.) injection every 24 hours for 5 consecutive days. Following this, transgene was maintained active by feeding the mice with a custom made Tamoxifen diet (Harlan, Madison, WI) for the duration of the experiments.
Cell culture and preparation of ShAkt1 stable cell lines
Human dermal (Telomerase-immortalized) microvascular endothelial cells (HMEC) (CRL-4025; ATCC, Manassas, VA) were maintained in Endothelial Cell Basal Medium-2 with a Growth Medium-2 Bullet Kit (Lonza; Walkersville, MD). All cultures were maintained in a humidified 5% CO2 incubator at 37 °C, and routinely passaged when 80–90% confluent.
Stable ShControl, ShAkt1 (ACGCTTAACCTTTCCGCTG) HMEC cells were generated using SMART vector 2.0 lentivirus particles (109 pfu) (Thermo Scientific, Waltham, MA). Lentivirus particles were mixed in 1ml Hyclone SFM4Transfx-293 (Fisher, Hanover Park, IL) and added along with 1 μl Polybrene (10mg/ml, American bioanalytical, Natick, MA). Three days later, transfection efficiency was tested through Turbo-GFP expression and subjected for 4 μg/ml puromycin (Life Technologies, Grand Island, NY) selection until all cells expressed GFP.
Quantitative RT-PCR arrays
Control and ShAkt1 cells were used for the qRT-PCR arrays. Briefly, cells were lysed and RNA was isolated using RNAase Mini plus Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Next, cDNA was generated by RT2 First Strand Kit (SABiosciences, Frederick, MD), mixed with qPCR SyberGreen master mix and loaded into Tight-junction and Adherens-junction RT2 Profiler PCR Array plates. Reading was completed in Eppendorf Mastercycler realplex-2 equipment (Hauppauge, NY).
Measurement of endothelial-barrier resistance
Endothelial-barrier integrity (measured as electrical resistance of the endothelial monolayer) was determined using ECIS equipment (Applied Biophysics, Troy, NY) as described previously [25,26]. To synchronize the HMEC before treatment, cells were cultured in serum free EBM-2 medium for 5 hours, followed by treatment with VEGF or Ang-1. Endothelial-barrier resistance was measured at multiple frequency modes.
Isolation of endothelial cells from mouse aorta
Aortae from vehicle (corn oil) and Tamoxifen (1mg/10g dose in corn oil) administered (i.p.) VECad-Cre mice were implanted in Matrigel and allowed to grow and sprout in the presence of 30 ng/ml of VEGF for 7 days, and the endothelial cells from the rings were harvested as standardized in our lab [3,4]. Cells were subjected for the Western analysis of Akt1 and β-actin.
Administration of adenoviral particles in mouse ears
Adenovirus particles (107 pfu) containing genes encoding VEGF-A, Ang-1, MyrAkt1, or green fluorescence protein (GFP; as negative control) (Abm, Richmond, BC, Canada) was introduced intra-dermally into mouse ears as previously described [3]. Expression of these proteins was confirmed three days post virus administration by Western analysis. In in vitro studies, HMEC were infected with Ad-GFP or Ad-MyrAkt1 virus particles at 107 pfu for 1 million cells plated in a culture dish along with 1μl Polybrene added on top of the monolayer. Three days following virus infection, expression of myrAkt1 was detected by western-blot analysis.
Immunofluorescence staining
For tissue immunofluorescent staining, sections were blocked with 10% goat serum followed by incubation with primary antibodies against Akt1 (1:100, rabbit anti-mouse, Cell Signaling, Danvers, MA) and CD31 (1:100, mouse anti-mouse, Abcam, Cambridge, MA) at 4 °C overnight. Immunofluorescent staining of HMEC monolayers was performed using the chamber slides. Cells were then washed twice with PBS, fixed using 2% paraformaldehyde for 30 min, permeabilized with 0.1% Triton X-100 for 15 min, and blocked with 2% BSA in sterile PBS. Cell monolayers were then incubated with antibodies against Claudin-5 (1:100, Rabbit antibody, Cell Signaling, Danvers, MA) at 4 °C overnight. Immunofluorescence was revealed using AlexaFlour secondary antibodies (1:2000 dilution of goat anti rabbit 488 and goat anti-mouse 594) obtained from Life Technologies, Grand Island, NY. Tissue sections or cells were mounted on to a glass slide using DAPI containing mounting medium (Vector Laboratories). Samples were observed under a confocal microscope equipped with argon and helium/neon lasers (LSM510, Zeiss, Germany). Controls were performed by omitting either one or both primary antibodies. All controls gave negative results with no detectable non-specific labeling.
Western blot analysis
Western blot analysis was performed as described previously [27,28]. Antibodies used include Akt1, β-catenin (ser675), FoxO1/3a and claudin-5 from Cell Signaling, anti- β-actin from Sigma, St. Louis, MO and Zo-1, Zo-2 from Abcam, Cambridge, MA.
Miles assay
Vascular permeability was assessed by visualizing and quantifying the leakage of Evan’s blue dye into the vascular wall according to the previously published protocol [3]. Briefly, Thirty μl each of 20 ng/ml VEGF or 50 ng/ml Ang-1 recombinant proteins (R&D Systems, Minneapolis, MN) as well as Ad-VEGF and/or Ad-Ang-1virus particles (Abm, Richmond, BC, Canada) were injected intra-dermally in the mouse ear skin for 30 min (for growth factors) or 3 days (for adenovirus particles), respectively. 1% (8μl/g) Evans blue dye solution in PBS was administered via the tail vein of Control and VECad-Cre-Akt1 mice. After 30 min of Evan’s blue injection, mice were perfused with sterile PBS using a pump to remove the excess dye from the vasculature (to specifically measure only dye leaked to the extravascular tissue in the ear). Circular ear tissues punches (Ø 8mm) from the injection sites were incubated in formamide for 24 hours at 60°C for the dye extraction. Extracted Evan’s blue dye was measured at 610 nm by spectrophotometer.
TUNEL assay
The TUNEL assay for in situ detection of apoptosis was performed using the ApopTag® Fluorescein in situ Apoptosis detection kit (Millipore, MA) as described previously [25,26]. Cells were analyzed for apoptosis using an inverted fluorescence microscope (Zeiss Axiovert100M, Carl Zeiss, Germany).
Statistical Analysis
All the data are presented as Mean + SD and were calculated from multiple independent experiments performed in quadruplicates. For normalized data analysis, data was confirmed that normality assumption was satisfied and analyzed using paired sample t test (dependent t test) and/or further confirmed with non-parametric test Wilcoxon signed rank test. For all other analysis, Student’s two-tailed t test or ANOVA test were used to determine significant differences between treatment and control values using the GraphPad Prism 4.03 software and SPSS 17.0 software.
Results
Akt1 is integral for recovery from VEGF-induced acute vascular permeability and Ang-1 mediated long-term endothelial-barrier protection
To explore how Akt1 is differentially utilized by growth factors in endothelial-barrier regulation, we generated stable ShAkt1 HMEC and VECad-Cre-Akt1 mice (Supplemental Figure 1A–B). ShAkt1 HMEC exhibited impaired barrier resistance in the real-time measurements from Electric Cell-substrate Impedance Sensing (ECIS) assay (Figure 1A). Since both VEGF and Ang-1 activate Akt1, we sought to compare the effect of Akt1 deficiency on VEGF and Ang-1-mediated endothelial-barrier function. Though treatment with VEGF significantly reduced control HMEC-barrier resistance, peaking between 2–4 h, Ang-1 treatment resulted in a dose and time-dependent increase in endothelial-barrier resistance (Figure 1C). Whereas VEGF-induced endothelial-barrier breakdown was recovered with time at lower doses (10 and 20 ng/ml), eventually reaching to the level of control monolayer, at higher doses (50 ng/ml), VEGF-mediated endothelial-barrier breakdown was irreversible (Supplemental Figure 1C and Figure 1D), likely due to the detachment of a few cells. In contrast, Ang-1 treatment elicited a dose-dependent increase in endothelial-barrier resistance peaking at 50 ng/ml (Supplemental Figure 1D). However, recovery from VEGF-induced endothelial permeability and barrier protection offered by Ang-1 were impaired in ShAkt1 HMEC (Figure 1C and D). Interestingly, endothelial-barrier breakdown due to high-dose VEGF (50 ng/ml) was reversed, starting from 30 minutes, by co-treatment with Ang-1 (50 ng/ml) (Figure 1D; left panel). However, the protective effect of Ang-1 over high-dose VEGF-induced endothelial-barrier injury at late hours, but not early hours, was blunted in ShAkt1 HMEC (Figure 1D; right panel), thus demonstrating the role of Akt1 in endothelial-barrier recovery following acute VEGF stimulation and Ang-1-mediated long-term protection.
