

Abstract
Cell-based neurotransmitter fluorescent engineered reporters (CNiFERs) provide a new tool for neuroscientists to optically detect the release of neurotransmitters in the brain in vivo. A specific CNiFER is created from a human embryonic kidney cell that stably expresses a specific G protein-coupled receptor, which couples to Gq/11 G proteins, and a FRET-based Ca2+-detector, TN-XXL – activation of the receptor leads to an increase in the FRET signal. Because a CNiFER clone utilizes the native receptor for a particular neurotransmitter (e.g. D2R for dopamine), it has nanomolar sensitivity and a temporal response of seconds. CNiFERs are directly implanted into the brain, enabling them to sense neurotransmitter release with a spatial resolution of less than one hundred micrometers, making them ideal to measure volume transmission in vivo. CNiFERs can also be used to screen other drugs for potential cross-reactivity in vivo. We recently expanded the family of CNiFERs to include GPCRs that couple to Gi/o G proteins. CNiFERs are available for detecting acetylcholine (ACh), dopamine (DA) and norepinephrine (NE). Given that any GPCR can be used to create a novel CNiFER and that there are approximately 800 GPCRs in the human genome, we describe here the general procedure to design, realize, and test any type of CNiFER.
Keywords: Optical imaging, FSCV, dialysis, volume transmission, neurotransmitters, neuropeptides, TPLSM, biosensor, GPCR
Introduction
To fully understand how neurons communicate in the brain, it is necessary to have a method to measure the release of neurotransmitters in vivo. There are several well-established techniques for measuring neurotransmitters in vivo. One commonly used technique is microdialysis, in which a cannula is inserted into the brain and a small volume of artificial cerebral spinal fluid is collected and analyzed using high-performance liquid chromatography and electrochemical detection1. Microdialysis has a spatial resolution on the order of a few diameters of the probe, or ∼ 0.5 mm for a 200 μm diameter microprobe. The temporal resolution of this technique, however, is slow due to sampling intervals that typically last ∼ 5 minutes or longer1. Moreover, analyses are not made in real-time. Another technique is fast scanning cyclic voltammetry (FSCV), which uses a carbon-fiber probe that is inserted into the brain. FSCV has excellent temporal resolution (subsecond), high sensitivity (nanomolar) and spatial resolution with probes of 5-30 μm in diameter2. However, FSCV is limited to transmitters that produce a characteristic oxidation profile with voltage on a carbon potentiometric probe2. A third technique to measure neurotransmitters is directly through genetically-encoded neurotransmitter (NT) biosensors3. With this method, a fusion protein is created that contains a ligand-binding domain for a transmitter coupled to a fluorescence resonance energy transfer (FRET)-based pair of fluorophores4 or a permutated GFP5. Unlike the previous two methods, these biosensors are genetically encoded and expressed on the surface of a host cell, such as a neuron, through the production of transgenic animals or acutely with the use of viral agents to infect cells. To date, genetically-encoded biosensors have been only developed for detecting glutamate and GABA3-5. A limitation with these techniques has been the inability to expand the detection to the large number of transmitters, e.g., classical neurotransmitters, neuropeptides and neuromodulators, that signal through G protein-coupled receptors (GPCRs). In fact, there are nearly 800 GPCRs represented in human genome6.
To address these shortfalls, we have developed an innovative tool to optically measure release of any neurotransmitter that signals through a GPCR and is released in vivo. CNiFERs (cell-based neurotransmitter fluorescent engineered reporters) are clonal HEK293 cells engineered to express a specific GPCR that, when stimulated, triggers an increase in intracellular [Ca2+] that is detected by a genetically encoded FRET-based Ca2+ sensor, TN-XXL. Thus, CNiFERs transform neurotransmitter receptor binding into a change in fluorescence, providing a direct and real-time optical read-out of local neurotransmitter activity. By utilizing the natural receptor for a given neurotransmitter, CNiFERs retain the chemical specificity, affinity and temporal dynamics of the endogenously expressed receptors. To date, we have created three types of CNiFERs: one for detecting acetylcholine using the M1R, one for detecting dopamine using the D2R, and one for detecting norepinephrine using the α1aR7,8. The CNiFER technology is readily expandable and scalable, making it amenable to any type of GPCR. In this JoVE article, we describe and illustrate the methodology to design, realize, and test CNiFERs for any application.
