

Abstract
Herein we report a CRISPR-Cas9-mediated loss-of-function kinase screen for cancer cell deformability and invasive potential in a high-throughput microfluidic chip. In this microfluidic cell separation platform, flexible cells with high deformability and metastatic propensity flowed out, while stiff cells remained trapped. Through deep sequencing, we found that loss of certain kinases resulted in cells becoming more deformable and invasive. High-ranking candidates identified included well-reported tumor suppressor kinases, such as chk2, IKK-α, p38 MAPKs, and DAPK2. A high-ranking candidate STK4 was chosen for functional validation and identified to play an important role in the regulation of cell deformability and tumor suppression. Collectively, we have demonstrated that CRISPR-based on-chip mechanical screening is a potentially powerful strategy to facilitate systematic genetic analyses.
Keywords: analytical methods, biomarkers, cell deformability, CRISPR-Cas9, microfluidics
Systematic loss-of-function genetic screening is an essential approach to identify genes and pathways involved in many biological processes and diseases.[1] The CRISPR (clustered regularly interspaced short palindromic repeats) Cas9 system represents an efficient tool for such screening and has been successfully utilized to identify genes that regulate cell survival, confer drug resistance, and/or drive tumor metastasis.[2] The CRISPR approach provides complete deletion of the target and yields cells with representative phenotypes.[3] A plethora of screening methods can then be used to sort such phenotypes and search for potential biomarkers.[4] CRISPR-based genetic screening has been achieved in both human cells and mouse models, which demonstrates that Cas9-based screening is a robust method to systematically assay gene functions.[2] The current cancer biomarker screening methods mainly rely on in vitro or in vivo cell proliferation and metastasis assays on the knockout cell phenotypes.[2,5] Such assays are often hindered by efficiency as well as extended time and effort, because of limited throughput and prolonged characterization and selection of cells. Integrated microfluidic chips are uniquely advantageous in improving the efficiency of CRISPR-based screening and may potentially recapitulate essential cellular activities spatially and temporally.[6] Such microfluidic chips have also been well-designed with micro and nanostructures to rapidly distinguish cell morphology and dynamics.[7] When applied to cell phenotype selection for biomarker development, these highly integrated microchips have great promise to link the cell phenotypes with their gene deletions, in a fast, efficient, and specific manner. Here we combined the CRISPR/Cas9 screen with cell-mechanics-based on-chip sorting to identify tumor-suppressor kinases. Compared with traditional screening for cellular activities, cell-mechanics-based sorting in a microfluidic chip is a label-free, high-throughput, cost-effective, and time-saving approach, which will likely accelerate the discovery of genes and pathways underlying key cellular processes. The presented method is the first CRISPR screening example developed in the field of microfluidics biotechnology.
Cell deformability is a promising label-free biomarker that indicates changes in cytoskeletal or nuclear organization. Research on the mechanical phenotyping of cancer cells has consistently revealed that high deformability is associated with increased tumor-initiating capacity and metastatic potential,[7a,8] which suggests that genetic screening based on cell deformability may allow for the discovery of new cancer biomarkers. The use of a microfluidics high-throughput assay to perform the CRISPR-based screening is new in the field and may provide higher efficiency and lower cost when studying gene functions. There are notable microfluidic cell deformability assays that are suitable for such screening. The deformability assays have been reported for the enrichment and isolation of cancer cells from blood and cytometry based on biophysics.[7a,8b,9] We herein continue our biophysical cytometry studies and provide a first example of CRISPR-based screening in a microfluidics system. A microfluidic cell deformability assay enables CRISPR-based screening with high efficiency, which can link the mechanical phenotypes of the cell with gene deletions in a fast, efficient, and specific manner. The mechanics-based biomarker-screening strategy will facilitate research into cancer cell metastasis as well as clinical therapy applications, especially in types of cells for which biomarkers have not yet been discovered. Our microfluidic method is able to handle the separation of millions of cells on the basis of their deformability in less than 30 minutes and provide unique opportunities for the screening of gene function.