Figure 1. Akt1 deficiency compromises endothelial-barrier integrity and modulates VEGF and Ang-1-mediated endothelial-barrier function.
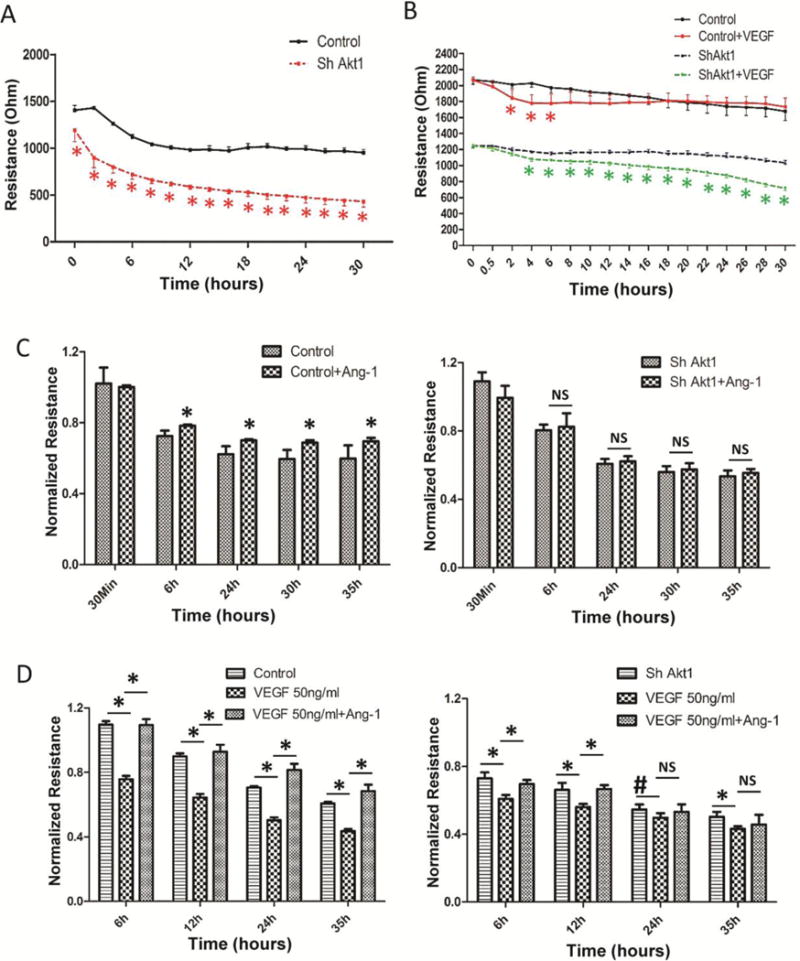
(A) Real-time changes in the barrier resistance of ShControl and ShAkt1 HMEC monolayers as measured using ECIS equipment showing differences in endothelial-barrier resistance between ShControl and ShAkt1 HMEC monolayers at various time points after plating equal amount of cells in array wells (n=3). (B) Graph showing quantification of a comparison between ShControl and ShAkt1 HMEC on the acute as well as chronic effects of optimal dose (20 ng/ml) of VEGF on barrier integrity (n=3). (C) Bar graph showing quantification of a comparison between control and ShAkt1 knockdown HMEC on the acute as well as chronic effects of optimal dose of Ang-1 (50 ng/ml) on their barrier-junction integrity (n=3). (D) Bar Graph showing quantification of a comparison between control and ShAkt1 knockdown HMEC on the acute as well as chronic effects of supra-optimal dose of VEGF (50 ng/ml) alone and in combination with Ang-1 (50 ng/ml) on their barrier-junction integrity (n=3). *P < 0.01.
In order to determine if SkAkt1 HMECs will form a monolayer similar to the ShControl HMEC, we performed immunostaining of respective monolayes with VE-cadherin staining. Our analysis indicated that although there is a modest, but significant decrease in VE-cadherin expression in ShAkt1 HMEC compared to ShControl, both these cells developed monolayers (Supplemental Figure 2A and B). Our further analysis of these monolayers indicated that Akt1 knockdown did result in increased apoptosis compared to control HMEC (Supplemental Figure 2C and D), together indicating that the effect of Akt1 on the endothelial-barrier is mostly due to its direct effect on protein turnover in the cell junctions.
Akt1 is necessary for endothelial-barrier protection and vascular leakage in vivo
Next, we tested vascular permeability in the ears of tamoxifen-administered WT and VECad-Cre-Akt1 mice through intra-venous administration of Evan’s blue. Akt1 deficiency in VECad-Cre-Akt1 mouse endothelial cells was confirmed by immunohistochemistry in mouse ears (Figure 2A) and Western blot analysis in mouse aortic endothelial cell lysates (Figure 2B). Endothelial Akt1 knockdown in VECad-Cre-Akt1 mice, but not expression WT mouse ears with Ad-myrAkt1 resulted in significantly increased vascular leakage (Figure 2C–D), indicating that endothelial-specific Akt1 gene ablation lead to endothelial-barrier breakdown and vascular leakage.
Figure 2. Endothelial specific Akt1 depletion lead to vascular leakage in vivo.
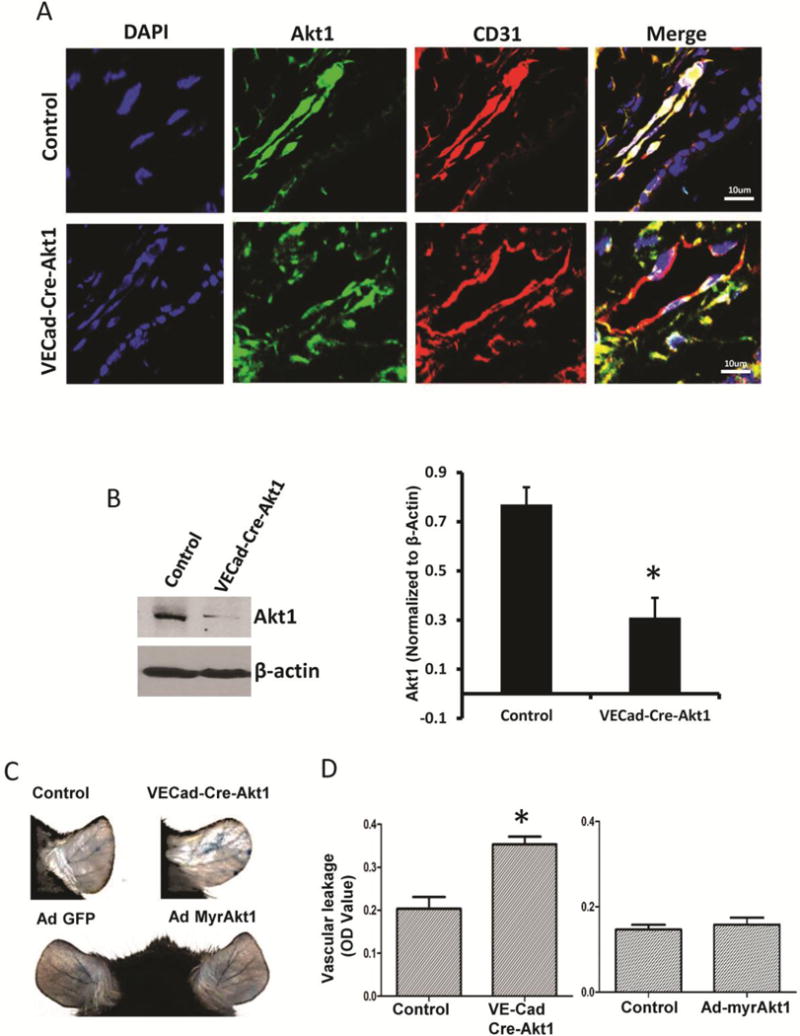
(A) Confocal images showing specific knockdown of Akt1 in mouse endothelial cells (earlobe section), evidenced by the loss of Akt1 (Green) in CD31 (Red) positive blood vessels (n=5). (B) Western blot images and optical densitometry analysis of aortic EC isolated from vehicle and tamoxifen treated VECad-Cre-Akt1 mice showing reduced Akt1 expression in tamoxifen-treated mice compared to untreated littermates (n=3). (C) Representative images of the tamoxifen-treated WT and VECad-Cre-Akt1 (above), and Ad-GFP and Ad-myrAkt1 expressing WT mice (below) ears showing leakage of Evan’s blue dye. (D) Graph showing the quantification of leaked Evan’s blue dye (Miles assay) in tamoxifen-treated VECad-Cre-Akt1 knockout (left) and Ad-myrAkt1 expressing mice ears (right), compared to respective untreated control mice (n=6). *P < 0.01. (Scale Bar: 10 μM).