Protocol
1. Generate GPCR lentivirus for transforming HEK293 cells
1.1
Purchase the cDNA for a specific GPCR, e.g., cdna.org, or PCR the cDNA from a cDNA library. Note G protein coupling specificity, i.e., Gi/o, Gq/11, or Gs, and species information, if appropriate. Confirm sequence by DNA sequencing.
1.2
Clone the GPCR cDNA into the MCS of a lentivirus-expressing vector. Use pCDH-CMV-MCS-EF1-Puro for generating lentivirus. Expand and purify DNA using an endotoxin-free maxi prep kit.
1.2.1 Example of cloning procedure
Design primers that contain restriction sites for cloning by PCR. In this example, the Gqi5 cDNA is being subcloned into the BamHI and NotI restriction sites in pCDH.
Order the following DNA primers.
5′-CATCGGGATCCATGACTCTGGAGTCCATCATGG -3′ (forward primer) 5′-GCGAGTGCGGCCGCTCAGAAGAGGCCACAGTCCTTAAGGTTCAG-3′ (reverse complement primer). The PCR reaction is performed in a total volume of 50 μl containing 10 ng of template DNA, 0.4 μM of each primers, 1 mM MgCl2, 0.2 mM deoxynucleotide triphosphate, 1× PCR buffer, and 2.5 (U/μl) of Taq polymerase. PCR machine is set to: 5′ at 94°C, followed by 25 cycles of denaturation at 95°C for 30 seconds, annealing at 57°C for 30 seconds, and extension at 72°C for one minute, then a final extension at 72°C for five minutes. 5 μl of the PCR product is analyzed on 0.8 % (W/v) agarose gel in 1× TAE buffer and rest of the PCR product is purified using QIAquick PCR purification kit according to the manufacturer's instructions. The purified PCR product and the PCDH vector are digested with BamHI and NotI and ligated to generate the recombinant plasmid PCDH-Gqi5.
1.3
Generate the lentivirus using a core facility that produces viral vectors, such as one at The Salk Institute, University of Pennsylvania, University of North Carolina, etc., or in-house. ∼ 25 μg (> 1 μg/μl) of endotoxin-free DNA is sufficient for a transfection of HEK cells in a T75 flask. It is important the DNA is high purity, having an absorbance ratio (A260/A280) of ∼ 1.8. Titers of virus ∼ 109-1010 work well for transduction of HEK293 cells.
Tip: Prior to submitting the DNA, digest an aliquot of the DNA with appropriate restriction enzymes to confirm correct size of insert and purity of DNA.
2. Culture HEK293 cells
2.1
Purchase HEK293 or HEK293T cells. If provided by another research laboratory, ensure that the cells are myco-free using a commercially available kit. For CNiFERs based on the TN-XXL backbone, HEK293/TN-XXL clone (#3g8) and HEK293/TN-XXL/Gqi5 clone (#qi5.6) are freely available upon request8.
2.2
Grow and expand HEK293 cells in culture media containing DMEM (high glucose) with GlutaMax; 10 % FBS (heat-inactivated) and 1× Pen/Step (P/S). Grow cells in a humidified incubator at 37°C with 5 % (v/v) CO2 in O2.
3. Lentiviral Infection of HEK293 cells
3.1
Seed HEK293 cells in a T25 (0.7×106 cells) or T75 (2.1×106) flask and grow until ∼ 50 % confluent (after approximately 1 day).
3.2
On day of infection, dilute the virus in media (DMEM_FBS_Glutamax_P/S) to a final concentration of about 109 GC/ml in a total volume of 5 ml for T25 and 10 ml for T75. Aspirate media from the T25 or T75 flask and replace with media + virus mixture. NOTE: High titers of lentivirus are biosafety level 2 (BSL-2).
3.3
After one day of infection, aspirate the media plus virus mixture and replace it with DMEM_FBS_Glutamax_P/S with puromycin (2-3 ug/ml). Culture cells until about ∼90% confluent (∼ 1-2 days). Save an aliquot of infected cells for freezing (see protocol below).