In this study, we combined the CRISPR-Cas9 screen with a cell-deformability-based on-chip sorting to identify tumor-suppressor kinases. Our research group has designed a unique cell purification system for sorting highly deformable cells in a high-throughput method.[7a] Here we have optimized the device according to the cell size. This microfluidic device allows flexible cells with high deformability and metastatic propensity to pass through the microbarriers and exit the separation chip under hydrodynamic forces, whereas stiff cells remain trapped. This unique purification system based on the mechanical properties of cells is combined with CRISPR-Cas9 knockout technologies to enrich highly deformable cell subpopulations. Through high-throughput sequencing analysis, we successfully identified potential kinases whose losses are involved in cell deformability and invasion regulation. Our highest ranking candidates cover well-reported tumor-suppressor kinases such as chk2 (checkpoint kinase 2), IKK-α (IkappaB kinase alpha), p38 MAPKs (p38 mitogen-activated protein kinases), and DAPK2 (death-associated protein kinase 2), as well as novel hits MAST1 (microtubule associated serine/threonine kinase 1) and STK4 (serine/threonine kinase 4). Functional validation of cellular and molecular phenotypes demonstrated STK4 to be a potential novel tumor suppressor in breast cancer. Compared with traditional screening for cellular activities, sorting on the basis of cell mechanics in a microfluidic chip is a label-free, high-throughput, cost-effective, and time-saving approach, which will likely accelerate the discovery of genes and pathways underlying key cellular processes.
We first designed and validated the cell-separation capability of the microfluidic deformability chip (named the mechanical separation chip (MS-Chip)). The MS-Chip utilizes artificial microbarriers to separate flexible cells from stiff ones by hydrodynamic forces, and the separating structure is composed of two million rectangular microposts 30 μm in height arrayed with gap distances decreasing from 15 μm to 6 μm (Figure 1A and Figure S1). As a proof of concept study, a 1:1 mixture of human breast cancer MDA-MB-231 cells treated with either a dimethylsulfoxide (DMSO) control or cytoskeleton-inhibiting drug Cytochalasin D were applied to the MS-Chip to validate the separation efficiency. Treatment with cytochalasin D inhibits actin polymerization, reduces F-actin bundling, and enhances flexibility,[10] as demonstrated by on-chip staining of trapped cells (Figure S2A–B). As a proof-of-concept study, MDA-MB-231 cells treated with Cytochalasin D and DMSO were stained with different fluorescent dyes and then mixed equally to a final density of 1 × 106 cellsmL−1. After perfusion of the cells through the MS-Chip, trapped cells were imaged by fluorescence microscopy. The distribution of cells treated with Cytochalasin D in the chip differed from the distribution of cells treated with DMSO in the chip. There were more Cytochalasin D treated cells than DMSO treated cells trapped in the small gaps further down the chip (Figure 1B). Statistical analysis of on-chip transport distance versus cell diameter reveals distinct separation efficiencies for the two treatments (Figure S2C). The average transport distances of cells treated with Cytochalasin D were about 1.7-fold greater than those of DMSO-treated cells. When a higher flow rate of 75 μL min−1 was applied, a comparison of the cell populations at the inlet and outlet (Figure 1C) showed that cells treated with Cytochalasin D accumulated at the outlet, and accounted for 88% of the cell population versus 50% of the inlet population (Figure 1D). It should be noted that cell heterogeneity, which includes characteristics such as cell size and cell-cycle phases, affects the separation efficiency. Nevertheless, the cells treated with Cytochalasin D were transported farther in the chip, and because no clear correlation between cell diameter and transport distance has been established (Figure S2C), these data indicate that changes in the cytoskeleton distribution induced by Cytochalasin D are responsible for the separation in the chip of cells treated with Cytochalasin D from those treated with DMSO.
Figure 1.