In order to determine the precise role of endothelial Akt1 in the regulation of vascular permeability in vivo, we utilized VECad-Cre-Akt1 mice. Administration of recombinant VEGF (30 μl of 20 ng/ml VEGF in sterile PBS) to mice ears resulted in short-term vascular permeability in both mice (Figure 3A and C). Interestingly, the reconstitution of VECad-Cre-Akt1 mice ears with Ad-myrAkt1 did not rescue VEGF-induced acute vascular permeability, compared to Ad-GFP controls (Figure 3A and C), thus demonstrating that VEGF-induced acute vascular permeability in vivo is independent of Akt1. Next, we determined the role of Akt1 in chronic endothelial-barrier regulation in vivo with VEGF treatment. Administration of Ad-VEGF in WT and VECad-Cre-Akt1 mice ears (Supplemental Figure 3) resulted in significantly increased vascular permeability compared to Ad-GFP controls (Figure 3B and D). As expected, VEGF-induced leakage in VECad-Cre-Akt1 was higher compared to WT (Figure 3B and D). Interestingly, reconstitution of VECad-Cre-Akt1 mice ears with Ad-myrAkt1 significantly rescued the VEGF-induced chronic vascular permeability, compared to Ad-GFP controls (Figure 3B and D) demonstrating that sustained Akt1 activity protects the endothelial-barrier and inhibits vascular permeability in vivo.
Figure 3. Akt1 augments VEGF-induced vascular permeability and Ang-1-induced vascular-barrier protection in vivo.
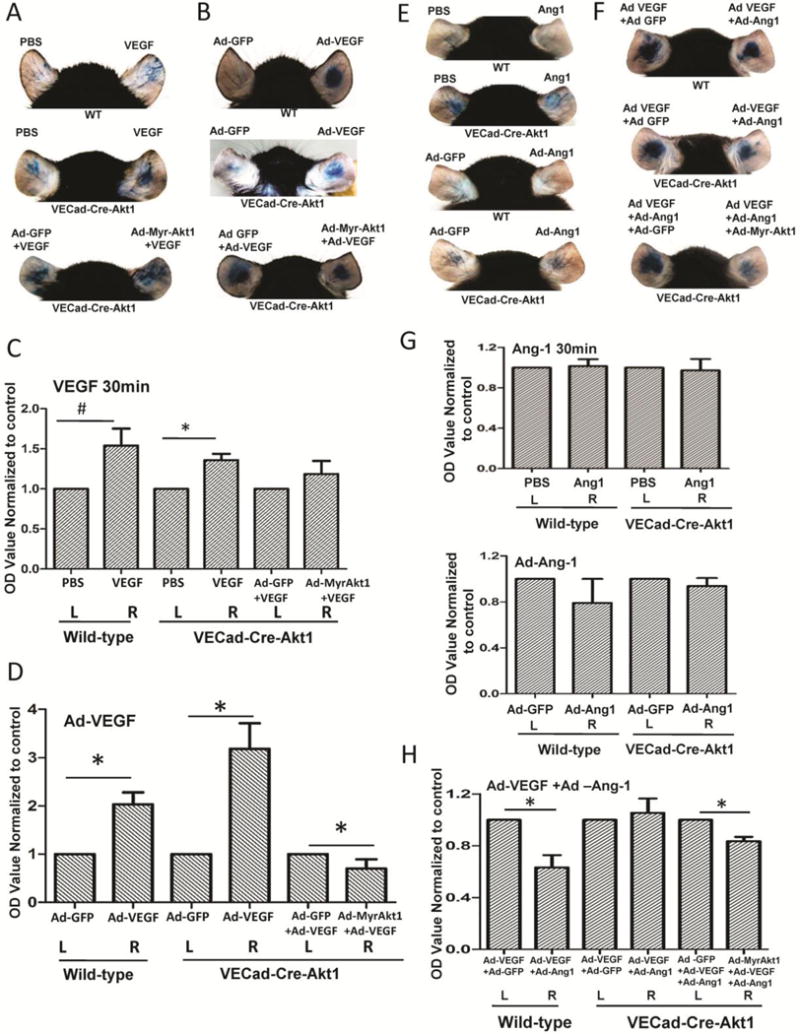
(A) Representative images of PBS and 30 μl of 20 ng/ml recombinant VEGF administered WT mice (top), tamoxifen-treated VECad-Cre-Akt1 mice administered with PBS and VEGF (middle) and tamoxifen-treated VECad-Cre-Akt1 mice administered with Ad-GFP + VEGF and Ad-MyrAkt1 + VEGF ears for 30 min (short-term) showing leakage of Evans blue dye (Miles assay). (B) Representative images of Ad-GFP and Ad-VEGF administered WT mice (top), tamoxifen-treated VECad-Cre-Akt1 mice administered with Ad-GFP and Ad-VEGF (middle) and tamoxifen-treated VECad-Cre-Akt1 mice ears expressing either Ad-GFP or Ad-myrAkt1 in the absence or presence of Ad-VEGF expression (long-term VEGF treatment) (bottom), showing leakage of Evans blue dye (Miles assay). (C) Histogram showing calorimetric quantification of the extravasated dye in the ears from PBS and 30 μl of 20 ng/ml recombinant VEGF administered WT mice (top), tamoxifen-treated VECad-Cre-Akt1 mice administered with PBS and VEGF (middle) and tamoxifen-treated VECad-Cre-Akt1 mice administered with Ad-GFP + VEGF and Ad-MyrAkt1 + VEGF after 30 min (n=6). (D) Histogram showing calorimetric quantification of the extravasated dye in the ears from Ad-GFP and Ad-VEGF administered WT mice (top), tamoxifen-treated VECad-Cre-Akt1 mice administered with Ad-GFP and Ad-VEGF (middle) and tamoxifen-treated VECad-Cre-Akt1 mice ears expressing either Ad-GFP or Ad-myrAkt1 in the absence or presence of Ad-VEGF expression (long-term VEGF treatment) (n=6). (E) Representative images of PBS and 30 μl of 50 ng/ml recombinant Ang1 administered WT mice and tamoxifen-treated VECad-Cre-Akt1 mice ears administered with PBS and 30 μl of 50 ng/ml recombinant Ang1 for 30 min (short-term) (top two panels), and representative images of mice ears expressing Ad-GFP or Ad-Ang-1 (long-term) in WT tamoxifen-treated VECad-Cre-Akt1 mice (bottom two panels) showing leakage of Evans blue dye (Miles assay). (F) Representative images of WT (top) and tamoxifen-treated VECad-Cre-Akt1 (middle) mice ears administered with combinations of either Ad-GFP/Ad-VEGF or Ad-VEGF/Ad-Ang-1, as well as VECad-Cre-Akt1 mice ears administered with a combination of either Ad-GFP/Ad-VEGF/Ad-Ang-1 or Ad-myrAkt1/Ad-VEGF/Ad-Ang-1 (bottom), showing leakage of Evans blue dye (Miles assay). (G) Histogram showing calorimetric quantification of the extravasated dye in the ears from WT and tamoxifen-treated VECad-Cre-Akt1 mice ears, with PBS or 30 μl PBS containing 50 ng/ml recombinant Ang-1 for 30 min, as well as WT and VECad-Cre-Akt1 mice ears, expressing either Ad-GFP or Ad-Ang-1 (n=6). (H) Histogram showing calorimetric quantification of the extravasated dye in the ears from WT and tamoxifen-treated VECad-Cre-Akt1 mice administered with combinations of either Ad-GFP/Ad-VEGF or Ad-VEGF/Ad-Ang-1, as well as tamoxifen-treated VECad-Cre-Akt1 mice ears administered with a combination of either Ad-GFP/Ad-VEGF/Ad-Ang-1 or Ad-myrAkt1/Ad-VEGF/Ad-Ang-1 (n=6). #P < 0.05, *P<0.01
We next determined that the role of Akt1 in endothelial-barrier protection offered by Ang-1. Recombinant Ang-1 (30 μl of 50 ng/ml VEGF in sterile PBS) administration to WT or VECad-Cre-Akt1 mice ears did not elicit any changes in the acute vascular permeability (Figure 3E and G). However, administration of Ad-Ang-1 in WT mice ears resulted in significantly decreased vascular permeability compared to Ad-GFP administered controls (Figure 3F and H). As expected, endothelial-barrier protection conferred by Ad-Ang-1 was blunted in VECad-Cre-Akt1 mice (Figure 3F and H). Furthermore, the reconstitution of VECad-Cre-Akt1 mice ears with Ad-myrAkt1 significantly restored the Ang-1-mediated endothelial-barrier protection (Figure 3F and H), demonstrating that acute vascular permeability is independent of Akt1, restoration of vascular integrity following VEGF treatment, and chronic effect of Ang-1 on blood-tissue barrier enhancement necessitates sustained Akt1 activity.