4. FACS and isolation of single clones
4.1
Before fluorescence activated cell sorting (FACS) analysis, confirm expression of GPCR following transduction by testing infected cells for agonist response on a fluorometric plate reader (see Section 6, below). Choose agonist concentrations that are above and below the published EC50 for the specific GPCR. If there is a significant change in the FRET ratio (ΔR/R) with receptor activation, then proceed onto the FACS analysis. If there is no GPCR at this stage or no FRET response with agonist, use a Ca2+ ionophore, e.g., A21387, to test the Ca2+ response and confrim that FRET-based sensor is working.
4.2
Day before: Prepare 96-well plates coated with fibronectin. Add 50 μl fibronectin (5μg/ml) per well in a 96 well plate. Incubate the plate at room temp for 1 hour. Rinse twice (5 minutes) with PBS. Add 50 μl of media and incubate at 37°C overnight. Also prepare PBS/5% BSA (5g/100ml) and filter (0.2 μm) into sterile bottle.
4.3
Aspirate media from T25 or T75 flask. Harvest cells with 0.05 % (w/v) trypsin/EDTA, following manufacture instructions. Centrifuge 5 minutes at 1000 g in cell culture centrifuge.
4.4
Aspirate media, resuspend pellet in 4 ml 5 % (w/v) BSA in PBS and centrifuge 5 minutes.
4.5
Aspirate media, resuspend pellet in 5 % (w/v) BSA in PBS to give a final dilution of ∼5 × 106 cells/ml. Note: check with your FACS core facility for specific requirements.
4.6
Filter the resuspended cells with a 40 μm cell strainer to remove clumps.
4.7
Transfer to a 5 ml polypropylene round bottom tube.
4.8
Place tube on ice for transfer to FACS facility.
4.9
Sort transduced HEK293 cells at a FACS facility. Set parameters on FACS for 4°C for the sample holder, 100 μm nozzle and 20 psi. Pre-sort analysis: select cells that have high expression level of FRET-based Ca2+ detector and the capability of undergoing FRET (see Results below).
4.10
Deposit sorted cells into a 96-well plate, one clone per well with 50 μl media. Plan to collect four 96-well plates for subsequent screening.
4.11
Add 50 μl of media (DMEM_FBS_Glutamax_P/S/Puro) for a total of 100 μl per well
Note: Media contains puromycin for selection of transduced cells.
5. Culturing and Expansion of clonal CNiFERs
Note: The overall goal of this step is to expand the single clones from a single well in a 96-well plate to a T75 flask. Proceed with this expansion gradually, moving to a slightly larger flask after each passage.
5.1
In the 96-well plate, maintain the cells by gently aspirating 50 μl of media from each well and replacing with 50 μl of fresh media (DMEM_FBS_Glutamax_P/S/Puro) every 5-7 days until wells are confluent.
5.2
When confluent (after 2-3 weeks), gently aspirate media. Add ∼ 20 μl of 0.05 % (w/v) trypsin/EDTA. Incubate for 1-2 minutes at 37°C. Add 250 μl of media to trypsin-treated cells and resuspend. Transfer contents to 24-well plate containing 750 μl fresh media.
Note: Do not rinse cells with PBS at this stage
5.3
In the 24-well plate, maintain cells by replacing 500 μl of media every 5-7 days until wells are confluent.
5.4
When confluent, gently aspirate media. Add ∼ 500 μl of 0.05 % (w/v) trypsin/EDTA. Incubate for 1-2 minutes at 37°C. Add 1000 μl of media to trypsin-treated cells and resuspend. Transfer contents to 12-well plate containing 1000 μl media (2 ml final volume).
Note: Do not rinse cells with PBS at this stage
5.5
In the 12-well plate, replace 750 μl of media every 5-7 days, repeat until confluent.
5.6
Before harvesting the cells aspirate media, rinse cells once with PBS. Add ∼ 500 μl 0.05 % (W/v) trypsin/EDTA. Incubate for 1-2 minutes at 37° C. Add 1000 μl of media to trypsin-treated cells and resuspend. Transfer to 6-well plate containing 2 ml media (3.5 ml final volume).