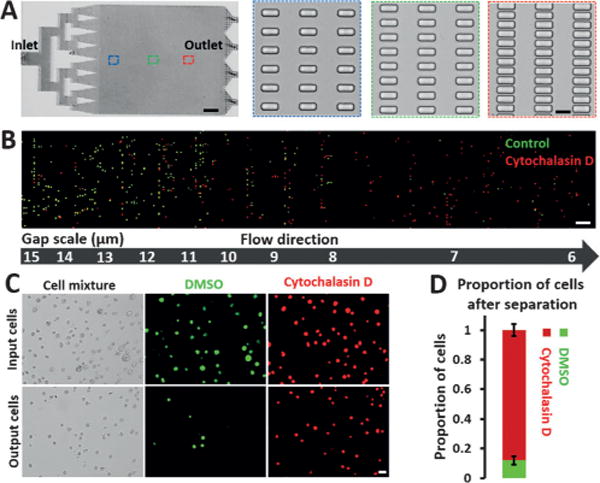
Performance of MS-Chips for cell separation. A) The complete structure of a mechanical separation chip (MS-Chip) (scale bar: 4 mm). Rectangular microposts are shown with gap widths that decrease from 15 μm to 6 μm (scale bar: 15 μm). B) Fluorescence images of DMSO and Cytochalasin D treated MDA-MB-231 cells trapped in an MS-Chip with a flow rate of 25 μLmin−1. DMSO and Cytochalasin D treated cells were stained with CellTracker Green CMFDA Dye and CellTracker Red CMTPX Dye, respectively (scale bar: 100 μm). C) Comparison of input and output cells in a typical DMSO and Cytochalasin D treated separation of MDA-MB-231 cells with a flow rate of 75 μLmin−1. Both bright-field and fluorescent images are presented (scale bar: 30 μm). D) The proportion of cells after separation in (C) was quantified. Error bars indicate standard error of the mean (SEM; n=3).
Since the MS-Chip enriches flexible cells at the end of the micropost array and the mechanical property of a cell is correlated with its metastatic potential,[7a] we explored the possibility of applying such a mechanical cell-sorting approach with the CRISPR-Cas9 knockout (KO) technology. As an initial test, a single-guide RNA (sgRNA) library targeting 507 kinase genes was screened for potential genes involved in the regulation of cell deformability (Figure 2A). First, we generated a derivative of the MDA-MB-231 cell line that stably expresses FLAG-Cas9 under a doxycycline-inducible promoter (Figure 2B). We transduced the Cas9-expressing cell line with a CRISPR kinase-KO lentivirus pool at a ratio of more than 500 cells per lentiviral CRISPR construct. After culturing the cells in vitro for 1 week, the transduced cells were loaded onto an MS-Chip for sorting. Nontransduced cells expressing FLAG-Cas9 were also loaded as a control. The statistical data demonstrated that the CRISPR kinase-KO cells were more heterogeneous when characterized by the on-chip transport distance, but there was no apparent change in the diameter of the cells (Figure 2C). As expected, a small portion of the transduced cells (ca. 15%) traveled further in the chip, and these cells with higher deformability were allowed to flow out of the MS-Chip. Based on our earlier investigation, we find that the collection of 15% of the cells at the outlet of the devices provides a significant difference in their deformability over the cells retained in the devices. We analyzed the cell size of CRISPR kinase-KO cells before and after separation by the MS-chip (Figure S3). There is no large difference in the average cell size before and after separation, thus suggesting that differences in cell deformability are responsible for the separation of the cells in the chip. Although the shear stress will affect gene expression, it won’t be able to change cell functions in less than 30 minutes. Our screening strategy is based on cell function versus loss of a specific gene applied earlier with the CRISPR-based knockout. The change in gene expression during the assay will not matter. The cell viability was also measured for CRISPR kinase-KO cells before and after separation (Figure S4). The results showed that on-chip processing of this cell separation had little affect on the cell viability. Then we sequenced the sgRNA barcodes of the sorted flexible cells (from the output), as well as the entire initial pool of cells (from the input). We did the cumulative distribution analysis of sgRNA counts and found the distribution was similar in two independent experiments (Figure S5). High correlations were achieved among replicates (Figure S6). The distribution of the fraction of detected sgRNA was analyzed for all the 507 kinase genes (Figure S7). In most of the genes, 8/9 pairs of the sgRNA can be detected, which suggested the reliability of our sgRNA pool as well as the consistency between same-gene-targeting sgRNAs. To identify gene hits, we evaluated the change for each sgRNA in the output and the input cell populations. Top sgRNA hits were identified by using the cutoff of log2(fold change) > 7 and a p value < 0.001 (Figure 2D). We identified 38 potential candidate genes with at least two independent sgRNAs among the top hits, whose function may be involved in the regulation of cell deformability and invasion (Table S1). The fraction of the detected sgRNA of top-ranked genes was also analyzed (Figure S8). According to our hypothesis, the loss of tumor-suppressor genes would drive cells to become more flexible and invasive. As expected, we identified 15 known kinase tumor suppressors, including chk2, IKK-α, p38 MAPKs, and DAPK2 (Table S1), thus confirming that our screening approach is effective. We hypothesize that the rest of the list would be new potential tumor suppressors.