Akt1 deficiency affects real-time changes in the expression of endothelial adherens- (AJ) and tight-junction (TJ) proteins in response to VEGF and Ang-1
Next, we determined if Akt1 deficiency has any effect on real-time changes in HMEC AJ and TJ protein expression with VEGF and Ang-1 treatment. No significant difference in VE-cadherin or β-catenin expression was observed between control and ShAkt1 HMEC with either VEGF or Ang-1 (Figure 4). However, whereas significant increase in the expression of Zo-1/2 and claudin-5 was observed in the long-term in HMEC with VEGF and Ang-1 treatment, this effect was significantly impaired in ShAkt1 HMEC (Figure 4 and Supplemental Figure 5), thus documenting the integral role of Akt1 in TJ formation.
Figure 4. Akt1 deficiency affects real-time changes in the expression of proteins in endothelial-barrier AJs and TJs in response to VEGF and Ang-1 treatments.
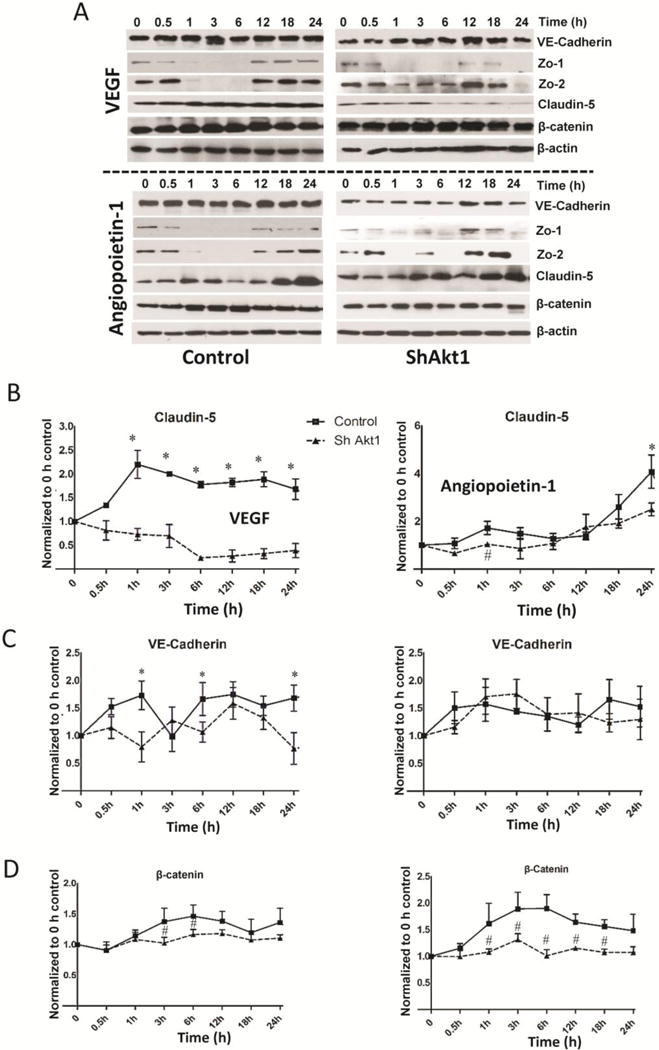
(A) Representative Western blot images of control (left) and ShAkt1 (right) HMEC lysates treated with 20 ng/ml VEGF and real-time changes in the expression levels of AJ proteins VE-Cadherin and β-catenin as well as TJ proteins Zo-1, Zo-2 and claudin-5 were determined, and compared to 0 h time point (above; n=3). Representative Western blot images of control (left) and ShAkt1 (right) HMEC lysates treated with 50 ng/ml Ang-1 and real-time changes in the expression levels of AJ proteins VE-Cadherin and β-catenin as well as TJ proteins Zo-1, Zo-2 and claudin-5 were determined, and compared to 0 h time point (below; n=3). (B) Densitometry analysis of Western blot images of control and ShAkt1 HMEC lysates treated with 20 ng/ml VEGF (left) and 50 ng/ml Ang-1 (right) showing changes in claudin-5 expression compared to 0 h (n=3). (C) Densitometry analysis of Western blot images of control and ShAkt1 HMEC lysates treated with 20 ng/ml VEGF (left) and 50 ng/ml Ang-1 (right) showing changes in VE-Cadherin expression compared to 0 h (n=3). (D) Densitometry analysis of Western blot images of control and ShAkt1 HMEC lysates treated with 20 ng/ml VEGF (left) and 50 ng/ml Ang-1 (right) showing changes in β-catenin expression compared to 0 h (n=3). *P < 0.01, #P < 0.05.
Endothelial-specific Akt1 deficiency results in impaired TJ protein expression
Since modulation of AJ has a direct effect on TJ protein expression [21,29] we performed PCR array analyses of control and ShAkt1 HMEC for AJ and TJ molecules. Our study revealed no significant effect of Akt1 deficiency on the expression of AJ proteins VE-cadherin and β-catenin, except modest changes in dynamin 2, notch-4, and talin-1 (Supplemental Figure 4). Interestingly, Akt1 deficiency resulted in 2 to 50-fold reduction in the mRNA levels of several TJ molecules, mainly the members of 20-gene claudin family (Figure 5A; Supplemental Figures 5), thereby demonstrating that Akt1 enhances endothelial-barrier integrity by stabilizing TJ through claudin expression. At the protein level, while expression of several claudins such as claudin-1, claudin-3, and claudin-17 were below detectable levels on Western blots, we observed significant reduction in claudin-5 expression in ShAkt1 HMEC; a predominant claudin isoform in endothelial cells [21] (Figure 5A–C). Next, we probed control and ShAkt1 HMEC monolayers for claudin-5 for expression changes at acute (30 min) and chronic (24 h) post-treatments with VEGF and Ang-1. Treatment with VEGF, but not Ang-1, significantly decreased claudin-5 expression in both control and ShAkt1 HMEC within 30 min (Figure 6). Although basal levels of Zo-1 and Zo-2 in ShControl and ShAkt1 HMECs were similar, there was significant difference in the turnover of Zo-1 and 2 in the long-term after treatment with VEGF or Ang1 (Supplemental Figure 6). Interestingly, whereas long-term stimulation with both VEGF and Ang-1 increased claudin-5 expression in control HMEC-barriers at 24 hours, this effect was blunted in ShAkt1 HMEC, thus demonstrating that long-term claudin-5 expression and endothelial-barrier protection by VEGF and Ang-1 is reliant on sustained Akt1 activity (Figure 6).
Figure 5. Long-term effect of Ang-1 and VEGF on endothelial-barrier protection is reliant on Akt1-mediated TJ stabilization.
