

Abstract
Dopamine D2 receptor (DRD2) deficiency increases renal inflammation and blood pressure in mice. We show here that long-term renal-selective silencing of Drd2 using siRNA increases renal expression of proinflammatory and profibrotic factors and blood pressure in mice. To determine the effects of renal-selective rescue of Drd2 expression in mice, the renal expression of DRD2 was first silenced using siRNA and 14 days later rescued by retrograde renal infusion of adeno-associated virus (AAV) vector with DRD2. Renal Drd2 siRNA treatment decreased the renal expression of DRD2 protein by 55%, and DRD2 AAV treatment increased the renal expression of DRD2 protein by 7.5- to 10-fold. Renal-selective DRD2 rescue reduced the expression of proinflammatory factors and kidney injury, preserved renal function, and normalized systolic and diastolic blood pressure. These results demonstrate that the deleterious effects of renal-selective Drd2 silencing on renal function and blood pressure were rescued by renal-selective overexpression of DRD2. Moreover, the deleterious effects of 45-minute bilateral ischemia/reperfusion on renal function and blood pressure in mice were ameliorated by a renal-selective increase in DRD2 expression by the retrograde ureteral infusion of DRD2 AAV immediately after the induction of ischemia/reperfusion injury. Thus, 14 days after ischemia/reperfusion injury, the renal expression of profibrotic factors, serum creatinine, and blood pressure were lower in mice infused with DRD2 AAV than in those infused with control AAV. These results indicate an important role of renal DRD2 in limiting renal injury and preserving normal renal function and blood pressure.
Introduction
The renal dopaminergic system plays a key role in salt homeostasis and blood pressure regulation. Dopamine, which is synthesized in the renal proximal tubule and acts in autocrine and paracrine fashion, is responsible for at least 50% of the net salt and water excretion when salt intake is increased (1). Genetic disruption in mice of any of the 5 dopamine receptor subtypes causes hypertension, and each subtype regulates blood pressure via dopamine receptor subtype-specific and -independent signal transduction pathways (2–6).
Inflammation, infiltration of immune cells, and oxidative stress in the kidney are involved in the development of renal injury and the induction and maintenance of hypertension (7). Renal tubule cells produce both proinflammatory and antiinflammatory cytokines and chemokines (8) that contribute to the development and progression of glomerular and tubular injury. However, the production and regulation of inflammatory factors in these cells are not well understood.
Dopamine and dopaminergic drugs have been shown to regulate the inflammatory reaction and immune response (9–11). Mice with intrarenal dopamine deficiency have increased oxidative stress and infiltration of inflammatory cells (12), and decreased renal dopamine production is associated with increased detrimental effects of angiotensin II on renal injury (13). The antiinflammatory effects of dopamine are mediated, at least in part, by the dopamine D2 receptor (DRD2) (14–18) that is expressed in proximal and distal convoluted tubules, collecting ducts, and glomerular mesangial cells (19). Lack or downregulation of DRD2 function in mice increases renal expression of proinflammatory cytokines/chemokines, resulting in histological and functional evidence of renal inflammation and injury, suggesting that the DRD2 has protective effects in the kidney by limiting the inflammatory reaction (15).
Deficient renal DRD2 function may be of clinical relevance. Some common SNPs in the noncoding region of the human DRD2 gene are associated with decreased DRD2 expression and function, and some of these SNPs are associated with increased blood pressure or hypertension (20–24). Renal proximal tubule cells from human subjects carrying these polymorphisms express elevated levels of proinflammatory and profibrotic factors and markers of epithelial mesenchymal transition indicating that the DRD2 has protective effects in these cells (16, 17). We now show that (a) prolonged silencing of Drd2 expression, selectively in the kidney, increases renal inflammation and injury and blood pressure; (b) rescue of Drd2 expression selectively in the Drd2-silenced kidney with adeno-associated virus (AAV) vectors provides sustained long-term DRD2 expression with minimal immunological consequences (25), reduces renal inflammation and injury, and normalizes the blood pressure; and (c) overexpression of DRD2 in the kidney ameliorates the renal injury induced by ischemia/reperfusion.
Results
Retrograde ureteral infusion of AAV-9 vector provides efficient gene transfer in the kidney
The efficiency of renal gene transfer by AAV-9 following systemic administration was determined using an AAV vector encoding firefly luciferase under the control of CMV promoter AAVLuc (Figure 1A). In vivo bioluminescence imaging of the mice showed luminescence signals in thoracic, abdominal, and pelvic cavities as well as in the hind legs and tail (Figure 1B). Luciferase activity assay on various organs 14 days following systemic administration of AAVLuc showed marked transduction in liver, pancreas, skeletal muscle, and heart; moderate transduction in stomach and lung; and minimal transduction in kidney, spleen, and brain (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/jci.insight.85888DS1). Transduction in the kidneys was 50-, 30-, 174-, and 137-fold lower relative to the liver, pancreas, skeletal muscle, and heart, respectively. Assessment of AAV genome copies in the AAVLuc-treated mice confirmed the transduction levels in various organs estimated by luciferase activity assay. AAV genome copies in the kidneys were 54-, 3.5-, 4.7-, and 4.3-fold lower relative to the liver, pancreas, skeletal muscle, and heart, respectively (Supplemental Figure 1B). These results show that systemic administration of AAV-9 vector does not provide efficient gene transfer and expression in the mouse kidney. Our results showing preferential transduction in the liver, pancreas, skeletal muscle, and heart by AAV-9 following systemic administration are consistent with previous reports (26–28).
Figure 1. Retrograde ureteral infusion of adeno-associated virus vectors provides renal-specific gene expression.

(A) Schematic representation of adeno-associated virus (AAV) vectors AAVLuc, AAVEGFP, and DRD2 AAV carrying the firefly luciferase cDNA, EGFP, or human dopamine receptor D2 (DRD2) cDNA driven by the CMV promoter. AAV-inverted terminal repeats (ITR) and SV40 polyadenylation signal (SV-pA) are also indicated. Mice were infused (B) systemically via the jugular vein or (C) retrogradely via the left ureter with ACMVLuc vector (1 × 1011 viral genome particles) carrying firefly luciferase under the control of CMV. Shown are representative bioluminescence images of the mouse in ventral and dorsal positions acquired on day 14 following vector administration. (D) Maximum and stable bioluminescence was achieved 14 days after injection. (E) Fourteen days following vector administration, the mice were euthanized and a panel of tissues (KR, right kidney; KL, left kidney; Sp, spleen; Lv, liver; Pa, pancreas; St, stomach; Sk, skeletal muscle; H, heart; Ln, lung; Br, brain) was collected. Luciferase activity in the indicated organs was determined using the in vitro luciferase assay kit (Promega Corporation) on protein extracts prepared as per the manufacturer’s guidelines. Luciferase activities are reported as RLU per mg protein. (F) AAV vector genome copy number in the indicated organs was determined by real-time quantitative PCR. n = 3/group.