Figure 2.
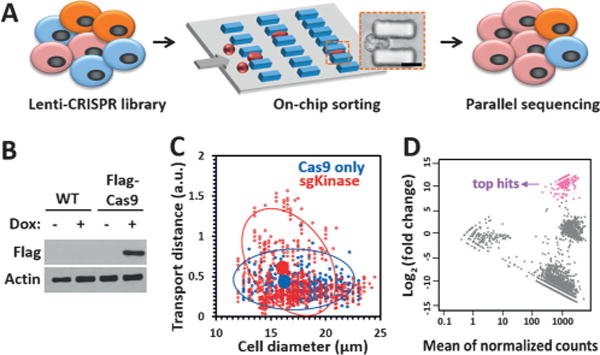
CRISPR-Cas9-mediated loss-of-function screen for cell deformability. A) Illustration of the CRISPR-Cas9 and microfluidic chip screening strategy. Cells were transduced with a lentiCRISPR kinase library and sorted by deformability in an MS-Chip. The flexible cells were allowed to flow out of the MS-Chip (the output) and collected for parallel sequencing together with the untreated whole cells (the input). Cell deformation was visualized by microscopy as a cell passed through a microconstriction (scale bar: 10 μm). B) Western blot analysis of nontransduced MDA-MB-231 cells and MDA-MB-231 cells transduced with a doxycycline-inducible FLAG-Cas9 construct upon doxycycline induction. Actin was used as the loading control. C) Statistical analysis of the on-chip transport distance (at a flow rate of 25 μLmin−1) versus cell diameter for the CRISPR kinase-KO cells. Cells expressing FLAG-Cas9 only were used as the control. The red and blue circles indicate 80% confidence intervals from the means. The means are depicted by solid circles. D) MA plot of mean normalized counts versus log2(fold change) for the output and input sgRNAs. The arrow indicates the top sgRNA hits with log2(fold change)> 7 and an adjusted p value of <0.001.
With increased cutoff criteria, two novel genes, MAST1 and STK4, were selected for further validation. Both MAST1 and STK4 showed three independent sgRNAs in the top hits. We generated two isogenic MDA-MB-231 cell lines with sgRNAs against MAST1 and STK4, and, consistent with our screening data, the MAST1 and STK4 KO cells were transported longer distances in MS-Chips than the wild-type (WT) cells at a lower flow rate of 25 μLmin−1 (Figure 3A and Figure S9). After on-chip-separation at a higher flow rate of 75 μLmin−1, the percentages of MAST1 and STK4 KO cells in the output increased about 1.6-fold and 2.4-fold, respectively (Figure 3B–E). The cells solely expressing FLAG-Cas9 were used as the control to rule out the effect of Cas9 expression on cell-transport ability in the chip. These data confirmed the reliability of the cell-deformability-based kinase screening.
Figure 3.