5.5
In the 6 well plate, maintain cells by replacing 750 μl of media every 5-7 days, repeat until confluent.
5.6
Aspirate media, rinse once with PBS. Add 1 ml 0.05 % (w/v) trypsin/EDTA. Incubate for 1-2 minutes. Add 1 ml of media to trypsin-treated cells and resuspend. Transfer contents to T25 flask containing 3 ml media (5 ml final volume).
5.7
In the T25 flask, maintain cells by replacing 5 ml of media every 2-3 days, repeat until confluent (∼ 5 days).
5.8
Aspirate media, wash 1× with PBS. Use 1 ml of 0.05 % (w/v) trypsin/EDTA and incubate for 1-2 minutes at 37°C. Add 5ml media and triturate to break up clumps. Centrifuge (1000 RPM) for 5 minutes, aspirate media, resuspend in 5 ml of fresh media. Repeat.
5.9
Use 1 ml of cell suspension and add to T75 with 9 ml of fresh media (10 ml final volume)
5.10
Use remaining 4 ml to freeze down for future use. Prepare eight 1.5 ml cryotubes on ice. Add 4 ml of 20 % (v/v) DMSO/media mixture for a final concentration of DMSO of 10 %, and triturate gently. Aliquot 1 ml in 8 tubes. Freeze in -80°C overnight in a foam insulated box. Transfer to liquid nitrogen for long-term storage.
5.11
In T75 flask, maintain cells by replacing 10 ml of media every 3-5 days, repeat until 70-80 % confluent (∼1-2 weeks).
5.12
Prepare 96-well fibronectin-coated plate for fluorometric plate reader.
5.13
Harvest cells from T75 flask (as described above). Resuspend cells in 14 ml of fresh media and triturate well to break up the clumps into single cells.
5.14
Transfer 100 μl of cells suspension into each well of a fibronectin-coated 96-well plate (see description above) using a multichannel pipette.
6. Identify candidate CNiFERs based on FRET response using fluorometric plate reader
Note: With the four 96-well plates, there will be at least 100 testable clones. Note: many fail to grow and expand. Identify potential candidate CNiFERs using a 3-point analysis for FRET response with cognate agonist.
6.1
Day before: For the 3-point analysis, seed (× × 106) each CNiFER clone into three wells, e.g., A1, B1, C1, etc., of a fibronectin-coated black clear bottom 96-well plate (see above).
6.2
Prepare a “3-fold” agonist test with three different concentrations of agonist, e.g., 0.3-, 3.0-, and 30-times the EC50 for the specific GPCR, in artificial cerebral spinal fluid (ACSF: 142 mM NaCl, 5 mM KCl, 10 mM D-glucose, 10 mM HEPES, 3.1 mM CaCl2, 1.3 mM MgCl2, pH 7.4).
Tip: Prepare different concentrations of drugs using a serial dilution method.
6.3
Aspirate media from the cell plate and add 100 μl ACSF.
Tip: Use a template to keep track of wells
6.4
Set up parameters on a 96-well fluorometric plate reader equipped to measure FRET and perform solution transfers. Set plate reader temperature to 37°C. For measuring FRET with TN-XXL, set excitation wavelength to 436 ± 4.5 nm and emission filters to 485 ± 7.5 nm for CFP (eCFP) and to 527 ± 7.5 nm for YFP (Citrine). Set the cutoffs to 475 and 515 nm for CFP and YFP, respectively. Set program for solution delivery of 50 μl from the 3-fold agonist test to 100 μl in each well after collecting 30 seconds of baseline fluorescence. Program reader to measure CFP and YFP emissions every 3.5-4 seconds for 180 seconds.
6.5
Equilibrate the plate of CNiFERs and drug plate in the plate reader for 30 minutes at 37°C before starting the program. Start program on plate reader.
6.6
When analyzing plate reader data, subtract background measurements (taken from wells without cells) from wells with CNiFERs. Normalize fluorescence intensities to pre-stimulus baselines (ΔF/F) and calculate the FRET ratio (ΔR/R) using the peak responses on the 527 and 485 nm channels (see Analysis below). Choose CNiFERs for full analysis that have appropriate sensitivity and largest FRET response.