In order to improve the gene transfer to the kidney by AAV-9, we infused the AAVLuc vector retrogradely via the left ureter. Luciferase expression in the mice was monitored by in vivo bioluminescence imaging at days 3, 7, and 14 after vector infusion. Bioluminescence signal was observed predominantly on the left side of the abdomen in ventral and dorsal positions (Figure 1C). Bioluminescence signal approached a steady-state plateau 2 weeks after vector infusion (Figure 1D). Luciferase activity assay on various organs collected 14 days following infusion of AAVLuc showed marked transduction in the left kidney (kidney infused with AAVLuc via the left ureter) as compared with the right kidney (unmanipulated), liver, heart, skeletal muscle, and pancreas (Figure 1E). Luciferase activity in the left kidney was 32-, 7.4-, 7.6-, 28-, and 20-fold that of the right kidney, liver, pancreas, skeletal muscle, and heart, respectively (Figure 1E). AAV genome copies in the left kidneys of mice treated with AAVLuc via the left ureter showed a similar trend. AAV genome copies in the left kidneys were 29-, 5-, 11-, 100-, and 13-fold that of the right kidney, liver, pancreas, skeletal muscle, and heart, respectively (Figure 1F).
Immunofluorescence analyses of the mouse left kidney infused with AAVEGFP via the left ureter showed EGFP expression from the collecting duct cells up to the proximal tubule cells (Figure 2).
Figure 2. Mice were retrogradely infused in the left kidney via the left ureter with AAVEGFP vector (1 × 1011 viral genome particles/mouse).

Fourteen days following vector administration the mice were euthanized and kidney sections were immunostained with EGFP antibody (green) and Lotus tetragonolobus agglutinin (red) that binds strongly to the ciliated renal proximal tubule cells and weakly to the renal collecting duct cells. Shown are the confocal images of cortex, medulla, and papilla of the immunostained kidney sections. Scale bar: 50 μm.
Prolonged renal Drd2 silencing increases the expression of inflammatory markers and arterial blood pressure
In order to test the role of DRD2 in renal inflammation and blood pressure (Figure 3), we silenced in mice the renal expression of DRD2 by left renal subcapsular infusion of Drd2 siRNA via osmotic minipumps. DRD2 expression was silenced only in the left kidney; the right kidney was unaffected. DRD2 expression was decreased by 64% in the left kidneys that were infused with Drd2 siRNA relative to those treated with nonsilencing siRNA (NSsiRNA) (Figure 3B). The Drd2 siRNA-treated kidneys showed increased mRNA expression, as quantified by qRT-PCR, of the inflammatory factors TNF-α (1.9-fold), MCP1 (2.1-fold), and IL-6 (1.4-fold) as well as the expression of TGF-β extracellular matrix proteins fibronectin 1 (FN1; 1.7-fold) and type 1 collagen (Col1a1; 1.3-fold) (Figure 3A). The protein expression of TNF-α, MCP1, and IL-6 was increased by 35%, 60%, and 200%, respectively, as determined by immunoblot (Figure 3B). By contrast, DRD2 protein expression and the mRNA and protein expression of proinflammatory factors were similar in the untreated (right) kidneys of mice treated by left renal subcapsular infusion with either Drd2 siRNA or NSsiRNA (Supplemental Figure 2).
Figure 3. Renal Drd2 silencing increases the expression of proinflammatory factors and blood pressure.

Mice were treated with nonsilencing siRNA (NSsiRNA) or dopamine receptor D2 (Drd2) siRNA by right renal subcapsular infusion via osmotic minipump for 28 days. (A) Renal cortical mRNA expression of TNF-α, monocyte chemoattractant protein 1 (MCP1), IL-6, fibronectin 1 (FN1), and collagen type 1a1 (Col1a1) was quantified by qRT-PCR. GAPDH mRNA was used for normalization of the data. n = 5–6/group, *P < 0.05 vs. NSsiRNA, Student’s t test. (B) Renal cortical protein expression of DRD2 (55 kDa), TNF-α (25 kDa), MCP1 (17 kDa), and IL-6 (25 kDa) was determined by immunoblot. One set of immunoblots is shown. Relative abundance of the proteins was normalized to GAPDH and expressed as a percentage of the NSsiRNA-treated group. n = 3–6/group, *P < 0.05 vs. NSsiRNA, Student’s t test. (C) Systolic blood pressures measured under pentobarbital anesthesia in mice before (Pre) and 28 days after (Post) siRNA infusion. n = 5–6/group, *P < 0.05 vs. all others, 1-way ANOVA and Tukey test.
Systolic blood pressure, measured under pentobarbital anesthesia, was similar in all mice before treatment. However, mice treated with left renal subcapsular infusion of Drd2 siRNA for 28 days had increased systolic blood pressure compared with the NSsiRNA-treated group (Figure 3C). Diastolic blood pressure was also increased in the Drd2 siRNA-treated mice (data not shown). These results indicate that prolonged reduction in DRD2 expression in one kidney causes renal inflammation and increases blood pressure and that these effects cannot be compensated for by the normal DRD2 expression in the contralateral untreated kidney.
DRD2 rescue abrogates the expression of proinflammatory and profibrotic factors induced by Drd2 silencing
DRD2 expression in the mice treated with left renal subcapsular infusion of Drd2 siRNA and left ureteral retrograde infusion of control AAV (CAAV) was decreased by 55% as compared with the mice treated with left renal subcapsular infusion of NSsiRNA and left ureteral retrograde infusion of CAAV. By contrast, the expression of DRD2 was increased by 7.5-fold in the mice treated with left renal subcapsular infusion of NSsiRNA and left ureteral retrograde infusion of DRD2 AAV and by 10-fold in mice treated with left renal subcapsular infusion of Drd2 siRNA and left ureteral retrograde infusion of DRD2 AAV as compared with the mice treated with left renal subcapsular infusion of NSsiRNA and left ureteral retrograde infusion of CAAV (Figure 4). Drd2 siRNA used in this experiment is species specific and designed to deplete only the mouse DRD2 protein. Therefore, mouse Drd2 siRNA did not attenuate the AAV-mediated human DRD2 expression in the mouse kidney.
Figure 4. Increased renal expression of DRD2 in mice treated with DRD2 AAV.