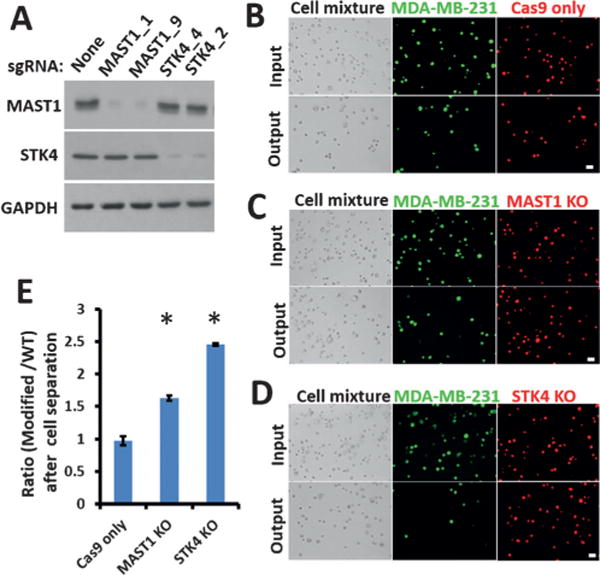
Validation of top hits on a chip assay. A) Western blot analysis of WT and sgRNA-modified MDA-MB-231 cells one week after infection. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the loading control. B–D) Comparisons of the input and the output cells before and after separation of WT and modified MDA-MB-231 cells at a flow rate of 75 μLmin−1. WT cells were stained with CellTracker Green CMFDA Dye and modified cells with CellTracker Red CMTPX Dye. Both bright-field and fluorescent images are presented (scale bar: 30 μm). E) After separation on the chip, the ratios of modified MDA-MB-231 cells to WT cells (B–D) were calculated. Error bars indicate the standard error of the mean (SEM; n=3). *: p values (p < 0.005) were determined by the Student t-test.
The cytoskeletal structure plays a major role in cell deformability and it is usually analyzed by measuring the expression of F-actin, cytokeratin 18, and vimentin.[11] We explored how STK4 regulates the cell deformability by analyzing the expression and distribution of these three molecules. STK4 is the human orthologue of Drosophila Hippo, the central constituent of a highly conserved pathway controlling cell growth and apoptosis.[12] STK4 deficiency is a novel human primary immunodeficiency syndrome.[13] STK4 has been reported to suppress the development of hepatocellular carcinoma through inactivation of the Hippo mediator YAP1.[14] In hematological cancers, the inhibition of STK4 genetically triggers YAP1-mediated apoptosis.[15] We generated three different STK4 KO breast cancer cell lines, analyzed them by the MS-Chip, and found that all three of them were present at higher percentages in the output compared to WT cells (Figure 4A). In addition, according to confocal imaging, STK4 appeared to colocalize with F-actin (Figure 4B); we did not observe any apparent colocalization of STK4 with CK18 or vimentin (Figure S10A, B). Furthermore, the bundling of F-actin was reduced in the STK4-depleted cells (Figure 4C); the distributions of CK18 and vimentin were also affected (Figure 4C and Figure S10C). However, we did not detect apparent changes in the protein level of F-actin, CK18, or vimentin (Figure 4D), thereby suggesting that the role of STK4 in cell deformability may be regulated through remodeling of the cytoskeleton. To conclude, the altered distribution and assembly rather than the amount of cytoskeletal structure markers may be responsible for the increased cell deformability in STK4 KO cells.
Figure 4.