7. Final selection of CNiFER clones
7.1
Day before: For the full plate analysis, seed (× × 106) each selected CNiFER clone into an entire 96-well plate (fibronectin-coated black clear bottom 96-well).
7.2
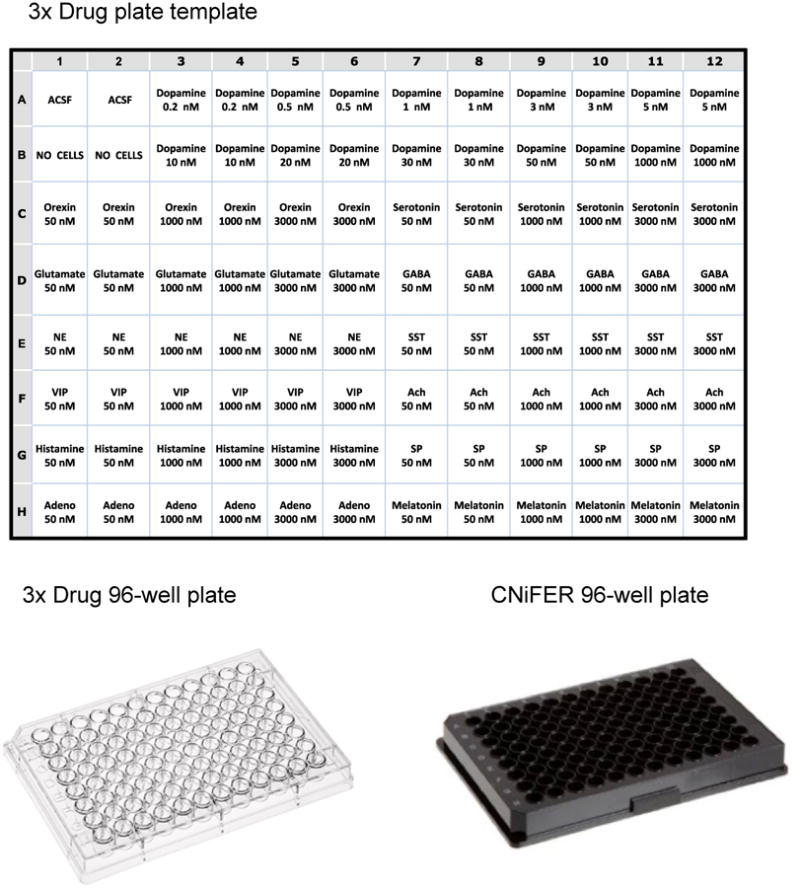
Prepare for final agonist testing. For generating a full dose-response curve, choose 10 different agonist concentrations. For determining non-specific responses, use the 3-fold agonist test concentrations for 12 different agonists for other receptors. Duplicate each measurement to guard against systematic errors. For example, the drug plate for testing a D2 CNiFER is 10 different concentrations of dopamine (10 concentrations) and three different concentrations of (acetylcholine, glutamate, orexin, VIP, adenosine, serotonin, norepinephrine, GABA, Substance P, melatonin).
7.3
Set up parameters on a 96-well fluorometric plate reader equipped to measure FRET and perform solution transfers. Set plate reader temperature to 37°C. For measuring FRET with TN-XXL, set excitation wavelength to 436 ± 4.5 nm and emission filters to 485 ± 7.5 nm for CFP (eCFP) and to 527 ± 7.5 nm for YFP (Citrine). Set the cutoffs to 475 and 515 nm for CFP and YFP, respectively. Set program for solution delivery of 50 μl from the 3-fold agonists to 100 μl in each well after collecting 30s of baseline fluorescence. Program reader to measure CFP and YFP emissions every 3.5-4 seconds for 180 seconds.
7.4
Equilibrate the plate of CNiFERs and drug plate in the plate reader for 30 minutes at 37°C before starting the program. Start program on plate reader.
7.5
When analyzing plate reader data, subtract background measurements (taken from wells without cells) from wells with CNiFERs. Normalize fluorescence intensities to pre-stimulus baselines (ΔF/F) and calculate the FRET ratio (ΔR/R) using the peak responses on the 527 and 485 nm channels (see Analysis below).