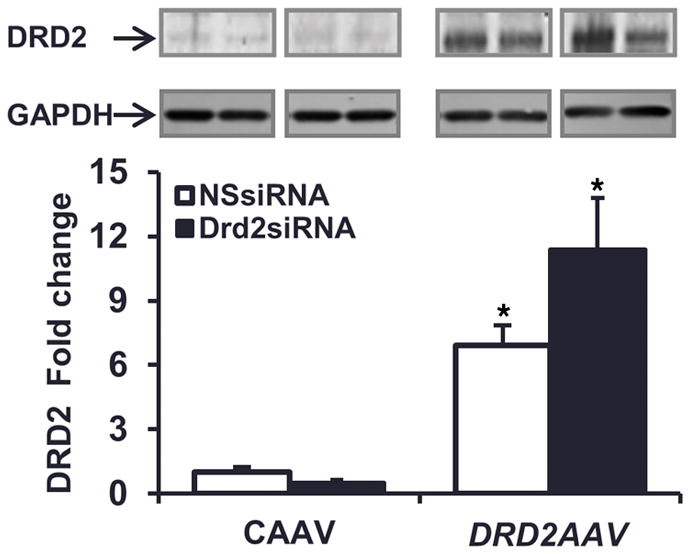
Renal cortical expression of dopamine D2 receptor (DRD2, 55 kDa) determined by immunoblot in mice treated with NSsiRNA or Drd2 siRNA by left renal subcapsular infusion via osmotic pump for 28 days and treated with control adeno-associated virus (CAAV) or DRD2 AAV (1 × 1011 viral genome particles) by retrograde left ureteral infusion for 14 days after starting the siRNA treatment. n = 3 per group, *P < 0.05 vs. CAAV+NSsiRNA, 1-way ANOVA and Tukey test. NSsiRNA, nonsilencing siRNA.
The mRNA expression of TNF-α (1.6-fold), TGF-β (2.8-fold), FN1 (4.5-fold), IL-6 (2.5-fold), and MCP1 (1.6-fold) was increased in the left kidneys of mice treated with Drd2 siRNA plus CAAV in comparison to those treated with NSsiRNA plus CAAV (Figure 5). Rescuing DRD2 expression blunted the increase or reversed to normal the mRNA expression of these molecules as well as the protein expression of TNF-α (Figure 5). Overexpression of DRD2 in kidneys treated with NSsiRNA decreased the expression of TNF-α, tended to decrease the expression of IL-6 mRNA, and had no effect on the mRNA expression of TGF-β, FN1, and MCP1.
Figure 5. Rescue of DRD2 expression reverses the increase in proinflammatory factors induced by Drd2 silencing in the left kidney.

Twenty-eight days after left renal subcapsular infusion of siRNA and following left ureteral retrograde adeno-associated virus (AAV) treatment from day 14 to day 28, the mice were euthanized. mRNA expression and protein (25 kDa) expression of TNF-α in the renal cortex were determined by qRT-PCR and immunoblot, respectively. One set of immunoblots is shown. Renal cortical mRNA expression of TGF-β, fibronectin 1 (FN1), IL-6, and monocyte chemoattractant protein 1 (MCP1) in the mouse kidneys from indicated groups was quantified by qRT-PCR. GAPDH mRNA was used for normalization of the data, n =3 per group. *P < 0.05 vs. all others, **P < 0.05 vs. CAAV+Drd2 siRNA group, 1-way ANOVA and Tukey test. DRD2, dopamine D2 receptor; NSsiRNA, nonsilencing siRNA.
DRD2 rescue minimizes the renal injury induced by Drd2 silencing
H&E staining of kidney sections from Drd2 siRNA-treated kidneys that were not rescued revealed areas of parenchymal scarring (interstitial fibrosis, tubular atrophy, infiltration of inflammatory cells, and interstitial mesenchymal cell proliferation), involving less than 5% to 25% of the cortex, while there was no frank parenchymal scaring in the kidneys treated with NSsiRNA or in DRD2-rescued kidneys, although these kidneys presented with very focal minimal interstitial fibrosis, tubular atrophy, and infiltration of inflammatory cells. Masson’s trichrome staining of the kidney sections confirmed the presence of multiple areas of scaring with excessive collagen accumulation in the Drd2-silenced kidneys (Figure 6A), as compared with the NSsiRNA-treated kidneys or the DRD2-rescued group, indicating marked renal fibrosis in the Drd2-silenced group. Immunostaining of renal sections for KIM-1, a marker of kidney injury, showed that the Drd2-silenced kidneys had a significantly higher number of KIM-1-positive cells as compared with the NSsiRNA-treated kidneys or DRD2-rescued kidneys (Figure 6, A and B). Immunohistochemical analysis for anti-CD3e, a marker for T cells, showed that the Drd2-silenced kidneys had a significantly higher number of infiltrating T cells as compared with the NSsiRNA-treated kidneys or DRD2-rescued kidneys. However, the number of T cells in the DRD2-rescued kidneys remained higher than in the control kidneys treated with NSsiRNA (Figure 6, A and C). Serum creatinine levels were similar in mice treated with NSsiRNA regardless of whether they were treated with CAAV or DRD2 AAV (0.25 ± 0.02 vs. 0.26 ± 0.03 mg/dl). However, the serum creatinine levels were lower in mice with Drd2 silencing and DRD2-rescued kidneys than in mice with Drd2 silencing and unrescued kidneys, i.e., kidneys treated with CAAV (0.27 ± 0.02 vs. 0.40 ± 0.03 mg/dl; P < 0.05). These results show that Drd2 silencing induces renal injury that can be partially prevented by rescue of DRD2 expression.
Figure 6. Rescue of DRD2 expression minimizes the renal injury induced by Drd2 silencing.

Twenty-eight days after left renal subcapsular infusion of siRNA and following left ureteral retrograde adeno-associated virus (AAV) treatment from day 14 to day 28, the mice were euthanized. Kidney sections from the indicated groups were stained with (A) H&E for morphology (scale bar: 100 μm) and Masson’s trichrome for collagen deposition (scale bar: 200 μm). These stains demonstrated areas of cortical scarring, appearing as clusters of smaller, atrophic tubules in association with increased intertubular connective tissue. Immunofluorescence/confocal microscopy for Ki-67 protein (scale bar: 50 μm) showed the proliferating renal tubule cells and T cells (scale bar: 100 μm). Quantification of (B) kidney injury molecule-1-positive (KIM-1-positive) cells and (C) T cells in the indicated kidney sections. n = 3/ group. *P < 0.05 vs. all others, **P < 0.05 vs. CAAV+Drd2 siRNA group, 1-way ANOVA and Tukey test. DRD2, dopamine D2 receptor; NSsiRNA, nonsilencing siRNA.