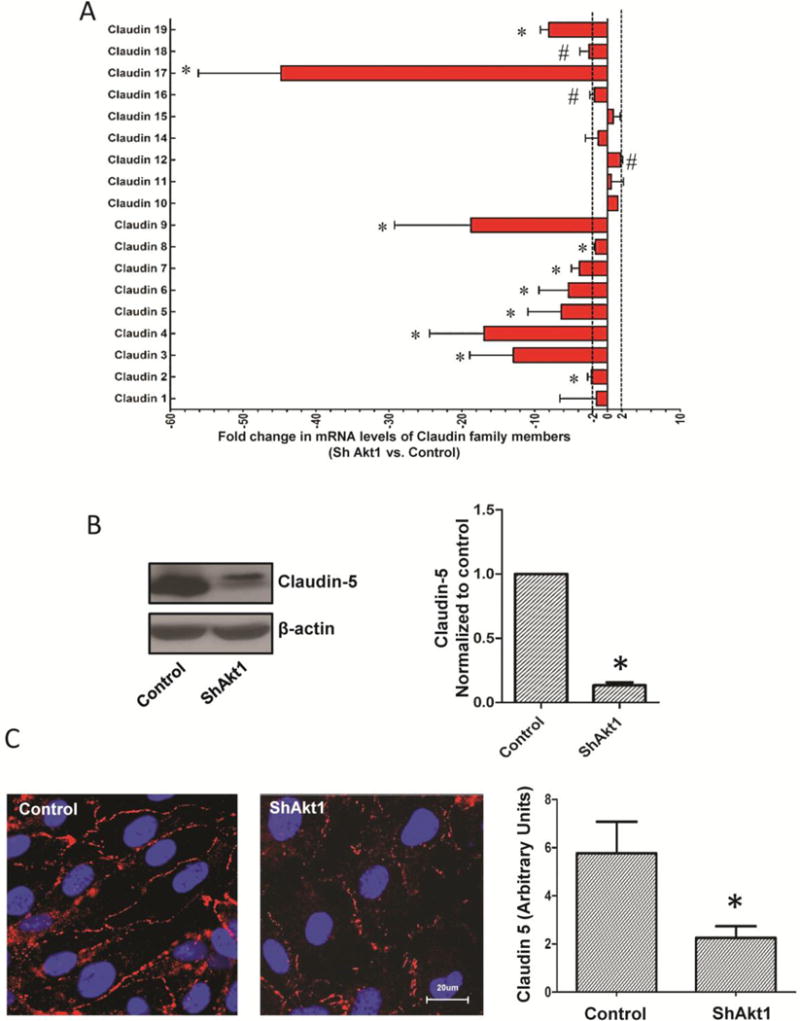
(A–B) Representative images of claudin-5 staining of ShControl and ShAkt1 HMEC monolayers after 30 min (short-term) treatment with 20 ng/ml VEGF or 50 ng/ml Ang-1, compared to PBS control, respectively. Quantification of the claudin-5 expression levels in HMEC monolayers following VEGF and Ang-1 treatment as analyzed using NIH-Image J software is provided in the right panels (n=10). (C–D) Representative images of claudin-5 staining of ShControl and ShAkt1 HMEC monolayers after 24 h (long-term) treatment with 20 ng/ml VEGF or 50 ng/ml Ang-1, compared to PBS control, respectively. Quantification of the claudin-5 expression levels in HMEC monolayers following VEGF and Ang-1 treatment as analyzed using NIH-Image J software is provided in the right panels (n=10). *P < 0.01. (Scale bars: 20 μM).
Figure 6. Long-term, not short-term changes in claudin-5 expression in HMEC is regulated by Akt1.
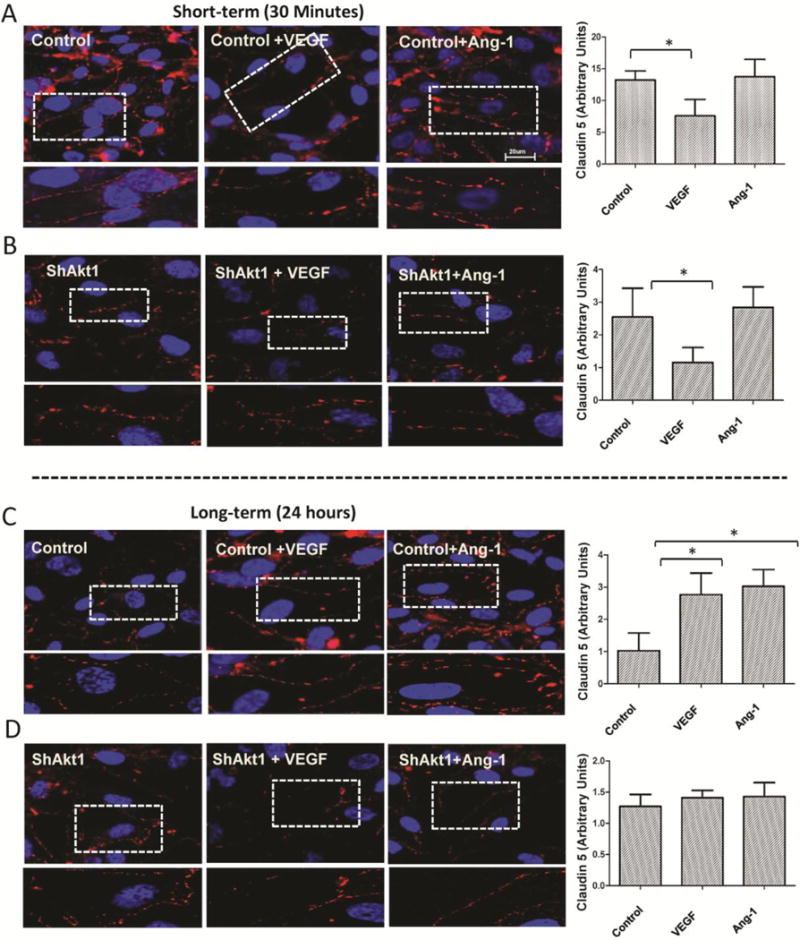
(A–B) Representative images of claudin-5 staining of ShControl and ShAkt1 HMEC monolayers after 30 min (short-term) treatment with 20 ng/ml VEGF or 50 ng/ml Ang-1, compared to PBS control, respectively. Quantification of the claudin-5 expression levels in HMEC monolayers following VEGF and Ang-1 treatment as analyzed using NIH-Image J software is provided in the right panels (n=10). (C–D) Representative images of claudin-5 staining of ShControl and ShAkt1 HMEC monolayers after 24 h (long-term) treatment with 20 ng/ml VEGF or 50 ng/ml Ang-1, compared to PBS control, respectively. Quantification of the claudin-5 expression levels in HMEC monolayers following VEGF and Ang-1 treatment as analyzed using NIH-Image J software is provided in the right panels (n=10). *P < 0.01. (Figure bars: 20 μM)
ShAkt1 HMECs exhibit increased nuclear localization of FoxO
In order to identify the downstream targets of Akt1 involved in the chronic endothelial-barrier regulation, we performed sub-cellular localization of VEGF and Ang-1 treated control and ShAkt1 HMEC lysates prepared after 24 h for β-catenin and FoxO transcription factors. Immunocytochemistry for FoxO3a revealed that at normal conditions (in the presence of serum), FoxO3a in the nucleus in ShAkt1 HMEC was significantly higher compared to ShControl (Figure 7A). In serum starved cells, the majority of FoxO3a was in the nucleus. Treatment with VEGF and Ang-1 increased FoxO3a in the cytosol with a corresponding reduction in the nucleus (Figure 7B–C). As expected, the effect of VEGF and Ang-1 on cytoplasmic sequestration of FoxO3a was blunted in ShAkt1 HMEC leading to its nuclear accumulation (Figure 7B–C).
Figure 7. Pharmacological inhibition of FoxO transcription factors restores endothelial-barrier function in ShAkt1 HMEC.