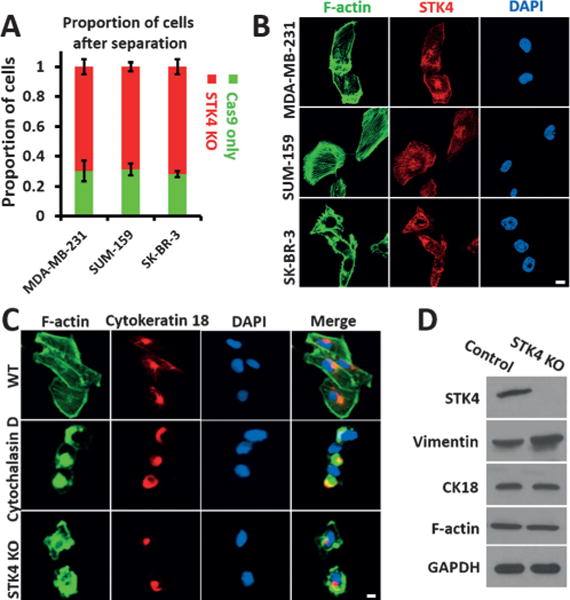
Effect of STK4 on cell deformability and cytoskeletal distribution. A) Quantification of cell proportions at the outlet after separating an equal mixture of STK4 KO cells and cells expressing FLAG-Cas9 only. Error bars indicate SEM (n=3). B) Co-staining of F-actin and STK4 with Alexa Fluor 488 Phalloidin and anti-STK4 (red) antibodies (scale bar: 5 μm). C) Staining of F-actin and CK18 in WT, Cytochalasin D treated, and STK4 KO MDA-MB-231 cells (scale bar: 5 μm). D) Western blot analysis of Vimentin, CK18, and F-actin in STK4 KO MDA-MB-231 cells. GAPDH was used as the loading control.
We next explored the function loss of STK4 in tumorigenicity by using a nontumor MCF-10A breast cell line. Consistent with our previous results, STK4 colocalized with F-actin in the MCF-10A cells (Figure 5A). Depletion of STK4 yielded an invasive phenotype of the MCF-10A cells, which has been associated with the up-regulation of multiple genes involved in the motility and metastasis of cancer cells (Figure 5B–D). Mammosphere formation assays revealed a significant increase in the size and numbers of mammospheres in the STK4 KO MCF-10A cells (Figure 5E). Additionally, the ratio of the CD44high/CD24low stem-cell-like subpopulation significantly increased from 1.9% in MCF-10A control cells to 18% in STK4 KO cells (Figure 5F). All these results demonstrate that STK4 deficiency enhances cell invasiveness in MCF-10A cells. The specific role of STK4 in the regulation of cell deformability and tumor suppression is thus confirmed in breast cancer through this genome-wide screening, which echoes the earlier finding in other cancer types.[12a,c,14]
Figure 5.
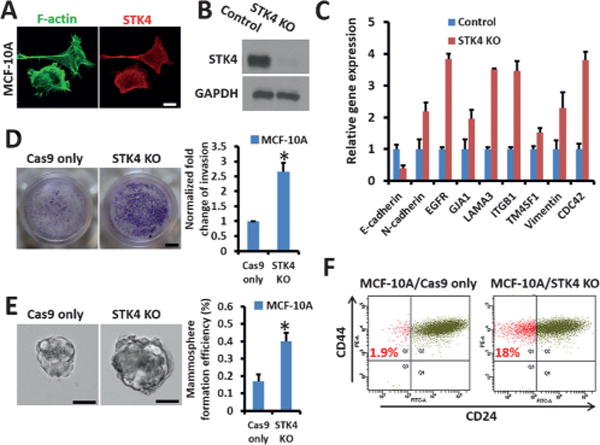
STK4 depletion drives MCF-10A cells to be more invasive. A) Co-staining of F-actin and STK4 in MCF-10A cells (scale bar: 5 μm). B) Western blot analysis of WT and sgSTK4-modified MCF-10A cells. C) Real-time PCR to measure gene expression in STK4 KO MCF-10A cells. Cells expressing FLAG-Cas9 only were used as the control. D) STK4 depletion promotes invasion of MCF-10A cells, as detected by a Matrigel-coated transwell invasion assay (scale bar: 2 mm). Quantification data is shown. E) Mammosphere formation in MCF-10A/Cas9 only and MCF-10A/STK4 KO cells (scale bar: 50 μm). Quantification data are shown. F) The ratio of CD44high/CD24low stem-like cells in MCF-10A expressing FLAG-Cas9 only and in STK4 KO MCF-10A cells.