DRD2 rescue in the mouse kidney normalizes the high blood pressure induced by Drd2 silencing
Systolic blood pressures at baseline and after siRNA plus AAV vector treatment were not different in the mice treated with NSsiRNA in combination with CAAV or DRD2 AAV (Figure 7). However, systolic blood pressures were significantly higher (121 ± 3 mm Hg; P < 0.05) in mice treated with Drd2 siRNA and CAAV in comparison with the baseline or systolic blood pressures in mice treated with NSsiRNA and CAAV (101 ± 4 mm Hg), NSsiRNA and DRD2 AAV (101 ± 1 mm Hg), or Drd2 siRNA and DRD2 AAV (99 ± 1.5 mm Hg) (Figure 7). Diastolic blood pressures were also significantly higher (91 ± 1.6 mm Hg) in the mice treated with Drd2 siR-NA and CAAV in comparison with the baseline or diastolic blood pressures in mice treated with NSsiRNA and CAAV (73 ± 2.26 mm Hg), NSsiRNA and DRD2 AAV (74 ± 2.9 mm Hg), or Drd2 siRNA and DRD2 AAV (74 ± 4 mm Hg) (Supplemental Figure 3). More importantly, the blood pressures, measured under pentobarbital anesthesia, in the DRD2-rescued group, i.e., mice treated with Drd2 siRNA and DRD2 AAV, were similar to those in mice treated with NSsiRNA and CAAV.
Figure 7. Rescue of left renal DRD2 expression normalizes the high blood pressure induced by Drd2 silencing in the left kidney.
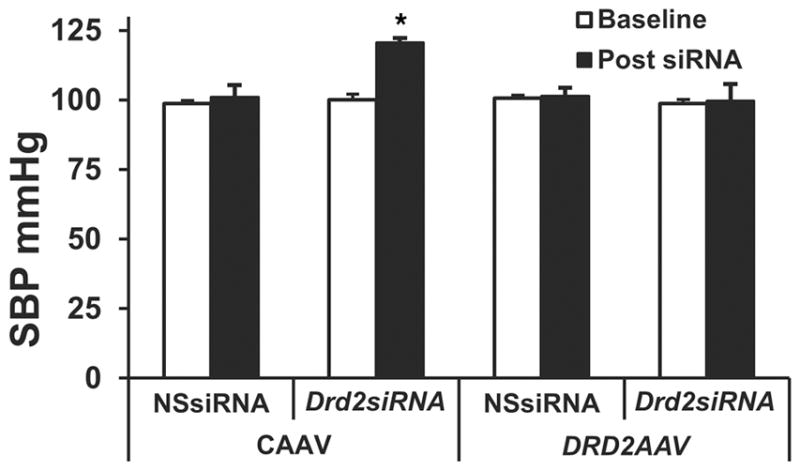
Systolic blood pressure (SBP) was measured under pentobarbital anesthesia in the mice prior to siRNA treatment (baseline) and 28 days after siRNA treatment and 14 days (from day 14 to day 28) after AAV treatment. n = 3/group. *P < 0.05 vs. all others, 1-way ANOVA and Tukey test. DRD2, dopamine D2 receptor; NSsiRNA, nonsilencing siRNA.
Increasing DRD2 expression in the mouse kidney ameliorates renal injury induced by ischemia/reperfusion
We further hypothesized that increasing DRD2 expression would be beneficial not only in conditions with reduced DRD2 expression but also in renal injury. To test this hypothesis, we determined whether DRD2 AAV treatment provides protection in a model of renal ischemia/reperfusion injury. We studied the effects of 45 minutes of bilateral renal ischemia immediately followed by infusion of CAAV or DRD2 AAV. The mice were studied 14 days later, a time when AAV-mediated DRD2 expression reaches maximum (Figure 1D). The mRNA expression of TGF-β, FN1, and Col1a1 was higher in mice infused with CAAV in comparison with those infused with DRD2 AAV (Figure 8A). H&E staining of the kidney sections showed that the mice treated with CAAV had more ischemia/reperfusion injury compared with those treated with DRD2 AAV (Figure 8B). Systolic blood pressures were significantly increased in mice treated with CAAV in comparison with their baseline systolic blood pressures. By contrast, the systolic blood pressures of mice treated with DRD2 AAV were similar to their baseline blood pressures and were not different from the baseline blood pressures of the CAAV-treated mice (Figure 8C). Serum creatinine levels, used as an index of renal function, were also higher in mice treated with CAAV than in those treated with DRD2 AAV (Figure 8D).
Figure 8. Effect of overexpression of dopamine D2 receptor on renal ischemia/reperfusion injury.

Mice were subjected to 45 minutes of bilateral renal ischemia, after which the ureters of both kidneys were retrogradely infused with control adeno-associated virus (CAAV) or dopamine D2 receptor adeno-associated virus (DRD2 AAV). (A) The renal cortex was obtained for mRNA determination of TGF-β, fibronectin 1 (FN1), and collagen type 1a1 (Col1a1) (qRT-PCR). (B) Images of kidney sections stained with H&E showing renal morphology. Scale bar: 200 μm. (C) Blood pressure was measured under anesthesia before starting the ischemia period and 14 days after the induction of ischemia/reper-fusion and retrograde ureteral infusion of CAAV or DRD2 AAV. SBP, systolic blood pressure. (D) Serum creatinine was measured from blood obtained prior to euthanasia. n = 3/group. (A and D) *P < 0.05 vs. CAAV, t test. (C) *P < 0.05 vs. all others, 1-way ANOVA followed by Tukey test.
Discussion
Our results show that, in mice, Drd2 silencing for 28 days in only one kidney, leaving the other kidney intact, increases the expression of proinflammatory and profibrotic factors, causes renal injury, and elevates blood pressure. These effects are completely or partially prevented by rescuing DRD2 expression in the silenced kidney. Furthermore our results also show that the renal injury and the increase in blood pressure resulting from ischemia/reperfusion is ameliorated by overexpression of the DRD2, demonstrating that an increase in DRD2 expression has protective effects against renal damage and high blood pressure.
We have shown that the lack of DRD2 expression, caused by germline deletion (Drd2−/− mice) or silencing of Drd2 in the remnant kidney of uninephrectomized mice, was associated with renal inflammation and elevated blood pressure that reached hypertensive levels (15, 29). However, silencing of Drd2 for 7 days in one kidney, leaving the other intact, did not increase blood pressure but was associated with an increase in inflammatory factors in the Drd2-silenced kidney (15). We now show that silencing Drd2 for 28 days in one kidney, leaving the other kidney intact, has the same effect as silencing Drd2 for 7 days in the remnant kidney of uninephrectomized mice, suggesting that the reduction in DRD2 expression in one kidney for an extended period cannot be compensated by the other kidney. These results also suggest that sustained renal inflammation results in kidney damage and as a consequence increases blood pressure.