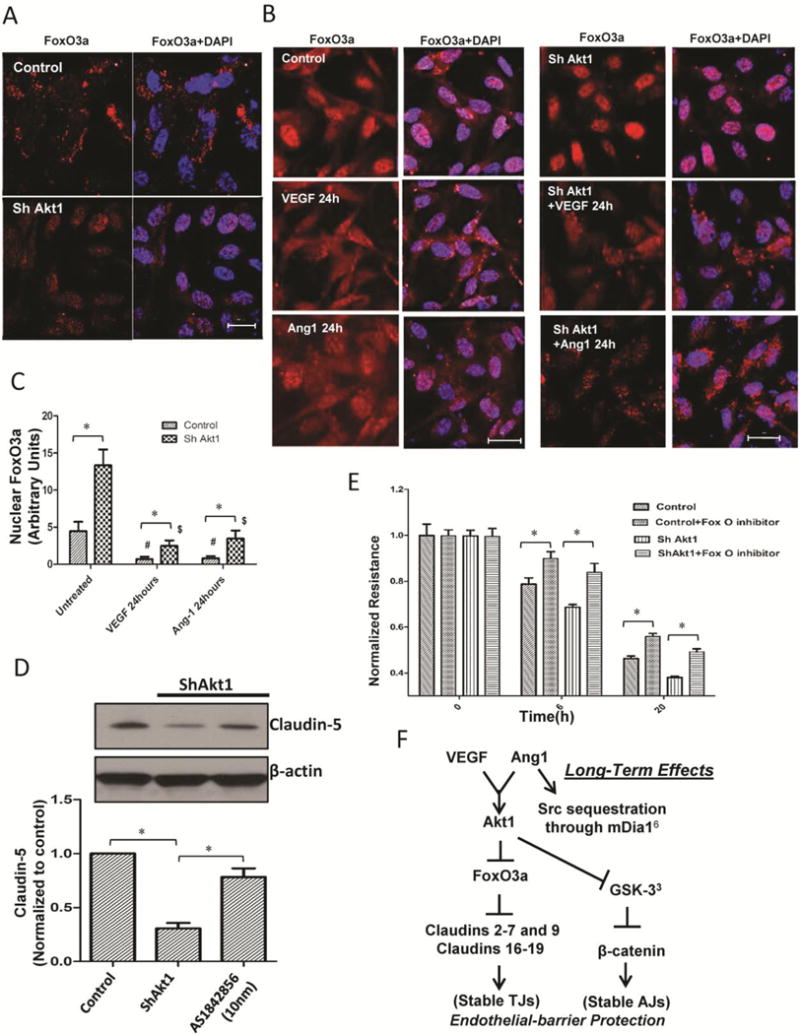
(A) Confocal images of ShControl and ShAkt1 HMEC stained with FoxO3a antibodies and DAPI in the presence of FBS. (B) Confocal images of serum starved ShControl (left panel) and ShAkt1 (right panel) HMEC, pre-treated with PBS (top), VEGF (middle) or Ang-1 (bottom) for 24 hours, and stained with FoxO3a antibodies and DAPI. (C) Bar graph showing quantification of the nuclear FoxO3a levels from the confocal images of ShControl and ShAkt1 HMEC, pre-treated with PBS, VEGF or Ang-1 for 24 h, and stained with FoxO3a antibodies and DAPI (n=6). (D) Western blot analysis of lysates from ShControl and ShAkt1 HMEC prepared after 12 hour treatment with 10 nM concentration of FoxO inhibitor AS1842856 (n=3). Bar graph showing quantification of Western blot analysis of ShControl and ShAkt1 HMEC prepared after 12 hour treatment with 10 nM concentration of FoxO inhibitor AS1842856 is shown below (n=4). (E) Bar graph showing the effect of FoxO inhibitor AS1842856 (10 nM) on endothelial-barrier resistance in ShControl and ShAkt1 HMEC as measured from the ECIS equipment (n=4). (F) Working hypothesis on the role of Akt1 on acute and chronic vascular permeability. *P < 0.01. (Scale bars: 20 μM).
Pharmacological inhibition of FoxO reverses aberrant vascular permeability in ShAkt1 HMEC monolayers
Next, we determined if pharmacological inhibition of FoxO can rescue the endothelial-barrier in ShAkt1 cells. Treatment with FoxO inhibitor AS1842856 (10 nM) significantly enhanced HMEC-barrier resistance and reversed impaired endothelial-barrier resistance in ShAkt1 HMEC (Figure 7D and E) indicating that cytosolic sequestration of FoxO transcription factors and removal of transcriptional repression of TJ proteins such as claudins are essential for the Akt1-mediated long-term endothelial-barrier enhancement in response to VEGF and Ang-1 (Figure 7F).
Pharmacological inhibition of glycogen synthase kinase-3 (GSK-3) reverses aberrant vascular permeability in ShAkt1 HMEC monolayers in the long-term
In order to determine the other contributing factors in Akt1-mediated endothelial barrier function, we analyzed the expression and phosphorylation levels of two other major Akt substrates, GSK-3 and eNOS. Our analysis indicated a basal decrease, and hence activation of GSK-3α/β Ser-9/21 phosphorylation in ShAkt1 HMEC compared to ShControl (Figure 8A and B). Our further analysis of phosphorylated β-catenin levels in these cells revealed increased phosphorylation of Ser-675 β-catenin (Figure 8C–D). Interestingly, higher dose of VEGF (50 ng/ml), but no other doses of VEGF or Ang-1 treatment maintained increased β-catenin phosphorylation 24 hours after treatment (Figure 8C–D), thus indicating that 50 ng/ml dose of VEGF elicits off-target effects in HMEC. On the other end, we did not observe a significant difference in the phospho-eNOS expression between ShControl and ShAkt1 HMEC, either in the presence or absence of VEGF and Ang-1 treatment (Supplemental Figure 7).
Figure 8. Pharmacological inhibition of GSK-3 in ShAkt1 HMEC partially restores the endothelial-barrier integrity in the long-term.
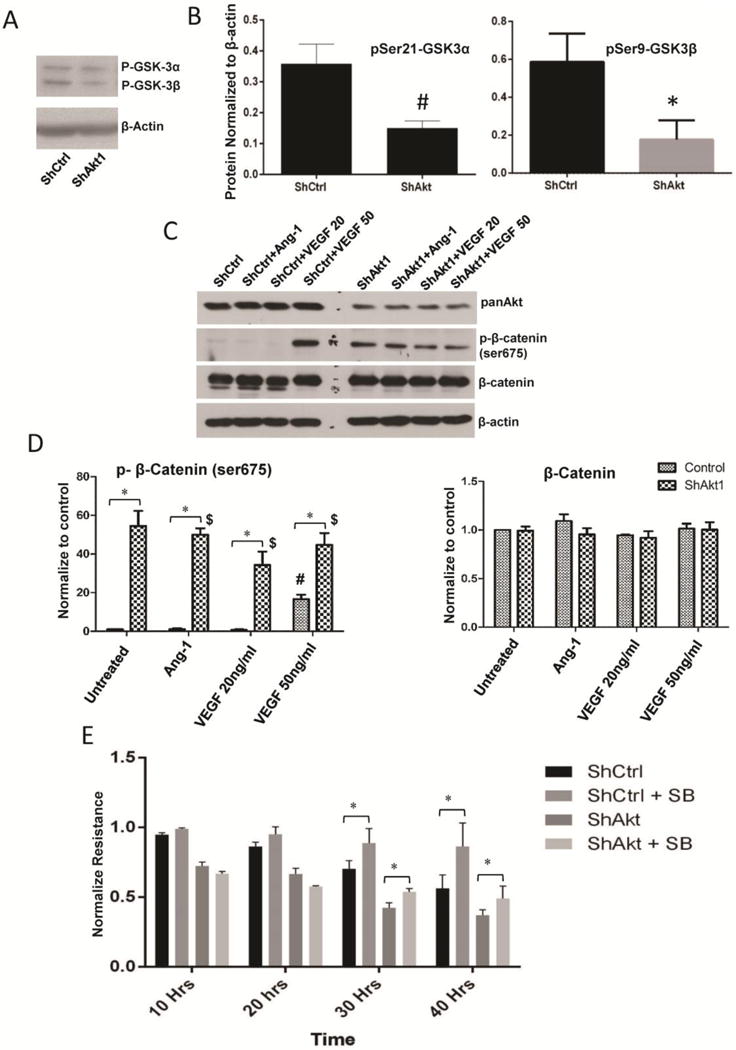
(A–B) Resperesentative Western blot images of ShControl and ShAkt1 HMEC lysates showing basal levels Ser9/21 phosphorylation of of GSK-3α/β (n=3). (C) Resperesentative Western blot images of ShControl and ShAkt1 HMEC lysates treated with PBS, 20 ng/ml VEGF, 50 ng/ml VEGF and 50 ng/ml Ang1 showing a comparison on phosphorylation and total expression of β-catenin, a GSK-3 substrate. (D) Densitometry analysis of Western blots of ShControl and ShAkt1 HMEC lysates treated with PBS, 20 ng/ml VEGF, 50 ng/ml VEGF and 50 ng/ml Ang1 showing a comparison on phosphorylation and total expression of β-catenin (n=3). (E) Bar graph showing the effect of GSK-3 inhibitor SB415286 (20 μM) on endothelial-barrier resistance in ShControl and ShAkt1 HMEC as measured from the ECIS equipment (n=4).*P < 0.01, #P < 0.05, $Compared to untreated control.