Interestingly, the correlation between STK4 gene expression in breast tumor biopsies and patient survival was analyzed by mining a publicly available database established by Clynes, Bertucci et al.[16] (R2 Genomics Analysis and Visualization Platform (http://r2.amc.nl); Figure S11). Low expression of STK4 in breast cancer samples predicted a poor overall patient survival rate, thus indicating that STK4 may be a novel suppressor of breast cancer tumors.
Our findings show that combining microfluidic sorting systems based on the mechanical properties of cells with CRISPR-Cas9 technologies is a novel genetic screening strategy that facilitates the rapid identification of genes that play roles in mechanical phenotypes, as well as in physiological and pathological processes. Our investigation provides the first lab-on-chip example to rapidly screen gene function based on the CRISPR knockout system. There are other well-established on-chip cell function assays such as cell adhesion, cell migration, proteomics, and viability assays. This study provides an example and all such assays can be integrated for the screening of gene function with proper device designs. Our study opens a new avenue for the large-scale integration of the on-chip study of cell function and search for such potential biomarkers.
Acknowledgments
This study was funded by NIH-R01 DA035868, R01 CA180083, R56 AG049714, and R21 CA191179.
Footnotes
Supporting information for this article can be found under: http://dx.doi.org/10.1002/anie.201601984.
Contributor Information
Xin Han, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA), Department of Cell and Developmental Biology, Weill Medical College of Cornell University, New York, NY 10065 (USA).
Zongbin Liu, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA), Department of Cell and Developmental Biology, Weill Medical College of Cornell University, New York, NY 10065 (USA).
Li Zhao, Department of Molecular and Human Genetics, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030 (USA).
Feng Wang, Department of Molecular and Human Genetics, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030 (USA).
Yang Yu, Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030 (USA).
Jianhua Yang, Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030 (USA).
Rui Chen, Department of Molecular and Human Genetics, Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030 (USA).
Lidong Qin, Email: LQin@houstonmethodist.org, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA), Department of Cell and Developmental Biology, Weill Medical College of Cornell University, New York, NY 10065 (USA).
References
- 1.a) Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R. Nature. 2004;428:431–437. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]; b) Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, Brummelkamp TR. Science. 2009;326:1231–1235. doi: 10.1126/science.1178955. [DOI] [PubMed] [Google Scholar]; c) Rad R, Rad L, Wang W, Cadinanos J, Vassiliou G, Rice S, Campos LS, Yusa K, Banerjee R, Li MA, de la Rosa J, Strong A, Lu D, Ellis P, Conte N, Yang FT, Liu P, Bradley A. Science. 2010;330:1104–1107. doi: 10.1126/science.1193004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang T, Wei JJ, Sabatini DM, Lander ES. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, Lee H, Zhang F, Sharp PA. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sander JD, Joung JK. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu PD, Lander ES, Zhang F. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Nat Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]; b) Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 6.Lecault V, Vaninsberghe M, Sekulovic S, Knapp DJ, Wohrer S, Bowden W, Viel F, McLaughlin T, Jarandehei A, Miller M, Falconnet D, White AK, Kent DG, Copley MR, Taghipour F, Eaves CJ, Humphries RK, Piret JM, Hansen CL. Nat Methods. 2011;8:581–586. doi: 10.1038/nmeth.1614. [DOI] [PubMed] [Google Scholar]