We hypothesized that rescuing renal DRD2 expression in mice in which renal Drd2 is silenced reduces renal inflammation and injury and prevents the increase in blood pressure. We used AAV vector-mediated gene transfer technology to demonstrate that the deleterious effect of renal Drd2 silencing could be minimized by the reexpression of DRD2 in the Drd2-silenced kidney. AAV vectors have become one of the most attractive tools for in vivo gene transfer in translational as well as basic research studies (30). AAV vectors provide sustained long-term gene expression with minimal immunological consequences (25). Systemic administration of AAV vector provides efficient transduction of various organs, such as the liver, heart, lung, and skeletal muscle but provides very low transduction in the kidney (26–28, 31). Systemic administration of AAV-9 carrying hepatocyte growth factor in combination with kidney-specific cadherin promoter provided gene expression in the mouse kidney and liver that attenuated tubulointerstitial fibrosis and repression of fibrotic markers in COL4A3-deficient mice (32). In neonatal mice, systemic administration of AAV-1 carrying G6Pase-A provided the gene expression in hepatocytes and kidneys that alleviated the metabolic abnormalities in mice with type 1A glycogen storage disease (33). In spite of the apparent lower transduction in the kidneys with systemic administration of AAV-9 vector, it provided the therapeutic benefits, largely due to superior hepatic gene expression that acted in endocrine fashion on the kidney (32, 33). Ito et al. demonstrated that intrapelvic injection of AAV-2 vector provides expression predominantly in the medulla (34). To improve transduction in the kidney epithelial cells, Chung et al. administered AAV vectors by retrograde ureteral infusion and showed that AAV-8 and AAV-9 transduce kidney cells efficiently compared with AAV-2 and -6 serotypes (35). We used retrograde ureteral infusion of AAV-9 vector carrying firefly luciferase or EGFP to determine the magnitude and distribution of gene expression in the infused kidney. Our results show that retrograde ureteral infusion of AAV-9 vector limited the gene expression to the infused kidney; there was minimal gene expression in the contralateral kidney, spleen, liver, pancreas, stomach, skeletal muscle, heart, lung, and brain. A time-course study showed that the AAV-9-mediated luciferase expression reached maximum by 2 weeks following vector infusion.
Treatment with DRD2 AAV for 14 days to reexpress DRD2 selectively in the Drd2-silenced kidney minimized the deleterious effects of Drd2 silencing by decreasing the expression of proinflammatory and profibrotic factors; limiting renal injury; preserving kidney function, as shown by the decrease in serum creatinine; and preventing the increase in blood pressure. The beneficial effects of the renal DRD2 are also demonstrated by the effects of its overexpression in ameliorating the increase in profibrotic factors, decrease in renal function, and increase in blood pressure in a model of ischemia/reperfusion injury.
The inhibitory effects of the DRD2 on renal inflammation are mediated by several mechanisms that include negative regulation of the Akt pathway through effects on PP2A activity/expression and the miR-217/Wnt5a/Ror2 pathway. These pathways mediate the increase in profibrotic factors in human renal proximal tubule cells bearing DRD2 SNPs associated with decreased DRD2 expression (17, 18).
The protective effects of DRD2 are not restricted to renal inflammation and may be organ specific (36, 37). Lack of DRD2 expression in astrocytes results in marked inflammation in multiple brain regions. The treatment of Drd2 wild-type mice with a selective DRD2 agonist partially suppressed 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-mediated inflammation in nigral dopaminergic neurons (36). In a mouse model of amyotrophic lateral sclerosis, a DRD2 agonist suppressed glial inflammation and moderated the progression of the disease (38). In the respiratory tract, DRD2 activation inhibited neurogenic inflammation (39). GLC756, a D1R antagonist/DRD2 agonist, inhibited TNF-α release from rat mast cells, suggesting that it reduced inflammation triggered by activated mast cells (40).
Genetic factors contribute to the susceptibility to renal disease associated with essential hypertension, and hypertension may cause progressive kidney disease only in genetically susceptible individuals (41, 42). The genetic predisposition to chronic kidney disease is polygenic, but to date only a few genes have been shown to be contributory (43–46). Renal susceptibility genes may determine the occurrence and severity of hypertension-induced progressive renal damage. Much of the research in this field has been directed to determining the detrimental factors involved in disease progression. However, the role of factors that prevent inflammation and may slow the progression of renal disease has received much less attention.
Our results provide support for the concept of increased susceptibility to renal disease resulting from decreased DRD2 expression and function. Along this line, a recent study in an Asian Indian population with type 2 diabetes found that a DRD2 polymorphism, resulting in decreased expression of the receptor, increases the susceptibility to chronic diabetic nephropathy (47).
This study suggests that the development of therapies directed to increase renal DRD2 expression/function may provide novel and effective approaches in the treatment of renal injury.
Methods
AAV vectors
The AAV vector harboring firefly luciferase cDNA (AAVLuc) with the expression controlled by the CMV was constructed using the plasmid pACS (48). To construct AAVLuc, the 2.2-kb firefly luciferase cDNA from plasmid pGL3 (Promega Corporation) was excised as a HindIII and BamHI fragment and inserted in between the same restriction sites of pACS. AAV vector AAVEGFP harboring EGFP cDNA under the control of CMV promoter was obtained from Penn Vector Core (University of Pennsylvania, Philadelphia, Pennsylvania, USA). To construct DRD2 AAV, the 1.7-kb human DRD2 cDNA from plasmid RC202476 (Origene Technologies Inc.) was excised as a KpnI and FseI fragment and inserted in between the same restriction sites of pACS. The amino acid sequence of human and mouse DRD2 share 95.5% identity. The CAAV used as CAAV vector, which does not code for any protein, contains an EGFP cDNA in reverse orientation under the control of CMV promoter. Recombinant AAV vector genomes were packaged into capsids from AAV-9 serotype by the triple transfection method in HEK 293 cells, as described previously (49, 50). The AAV vectors were purified by ammonium sulfate fractionation and iodixanol gradient centrifugation. Titers of the AAV vectors (viral genome particles/ml) were determined by quantitative real-time PCR (50, 51).
Animal procedures
C57BL/6 mice (8–10 weeks old, weighing ~20 g) were purchased from Jackson Laboratory. The mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg) and placed in a supine position, and legs were taped down on a heated board to maintain their body temperature at 37°C. For all procedures, depth of anesthesia was monitored by foot-pinch reflex. The mice were treated with NSsiRNA or Drd2 siRNA by subcapsular infusion (3 μg/day) using osmotic minipump (15, 52, 53) for 28 days in the left kidney, leaving the contralateral kidney intact (Supplemental Figure 4A). Briefly, the siRNA, prepared using an in vivo transfection reagent, was loaded into the osmotic minipump (ALZET Osmotic Pumps). A polyethylene tube connected to the osmotic minipump was inserted underneath the kidney capsule for continuous delivery of the siRNA at the rate of 3 μg/day for 28 days. The tube was secured underneath the kidney capsule using surgical glue, and the osmotic minipump was secured onto the abdominal wall by a suture.