Interestingly, although treatment with GSK-3 inhibitor SB415286 (20 μM) did not have any significant effect on the endothelial-barrier resistance during the initial 20 hrs after the treatment, prolonged treatment to 30 and 40 hours significantly enhanced HMEC-barrier resistance and reversed impaired endothelial-barrier resistance in ShAkt1 HMEC (Figure 8E) indicating that GSK-3, at least in part, does contribute in the Akt1-mediated endothelial-barrier regulation.
Discussion
Signaling pathways regulated by the Src family of tyrosine kinases (SFKs) are the most studied in VEGF-induced acute endothelial-barrier breakdown [4,11]. In several malignancies, pathological levels of VEGF induces chronic vascular leakage through Src-mediated degradation of VE-cadherin [30]. Apart from SFKs, another important molecule that is currently being investigated in endothelial-barrier regulation is protein kinase B (Akt). Increased Akt phosphorylation in endothelial cells upon VEGF stimulation [3,14], probably through a Src-Axl pathway [31], was indicative of its possible role in promoting VEGF-induced acute vascular permeability via eNOS [17]. Although studies have demonstrated the importance of eNOS in transcellular transport of components across the endothelial-barrier through caveolin1-positive vesicles [32], its role in paracellular endothelial-barrier regulation is not clear. Activation of Akt by both agents that induce (e.g. VEGF, Ang-2) or inhibit (e.g. Ang-1, Robo4, S1P) vascular permeability [5,12,14–16] further questioned the Akt-eNOS theory, and demanded a reinvestigation on Akt’s role in the regulation of endothelial-barrier function.
Our previous study utilizing Akt1−/− mice revealed enhanced VEGF-induced vascular permeability in vivo [3] and provided the first direct evidence for the involvement of Akt1 in endothelial-barrier protection. This was later supported by other laboratories [20–22] and challenged the ‘dogma’ on the role of Akt1 in promoting vascular permeability. Also, these studies opened up a series of new questions for further investigation such as how VEGF and Ang-1 both activate Akt1, but exert opposing effects on endothelial-barrier? What is the short-term vs long-term effect of Akt1 activity on endothelial-barrier function? If co-treatment with Ang-1 inhibits VEGF-induced endothelial-barrier breakdown, both via Akt1, what molecular mechanisms downstream of Akt1 mediate this net effect of co-stimulation?
Existing controversies on the role of Akt1 on vascular permeability may be due to the overcompensating mechanisms in the gene knockout mice and aberrant post-translational modifications induced by protein over-expression in gene-overexpressing mice. Hence, we generated a new tamoxifen-inducible, endothelial-specific Akt1−/− (VECad-Cre-Akt1) mice to rule out any chance of embryonically developed compensating mechanisms. The results were then compared with stable Lenti-ShAkt1 HMEC in vitro analyzed using ECIS technology. Our studies revealed that Akt1 deficiency in HMEC prevents restoration of basal endothelial-barrier resistance after acute VEGF-induced permeability and inhibits Ang-1-mediated endothelial-barrier enhancement. Although co-treatment with Ang-1 abolished the endothelial-barrier disruption in vitro and vascular permeability in vivo by VEGF, this effect was blunted in Akt1-null HMEC and VECad-Cre-Akt1 mice. Gene array and subsequent Western blot analyses revealed that Akt1 has a significant effect on the expression of claudin family of TJ proteins. The effect was more profound on claudin-5 expression at the protein levels, the predominant endothelial claudin [21]. Together, these results demonstrate that while acute effects of VEGF and Ang-1 on endothelial-barrier is independent of Akt1, the recovery following VEGF-induced vascular permeability and chronic Ang-1 mediated endothelial-barrier enhancement is dependent of Akt1. Thus, our study provides the first concrete evidence on the integral role of Akt1 in endothelial-barrier protection.
An important question that perplexed scientists for a very long time is how Akt, which is activated by both vascular permeability stimulating and inhibiting agents, is involved in endothelial-barrier regulation. An earlier study pointed to the importance of Ang-1 in endothelial-barrier protection via transcriptional repression of Ang-2 that antagonizes Ang-1 action, a process necessitating Akt-FoxO pathway [33]. Surprisingly, two independent studies utilizing different systemic Akt1−/− mouse models, argued on the role of Akt1 in vascular permeability; one indicating a role for Akt1 in vascular protection via a thrombospondin-mediated, eNOS-independent mechanism [3], and the other showing a role for Akt1 in augmenting vascular permeability via activation of eNOS [17]. Interestingly, another study reported vascular protection by Akt1 via an eNOS-independent, but mTOR-dependent mechanism concluding that enhancing Akt activity alone could have therapeutic benefits after vascular injury [20]. Based on these reports, scientists also postulated that controversy over the role of Akt1 in the regulation of vascular permeability might be due to its acute vs. chronic effects on the endothelial-barrier protein turnover [34]. Most recent studies conducted in various models of endothelial-barrier function and vascular permeability further supported the hypothesis that Akt1 may have a net vascular protective role in vivo [16,22]. However, none of these studies explain how Akt1 modulated the endothelial-barrier.
The initial report on the long-term effects of Akt1 on endothelial-barrier protein turnover came from an elegant study from Dejana and colleagues [21]. This study reported the effect of VE-Cadherin activation and AJ stability on claudin-5 expression and TJ formation via an Akt-FoxO pathway, an effect that was not perturbed by acute stimulation with VEGF. However, long-term effects of sustained Akt1 activation by VEGF and Ang-1 and the molecular mechanisms downstream of Akt1 in endothelial-barrier regulation were not addressed in this study. Therefore, our study constitutes the first to investigate the effect of transient and sustained Akt1 activation by VEGF and Ang-1 and to test the involvement of FoxO signaling pathways in Akt1-mediated endothelial-barrier protection. Multiple gene array analysis in HMEC revealed that Akt1 deficiency does not elicit any discernible effect on the mRNA expression of AJ proteins. Surprisingly, significant changes were observed in the mRNA levels of TJ proteins, predominantly claudins. Although the basal levels of zona occludens (Zo1 and Zo2) were similar in ShControl and ShAkt1 HMEC, increase in the expression of these were observed with VEGF and Ang-1 treatment in ShControl HMEC, an effect that was blunted in ShAkt1 HMEC. Thus, in the long-term, absence of Akt1 will lead to weaker TJ resulting in endothelial-barrier breakdown. The importance of Akt1 in the acute and chronic vascular permeability in vivo was also confirmed in WT and VECad-Cre-Akt1 mice by administering recombinant proteins (acute) and adenoviral particles (chronic) of VEGF and Ang-1 in the ears, thus demonstrating the integral role of Akt1 in long-term protection of endothelial-barrier.
Increased nuclear localization of FoxO in ShAkt1 HMEC suggested the potential role of FoxO in Akt1-mediated endothelial-barrier protection. FoxO transcription factors have been implicated in the regulation of angiogenesis [35]. Decreased phosphorylation and increased activation of GSK-3, a known Akt substrate [17] in ShAkt1 HMEC also suggested its potential role in endothelial-barrier regulation. The role of FoxOs and GSK-3 in VEGF- and Ang-1-induced vascular permeability is not known. Our immunocytochemistry and gene array analysis, provide the primary evidence on the predominant role of FoxO3a in the expression of claudin family of TJ proteins in the long-term endothelial-barrier protection. In our analysis, we also identified novel Akt1-regulated endothelial claudin isoforms. Akt1 deficiency resulted in decreased mRNA of claudin-3, 4, 5, 6, 7, and 9 isoforms. Interestingly, significant decrease in claudin-17, 18 and 19 that are involved in TJ-regulated anion selective transport of molecules [36], was also observed in ShAkt1 HMECs. Whereas, our study identifies a number of novel genes involved in Akt1-mediated endothelial-barrier function, our data on the ability of FoxO inhibitor (10 nM AS1842856) and GSK-3 inhibitor (20 μM SB415286) to rescue the endothelial-barrier defect in ShAkt1 HMEC independently indicates the therapeutic potential of targeting FoxO and GSK-3 for vascular permeability-related disorders.