- 7.a) Zhang W, Kai K, Choi DS, Iwamoto T, Nguyen YH, Wong H, Landis MD, Ueno NT, Chang J, Qin L. Proc Natl Acad Sci USA. 2012;109:18707–18712. doi: 10.1073/pnas.1209893109. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Han X, Liu Z, Jo MC, Zhang K, Li Y, Zeng Z, Li N, Zu Y, Qin L. Sci Adv. 2015;1:e1500454. doi: 10.1126/sciadv.1500454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Byun S, Son S, Amodei D, Cermak N, Shaw J, Kang JH, Hecht VC, Winslow MM, Jacks T, Mallick P, Manalis SR. Proc Natl Acad Sci USA. 2013;110:7580–7585. doi: 10.1073/pnas.1218806110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gossett DR, Tse HT, Lee SA, Ying Y, Lindgren AG, Yang OO, Rao J, Clark AT, Di Carlo D. Proc Natl Acad Sci USA. 2012;109:7630–7635. doi: 10.1073/pnas.1200107109. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wirtz D, Konstantopoulos K, Searson PC. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Mohamed H, Murray M, Turner JN, Caggana M. J Chromatogr A. 2009;1216:8289–8295. doi: 10.1016/j.chroma.2009.05.036. [DOI] [PubMed] [Google Scholar]; b) Hur SC, Henderson-MacLennan NK, McCabe ER, Di Carlo D. Lab Chip. 2011;11:912–920. doi: 10.1039/c0lc00595a. [DOI] [PubMed] [Google Scholar]
- 10.Otto O, Rosendahl P, Mietke A, Golfier S, Herold C, Klaue D, Girardo S, Pagliara S, Ekpenyong A, Jacobi A, Wobus M, Topfner N, Keyser UF, Mansfeld J, Fischer-Friedrich E, Guck J. Nat Methods. 2015;12:199–202. doi: 10.1038/nmeth.3281. [DOI] [PubMed] [Google Scholar]
- 11.a) Eckes B, Dogic D, Colucci-Guyon E, Wang N, Maniotis A, Ingber D, Merckling A, Langa F, Aumailley M, Delouvee A, Koteliansky V, Babinet C, Krieg T. J Cell Sci. 1998;111:1897–1907. doi: 10.1242/jcs.111.13.1897. [DOI] [PubMed] [Google Scholar]; b) Beil M, Micoulet A, von Wichert G, Paschke S, Walther P, Omary MB, Van Veldhoven PP, Gern U, Wolff-Hieber E, Eggermann J, Waltenberger J, Adler G, Spatz J, Seufferlein T. Nat Cell Biol. 2003;5:803–811. doi: 10.1038/ncb1037. [DOI] [PubMed] [Google Scholar]; c) Gardel ML, Shin JH, MacKintosh FC, Mahadevan L, Matsudaira P, Weitz DA. Science. 2004;304:1301–1305. doi: 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- 12.a) Hergovich A, Kohler RS, Schmitz D, Vichalkovski A, Cornils H, Hemmings BA. Curr Biol. 2009;19:1692–1702. doi: 10.1016/j.cub.2009.09.020. [DOI] [PubMed] [Google Scholar]; b) Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y. Proc Natl Acad Sci USA. 2010;107:1431–1436. doi: 10.1073/pnas.0911409107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Thompson BJ, Sahai E. J Cell Biol. 2015;210:871–882. doi: 10.1083/jcb.201507005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdollahpour H, Appaswamy G, Kotlarz D, Diestelhorst J, Beier R, Schaffer AA, Gertz EM, Schambach A, Kreipe HH, Pfeifer D, Engelhardt KR, Rezaei N, Grimbacher B, Lohrmann S, Sherkat R, Klein C. Blood. 2012;119:3450–3457. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, Ponzoni M, Marcatti M, Richardson PG, Carrasco R, Kimmelman AC, Wong KK, Caligaris-Cappio F, Blandino G, Kuehl WM, Anderson KC, Tonon G. Nat Med. 2014;20:599–606. doi: 10.1038/nm.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Clarke C, Madden SF, Doolan P, Aherne ST, Joyce H, O’Driscoll L, Gallagher WM, Hennessy BT, Moriarty M, Crown J, Kennedy S, Clynes M. Carcinogenesis. 2013;34:2300–2308. doi: 10.1093/carcin/bgt208. [DOI] [PubMed] [Google Scholar]; b) Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, Tallet A, Chabannon C, Extra JM, Jacquemier J, Viens P, Birnbaum D, Bertucci F. Breast Cancer Res Treat. 2011;126:407–420. doi: 10.1007/s10549-010-0897-9. [DOI] [PubMed] [Google Scholar]