For retrograde ureteral infusion of AAV vectors, 14 days following the initiation of siRNA treatment (Supplemental Figure 4B), the mice were anesthetized as mentioned above and an abdominal incision was made between the point of the xiphoid cartilage and the navel to expose the kidneys. Then, the ureter, corresponding to the treated kidney, was located and gently dissected out. The distal ureteral portions closest to the bladder and the renal artery supplying the target kidney were clamped off using a microvenous clip. Using a tuberculin syringe fitted with a 33-gauge needle, the ureter was then punctured. The needle was temporarily ligated in place by a snugly tied 6-0 silk suture to prevent leakage. The urine was then gently aspirated out. The tuberculin syringe was then removed and replaced with another containing approximately 100 μl of the AAV vector (1 × 1011 viral genome particles), and the solution was then retrogradely infused into the ureter. The needle was withdrawn and a microvenous clip was placed proximal to the injection site on the ureter to prevent leakage and to attain maximum exposure to the infusion. Fifteen minutes later, the arterial and ureteral clips were sequentially removed and the ureter was inspected for any evidence of leakage. The abdominal contents were replaced in reverse order and the incision site was closed using a double layer of 6-0 silk sutures for the muscle and skin.
Renal ischemia/reperfusion
The mice were anesthetized with an intraperitoneal injection of pentobarbital sodium (50 mg/kg), and blood pressure was measured as described below. The kidneys of the mice were exposed as described above. Both right and left renal vessels (artery and vein) were clamped with arterial clamps for 45 minutes to stop blood flow to both kidneys. Then, the clamps were removed, and, immediately thereafter, the mice were subjected to bilateral retrograde ureteral infusion of CAAV or DRD2 AAV, as described above. The abdominal incision was closed, and the mice were allowed to recover. Fourteen days later, the mice were anesthetized and blood pressure measured. Blood, kidney, and other selected organs were harvested before the mice were euthanized with an overdose of pentobarbital sodium (100 mg/kg).
Blood pressure measurement
Systolic and diastolic blood pressures were recorded, under anesthesia, at baseline (prior to siRNA infusion) and 14 days after AAV treatment using the femoral artery or carotid artery using Cardiomax II (Columbus Instruments). The organs (see below) were harvested (properly stored for future analyses) before the mice were euthanized with an overdose of pentobarbital (100 mg/kg).
In vivo bioluminescence imaging
Luciferase expression in live, anesthetized mice was detected noninvasively by in vivo bioluminescence imaging using previously reported methods (50, 54). The mice were anesthetized and maintained on 0.5%–1% isoflurane in oxygen. D-luciferin (150 μg/g body weight, Gold Biotechnology Inc.) was administered to the mice by intraperitoneal injection. Five minutes following D-luciferin administration, the mice were imaged using an IVIS-200 imaging system (Caliper Life Sciences). Photons emitted from the mice were collected and integrated for a period of 1 minute. Images were processed using Living Image software (Caliper Life Sciences).
Luciferase activity assay
Luciferase activity in organs was measured using a kit from the Promega Corporation (E4030, Luciferase Assay System with Reporter Lysis Buffer). Kidneys, spleen, liver, pancreas, stomach, skeletal muscle, heart, lungs, and brain were collected from mice after bioluminescence imaging 14 days after vector injection. Protein was obtained from the organs as recommended by the manufacturer, and protein concentrations were determined using the Bio-Rad DC BCA protein assay kit. Luciferase activity in the protein samples from each organ was determined using a Centro LB 960 microplate reader (Berthold Technologies USA) and expressed as RLU per mg protein.
Quantitative RT-PCR
Total RNA was purified using the RNeasy Plus Mini kit (Qiagen). cDNA was prepared using an RT2 First Strand kit per the manufacturer’s protocol (SABiosciences-Qiagen). Quantitative gene expression was analyzed by real-time qPCR using an ABI Prism 7900 HT (Applied Biosystems). Quantitative PCR assay was performed using gene-specific primers (TNF-α: PPM03113F; MCP-1: PPM03151F; IL-6: PPM03015A; FN1: PPM03786A; Col1a1: PPM-3845F; TGF-β: PPM02991B; and GAPDH: PPM02946E) for mice from SABiosciences-Qiagen and RT2 SYBR Green ROX qPCR Master-mix (Qiagen). Data were analyzed using the ΔΔCt method (55).
Immunoblotting
Flash-frozen tissue samples were homogenized in RIPA buffer, and equal amounts of protein (20 μg) were electrophoresed under reducing conditions on 4%–20% gradient polyacrylamide gels and then transferred onto polyvinylidene difluoride membranes. DRD2 and TNF-α were detected by standard protocol using rabbit polyclonal anti-DRD2 (sc-9113, Santa Cruz Biotechnology Inc.) and rat anti mouse anti-TNF-α (Biolegend). Results were normalized to GAPDH and expressed as fold change, relative to the average signal intensity obtained from the kidneys of the respective controls (NSsiRNA- or NSsiRNA plus CAAV-treated mice).
Immunohistochemistry
EGFP and KIM-1 expression and infiltrating T cells in mouse kidneys were documented by immunohistochemical analysis. For EGFP, 2 weeks following vector administration, the kidneys were collected and fixed in 3.7% paraformaldehyde for 1 hour at 4°C. After washing in phosphate-buffered saline (3 times, 5 minutes each), the tissues were equilibrated with 30% sucrose in phosphate-buffered saline overnight. Six-micrometer cryosections were prepared. The sections were immunostained for EGFP using chicken anti-GFP antibody (Aves Labs Inc.). Lotus tetragonolobus agglutinin, which binds to the apical membrane of the proximal tubule and collecting duct cells, was used to distinguish proximal tubules and collecting ducts from other nephron segments.
The expression of KIM-1 protein, also known as TIM1, a kidney injury marker, was detected using a goat polyclonal anti-mTIM-1 antibody (R&D Systems; AF1817). The location of KIM-1 expression was detected by double immune staining using anti-CD15 antibody, which is specific for renal proximal tubule cells. T cells in the mouse kidney sections were detected by immunohistochemistry using rabbit anti-CD3e monoclonal antibody (Thermo Scientific; MA5-14524) and the EXPOSE Mouse and Rabbit AP (red) Detection IHC Kit (Abcam). T cells were counted in 3 microscopy fields from each section at ×10 magnification (n = 3 per group).