Supplementary Material
Acknowledgments
Funds were provided by the National Institutes of Health grant (R01HL103952) to PRS. This material is the result of work supported with resources and the use of facilities at the Charlie Norwood VAMC, Augusta, GA. The funders had no role in the study design, data collection, analysis and decision to publish. Preparation of the manuscript and the contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations
- VEGF
Vascular endothelial growth factor
- Ang-1
Angiopoietin-1
- VECad-Cre-Ak1
Vascular endothelial cadherin-cre recombinase-Akt1 knockdown
- HMEC
Human microvascular endothelial cells
- FoxO
Forkhead box protein O
- ECIS
Electric cell substrate impedance sensing
- WT
Wild type
- GFP
Green fluorescent protein
- eNOS
Endothelial nitric oxide synthase
- AJ
Adherens junction
- TJ
Tight junction
- Zo
Zona occludens
Footnotes
Conflicts of interest: Authors declare that there are no financial or conflicts of interests exist.
References
- 1.Nagy JA, Dvorak AM, Dvorak HF. Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harbor perspectives in medicine. 2012;2:a006544. doi: 10.1101/cshperspect.a006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goddard LM, Iruela-Arispe ML. Cellular and molecular regulation of vascular permeability. Thrombosis and haemostasis. 2013;109:407–415. doi: 10.1160/TH12-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Somanath PR, Razorenova O, et al. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nature medicine. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ha CH, Bennett AM, Jin ZG. A novel role of vascular endothelial cadherin in modulating c-Src activation and downstream signaling of vascular endothelial growth factor. The Journal of biological chemistry. 2008;283:7261–7270. doi: 10.1074/jbc.M702881200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daly C, Wong V, Burova E, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1) Genes & development. 2004;18:1060–1071. doi: 10.1101/gad.1189704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Developmental cell. 2008;14:25–36. doi: 10.1016/j.devcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Weis SM. Vascular permeability in cardiovascular disease and cancer. Current opinion in hematology. 2008;15:243–249. doi: 10.1097/MOH.0b013e3282f97d86. [DOI] [PubMed] [Google Scholar]
- 8.Augustin HG, Koh GY, Thurston G, et al. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nature reviews Molecular cell biology. 2009;10:165–177. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 9.Murakami M, Nguyen LT, Zhuang ZW, et al. The FGF system has a key role in regulating vascular integrity. The Journal of clinical investigation. 2008;118:3355–3366. doi: 10.1172/JCI35298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nature cell biology. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 11.Esser S, Lampugnani MG, Corada M, et al. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. Journal of cell science. 1998;111(Pt 13):1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 12.Jones CA, London NR, Chen H, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nature medicine. 2008;14:448–453. doi: 10.1038/nm1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ngok SP, Geyer R, Liu M, et al. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. The Journal of cell biology. 2012;199:1103–1115. doi: 10.1083/jcb.201207009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Somanath PR, Kandel ES, Hay N, et al. Akt1 signaling regulates integrin activation, matrix recognition, and fibronectin assembly. The Journal of biological chemistry. 2007;282:22964–22976. doi: 10.1074/jbc.M700241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Palma C, Meacci E, Perrotta C, et al. Endothelial nitric oxide synthase activation by tumor necrosis factor alpha through neutral sphingomyelinase 2, sphingosine kinase 1, and sphingosine 1 phosphate receptors: a novel pathway relevant to the pathophysiology of endothelium. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:99–105. doi: 10.1161/01.ATV.0000194074.59584.42. [DOI] [PubMed] [Google Scholar]
- 16.Singleton PA, Chatchavalvanich S, Fu P, et al. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circulation research. 2009;104:978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ackah E, Yu J, Zoellner S, et al. Akt1/protein kinase Balpha is critical for ischemic and VEGF-mediated angiogenesis. The Journal of clinical investigation. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue Q, Hopkins B, Perruzzi C, et al. Palomid 529, a novel small-molecule drug, is a TORC1/TORC2 inhibitor that reduces tumor growth, tumor angiogenesis, and vascular permeability. Cancer research. 2008;68:9551–9557. doi: 10.1158/0008-5472.CAN-08-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Somanath PR, Razorenova OV, Chen J, et al. Akt1 in endothelial cell and angiogenesis. Cell cycle. 2006;5:512–518. doi: 10.4161/cc.5.5.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukai Y, Rikitake Y, Shiojima I, et al. Decreased vascular lesion formation in mice with inducible endothelial-specific expression of protein kinase Akt. The Journal of clinical investigation. 2006;116:334–343. doi: 10.1172/JCI26223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taddei A, Giampietro C, Conti A, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nature cell biology. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- 22.Ashida N, Senbanerjee S, Kodama S, et al. IKKbeta regulates essential functions of the vascular endothelium through kinase-dependent and -independent pathways. Nature communications. 2011;2:318. doi: 10.1038/ncomms1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qi X, Okamoto Y, Murakawa T, et al. Sustained delivery of sphingosine-1-phosphate using poly(lactic-co-glycolic acid)-based microparticles stimulates Akt/ERK-eNOS mediated angiogenesis and vascular maturation restoring blood flow in ischemic limbs of mice. European journal of pharmacology. 2010;634:121–131. doi: 10.1016/j.ejphar.2010.02.038. [DOI] [PubMed] [Google Scholar]
- 24.Kogata N, Arai Y, Pearson JT, et al. Cardiac ischemia activates vascular endothelial cadherin promoter in both preexisting vascular cells and bone marrow cells involved in neovascularization. Circulation research. 2006;98:897–904. doi: 10.1161/01.RES.0000218193.51136.ad. [DOI] [PubMed] [Google Scholar]
- 25.Gao F, Al-Azayzih A, Somanath PR. Discrete functions of GSK3alpha and GSK3beta isoforms in prostate tumor growth and micrometastasis. Oncotarget. 2015;6:5947–5962. doi: 10.18632/oncotarget.3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goc A, Al-Azayzih A, Abdalla M, et al. P21 activated kinase-1 (Pak1) promotes prostate tumor growth and microinvasion via inhibition of transforming growth factor beta expression and enhanced matrix metalloproteinase 9 secretion. The Journal of biological chemistry. 2013;288:3025–3035. doi: 10.1074/jbc.M112.424770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Azayzih A, Gao F, Somanath PR. P21 activated kinase-1 mediates transforming growth factor beta1-induced prostate cancer cell epithelial to mesenchymal transition. Biochimica et biophysica acta. 2015;1853:1229–1239. doi: 10.1016/j.bbamcr.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abdalla M, Goc A, Segar L, et al. Akt1 mediates alpha-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor. The Journal of biological chemistry. 2013;288:33483–33493. doi: 10.1074/jbc.M113.504290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gavard J, Gutkind JS. VE-cadherin and claudin-5: it takes two to tango. Nature cell biology. 2008;10:883–885. doi: 10.1038/ncb0808-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Frontiers in oncology. 2013;3:211. doi: 10.3389/fonc.2013.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruan GX, Kazlauskas A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. The EMBO journal. 2012;31:1692–1703. doi: 10.1038/emboj.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Predescu D, Predescu S, Shimizu J, et al. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. American journal of physiology Lung cellular and molecular physiology. 2005;289:L371–381. doi: 10.1152/ajplung.00175.2004. [DOI] [PubMed] [Google Scholar]
- 33.Potente M, Urbich C, Sasaki K, et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. The Journal of clinical investigation. 2005;115:2382–2392. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? The Journal of clinical investigation. 2005;115:2059–2064. doi: 10.1172/JCI25900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oellerich MF, Potente M. FOXOs and sirtuins in vascular growth, maintenance, and aging. Circulation research. 2012;110:1238–1251. doi: 10.1161/CIRCRESAHA.111.246488. [DOI] [PubMed] [Google Scholar]
- 36.Krug SM, Gunzel D, Conrad MP, et al. Claudin-17 forms tight junction channels with distinct anion selectivity. Cellular and molecular life sciences: CMLS. 2012;69:2765–2778. doi: 10.1007/s00018-012-0949-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.