Serum creatinine
Serum creatinine was quantified using a full enzymatic method with substantially reduced interference compared with the Jaffe method (Crystal Chem Inc.).
Statistics
Data are expressed as mean ± SEM. Comparisons between 2 groups used the 2-tail Student’s t test. One-way ANOVA followed by Tukey multiple comparison test was used to assess significant differences in greater than 2 groups. P < 0.05 was considered significant.
Study approval
All animal protocols used in this study were conducted in accordance with the NIH and were approved by the Institutional Animal Care and Use Committee at the University of Maryland.
Supplementary Material
Acknowledgments
This work was funded by grants from the NIH: R01DK090918, P01HL074940, P01HL068686, R01HL092196, R37HL023081, and R01DK039308.
Footnotes
Author contributions
PRK designed the experiments, acquired and interpreted the data, and drafted the manuscript. LDA, YZ, YY, CD, XZ, and FH acquired and interpreted the data. CD and PAJ revised the manuscript. IA designed the experiments and drafted and revised the manuscript.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Zeng C, Sanada H, Watanabe H, Eisner GM, Felder RA, Jose PA. Functional genomics of the dopaminergic system in hypertension. Physiol Genomics. 2004;19(3):233–246. doi: 10.1152/physiolgenomics.00127.2004. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht FE, et al. Role of the D1A dopamine receptor in the pathogenesis of genetic hypertension. J Clin Invest. 1996;97(10):2283–2288. doi: 10.1172/JCI118670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asico LD, et al. Disruption of the dopamine D3 receptor gene produces renin-dependent hypertension. J Clin Invest. 1998;102(3):493–498. doi: 10.1172/JCI3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson TL, Tulis DA, Keeler BE, Virag JA, Lust RM, Clemens S. The dopamine D3 receptor knockout mouse mimics aging-related changes in autonomic function and cardiac fibrosis. PLoS One. 2013;8(8):e74116. doi: 10.1371/journal.pone.0074116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollon TR, et al. Mice lacking D5 dopamine receptors have increased sympathetic tone and are hypertensive. J Neurosci. 2002;22(24):10801–10810. doi: 10.1523/JNEUROSCI.22-24-10801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li XX, et al. Adrenergic and endothelin B receptor-dependent hypertension in dopamine receptor type-2 knockout mice. Hypertension. 2001;38(3):303–308. doi: 10.1161/01.hyp.38.3.303. [DOI] [PubMed] [Google Scholar]
- 7.Harrison DG, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segerer S, Schlöndorff D. Role of chemokines for the localization of leukocyte subsets in the kidney. Semin Nephrol. 2007;27(3):260–274. doi: 10.1016/j.semnephrol.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Bendele AM, Spaethe SM, Benslay DN, Bryant HU. Anti-inflammatory activity of pergolide, a dopamine receptor agonist. J Pharmacol Exp Ther. 1991;259(1):169–175. [PubMed] [Google Scholar]
- 10.Haskó G, Szabó C, Németh ZH, Deitch EA. Dopamine suppresses IL-12 p40 production by lipopolysaccharide-stimulated macrophages via a beta-adrenoceptor-mediated mechanism. J Neuroimmunol. 2002;122(1–2):34–39. doi: 10.1016/s0165-5728(01)00459-3. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh MC, et al. Dopamine inhibits cytokine release and expression of tyrosine kinases, Lck and Fyn in activated T cells. Int Immunopharmacol. 2003;3(7):1019–1026. doi: 10.1016/S1567-5769(03)00100-0. [DOI] [PubMed] [Google Scholar]
- 12.Zhang MZ, et al. Intrarenal dopamine deficiency leads to hypertension and decreased longevity in mice. J Clin Invest. 2011;121(7):2845–2854. doi: 10.1172/JCI57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang S, Yao B, Zhou Y, Yin H, Zhang MZ, Harris RC. Intrarenal dopamine modulates progressive angiotensin II-mediated renal injury. Am J Physiol Renal Physiol. 2012;302(6):F742–F749. doi: 10.1152/ajprenal.00583.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armando I, Konkalmatt P, Felder RA, Jose PA. The renal dopaminergic system: novel diagnostic and therapeutic approaches in hypertension and kidney disease. Transl Res. 2015;165(4):505–511. doi: 10.1016/j.trsl.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, et al. Deficient dopamine D2 receptor function causes renal inflammation independently of high blood pressure. PLoS One. 2012;7(6):e38745. doi: 10.1371/journal.pone.0038745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, et al. Single-nucleotide polymorphisms of the dopamine D2 receptor increase inflammation and fibrosis in human renal proximal tubule cells. Hypertension. 2014;63(3):e74–e80. doi: 10.1161/HYPERTENSIONAHA.113.02569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han F, et al. MiR-217 mediates the protective effects of the dopamine D2 receptor on fibrosis in human renal proximal tubule cells. Hypertension. 2015;65(5):1118–1125. doi: 10.1161/HYPERTENSIONAHA.114.05096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Jiang X, Qin C, Cuevas S, Jose PA, Armando I. Dopamine D2 receptors’ effects on renal inflammation are mediated by regulation of PP2A function. Am J Physiol Renal Physiol. 2016;310(2):F128–F134. doi: 10.1152/ajprenal.00453.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin Y, Kumar U, Patel Y, Patel SC, Sidhu A. Differential expression of D2-like dopamine receptors in the kidney of the spontaneously hypertensive rat. J Hypertens. 2003;21(1):199–207. doi: 10.1097/00004872-200301000-00030. [DOI] [PubMed] [Google Scholar]
- 20.Thomas GN, Tomlinson B, Critchley JA. Modulation of blood pressure and obesity with the dopamine D2 receptor gene TaqI polymorphism. Hypertension. 2000;36(2):177–182. doi: 10.1161/01.hyp.36.2.177. [DOI] [PubMed] [Google Scholar]
- 21.Duan J, et al. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet. 2003;12(3):205–216. doi: 10.1093/hmg/ddg055. [DOI] [PubMed] [Google Scholar]
- 22.Noble EP, Blum K, Ritchie T, Montgomery A, Sheridan PJ. Allelic association of the D2 dopamine receptor gene with receptor-binding characteristics in alcoholism. Arch Gen Psychiatry. 1991;48(7):648–654. doi: 10.1001/archpsyc.1991.01810310066012. [DOI] [PubMed] [Google Scholar]
- 23.Thompson J, et al. D2 dopamine receptor gene (DRD2) Taq1 A polymorphism: reduced dopamine D2 receptor binding in the human striatum associated with the A1 allele. Pharmacogenetics. 1997;7(6):479–484. doi: 10.1097/00008571-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Jönsson EG, et al. Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry. 1999;4(3):290–296. doi: 10.1038/sj.mp.4000532. [DOI] [PubMed] [Google Scholar]
- 25.Hernandez YJ, Wang J, Kearns WG, Loiler S, Poirier A, Flotte TR. Latent adeno-associated virus infection elicits humoral but not cell-mediated immune responses in a nonhuman primate model. J Virol. 1999;73(10):8549–8558. doi: 10.1128/jvi.73.10.8549-8558.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inagaki K, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bostick B, Ghosh A, Yue Y, Long C, Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007;14(22):1605–1609. doi: 10.1038/sj.gt.3303029. [DOI] [PubMed] [Google Scholar]
- 28.Konkalmatt PR, Beyers RJ, O’Connor DM, Xu Y, Seaman ME, French BA. Cardiac-selective expression of extracellular super-oxide dismutase after systemic injection of adeno-associated virus 9 protects the heart against post-myocardial infarction left ventricular remodeling. Circ Cardiovasc Imaging. 2013;6(3):478–486. doi: 10.1161/CIRCIMAGING.112.000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armando I, et al. Reactive oxygen species-dependent hypertension in dopamine D2 receptor-deficient mice. Hypertension. 2007;49(3):672–678. doi: 10.1161/01.HYP.0000254486.00883.3d. [DOI] [PubMed] [Google Scholar]
- 30.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20(4):699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23(3):321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 32.Schievenbusch S, et al. Combined paracrine and endocrine AAV9 mediated expression of hepatocyte growth factor for the treatment of renal fibrosis. Mol Ther. 2010;18(7):1302–1309. doi: 10.1038/mt.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh A, et al. Long-term correction of murine glycogen storage disease type Ia by recombinant adeno-associated virus-1-mediated gene transfer. Gene Ther. 2006;13(4):321–329. doi: 10.1038/sj.gt.3302650. [DOI] [PubMed] [Google Scholar]
- 34.Ito K, et al. Adeno-associated viral vector transduction of green fluorescent protein in kidney: effect of unilateral ureteric obstruction. BJU Int. 2008;101(3):376–381. doi: 10.1111/j.1464-410X.2007.07313.x. [DOI] [PubMed] [Google Scholar]
- 35.Chung DC, et al. Adeno-associated virus-mediated gene transfer to renal tubule cells via a retrograde ureteral approach. Nephron Extra. 2011;1(1):217–223. doi: 10.1159/000333071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shao W, et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via αB-crystallin. Nature. 2013;494(7435):90–94. doi: 10.1038/nature11748. [DOI] [PubMed] [Google Scholar]
- 37.Delis F, et al. Loss of dopamine D2 receptors induces atrophy in the temporal and parietal cortices and the caudal thalamus of ethanol-consuming mice. Alcohol Clin Exp Res. 2012;36(5):815–825. doi: 10.1111/j.1530-0277.2011.01667.x. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka K, et al. Bromocriptine methylate suppresses glial inflammation and moderates disease progression in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2011;232(1):41–52. doi: 10.1016/j.expneurol.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 39.Peiser C, et al. Dopamine type 2 receptor expression and function in rodent sensory neurons projecting to the airways. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L153–L158. doi: 10.1152/ajplung.00222.2004. [DOI] [PubMed] [Google Scholar]
- 40.Laengle UW, Markstein R, Pralet D, Seewald W, Roman D. Effect of GLC756, a novel mixed dopamine D1 receptor antagonist and dopamine D2 receptor agonist, on TNF-α release in vitro from activated rat mast cells. Exp Eye Res. 2006;83(6):1335–1339. doi: 10.1016/j.exer.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 41.McKnight AJ, Currie D, Maxwell AP. Unravelling the genetic basis of renal diseases; from single gene to multifactorial disorders. J Pathol. 2010;220(2):198–216. doi: 10.1002/path.2639. [DOI] [PubMed] [Google Scholar]
- 42.Garrett MR, Pezzolesi MG, Korstanje R. Integrating human and rodent data to identify the genetic factors involved in chronic kidney disease. J Am Soc Nephrol. 2010;21(3):398–405. doi: 10.1681/ASN.2009080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Köttgen A, et al. TCF7L2 variants associate with CKD progression and renal function in population-based cohorts. J Am Soc Nephrol. 2008;19(10):1989–1999. doi: 10.1681/ASN.2007121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma RC, et al. Genetic variants of the protein kinase C-beta 1 gene and development of end-stage renal disease in patients with type 2 diabetes. JAMA. 2010;304(8):881–889. doi: 10.1001/jama.2010.1191. [DOI] [PubMed] [Google Scholar]
- 45.Dummer PD, et al. APOL1 kidney disease risk variants: an evolving landscape. Semin Nephrol. 2015;35(3):222–236. doi: 10.1016/j.semnephrol.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia Y, Entman ML, Wang Y. Critical role of CXCL16 in hypertensive kidney injury and fibrosis. Hypertension. 2013;62(6):1129–1137. doi: 10.1161/HYPERTENSIONAHA.113.01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prasad P, Kumar KM, Ammini AC, Gupta A, Gupta R, Thelma BK. Association of dopaminergic pathway gene polymorphisms with chronic renal insufficiency among Asian Indians with type-2 diabetes. BMC Genet. 2008;9:26. doi: 10.1186/1471-2156-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prasad KM, Yang Z, Bleich D, Nadler JL. Adeno-associated virus vector mediated gene transfer to pancreatic β cells. Gene Ther. 2000;7(18):1553–1561. doi: 10.1038/sj.gt.3301279. [DOI] [PubMed] [Google Scholar]
- 49.Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24(4):596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18(1):43–52. doi: 10.1038/gt.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ried MU, Girod A, Leike K, Büning H, Hallek M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J Virol. 2002;76(9):4559–4566. doi: 10.1128/JVI.76.9.4559-4566.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cuevas S, et al. Role of renal DJ-1 in the pathogenesis of hypertension associated with increased reactive oxygen species production. Hypertension. 2012;59(2):446–452. doi: 10.1161/HYPERTENSIONAHA.111.185744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villar VA, et al. Sorting nexin 1 loss results in D5 dopamine receptor dysfunction in human renal proximal tubule cells and hypertension in mice. J Biol Chem. 2013;288(1):152–163. doi: 10.1074/jbc.M112.428458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu JC, Inubushi M, Sundaresan G, Schelbert HR, Gambhir SS. Optical imaging of cardiac reporter gene expression in living rats. Circulation. 2002;105(14):1631–1634. doi: 10.1161/01.cir.0000014984.95520.ad. [DOI] [PubMed] [Google Scholar]
- 55.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.