7.6
For the cognate agonist, plot the peak ΔR/R as a function of agonist concentration and fit with the Hill equation (see Analysis below). Determine the EC50, Hill coefficient and maximal FRET ratio. For the 10 other agonists, plot peak ΔR/R as a function of agonist concentration. Choose ∼ 10 CNiFER clones that have a large FRET ratio, an appropriate EC50 and little or no background responses to other neurotransmitters (non-specific response).
8. Determine receptor specificity and desensitization of specific CNiFER clone
8.1
Day before testing, harvest CNiFERs with 0.05 % (w/v) trypsin/EDTA (described above) and plate 1.25×106 cells/well on fibronectin-coated glass (25 mm) coverslips in a 24-well plate. Culture CNiFERs for 24 hours.
8.2
Set up inverted fluorescence microscope with 20× air objective for FRET-based imaging. Set filters for measuring CFP/YFP FRET: (470/30 nm - CFP; 535/50 nm – YFP, dual dichroic), excitation for CFP, e.g., 442 nm laser, and CCD camera for collecting fluorescent images.
8.3
Set up continuous perfusion using a laminar flow perfusion chamber with gravity-fed solutions containing ACSF and drug solutions. Collect waste solutions with vacuum and vacuum flask.
8.4
Set in line temperature controller to 35°C.
Note: Measure the temperature in the bath with the thermistor. Adjust heating temperature so that bath is at 35°C (depending on the chamber, there is some cooling of solution during flow).
8.5
Transfer one glass coverslip with CNiFERs to perfusion chamber. Start perfusion with 35°C ACSF. Manually focus on cluster of CNiFER cells.
8.6
Start acquisition of images with a system that has at least a 1 Hz temporal resolution. The FRET response, i.e., CFP excitation and CFP and YFP emission, is meaured with following solutions: 60 seconds ACSF only (baseline), 60 seconds of agonist/ACSF, 60 seconds of antagonist/ACSF, 60 seconds of (antagonist + agonist)/ACSF and 60 seconds of agonist/ACSF. Calculate the change in FRET by subtracting the peak FRET response during the second agonist presentation (in the presence of antagonist) from the first (agonist only).
8.7
Determine specificity of CNiFER activation by calculating fractional change in FRET (antagonist+agonist divided by agonist alone).
8.8
Transfer a new glass coverslip with CNiFERs to perfusion chamber. Start perfusion with 35°C ACSF. Manually focus on a cluster of CNiFER cells.
8.9
Start acquisition of images for FRET response (CFP excitation, CFP and YFP emission) Measure the FRET response during 10 consecutive pulses of agonist (at saturating dose) with a 180 seconds wash with ACSF between pulses.
9. Determine temporal resolution of CNiFER response
9.1
Determine the temporal resolution of CNiFERs by imaging CNiFERs in a chamber equipped with a fast perfusion stepper for rapidly switching between two solutions. The imaging system should, ideally, have a 5 Hz acquisition rate or better and can be either a low-noise camera, a confocal microscope, or a two-photon microscope.
Tip: Mix Alexa-594 with the agonist to provide an optical read-out of the time course of agonist perfusion, using a third channel.
9.2
Set up continuous perfusion using a laminar flow perfusion chamber with gravity-fed solutions containing either ACSF or ACSF and saturating agonist concentration. Connect to theta tub and attach to stepper motor.
9.3
Transfer one glass coverslip with CNiFERs to perfusion chamber. Start perfusion with 35°C ACSF. Manually focus on cluster of CNiFER cells.
9.4
Image the CNiFERs. Use 820 nm excitation light to excite the eCFP and split emission with a 505 dichroic mirror into two channels: 470 ± 20 nm for eCFP and 535 ± 20 nm for YFP (Citrine). Use a 10× air objective or 10× water. Measure the FRET response while changing the delay between two 2.5 second pulses of agonist.
9.3
Determine shortest time between FRET ratio peaks with two consecutive agonist pulses
10. Freeze-back selected CNiFER clones
10.1
Prepare a T75 flask of an individual CNiFER clone. Harvest cells as above and resuspend in 5 ml.
10.2
Prepare ten 1.5 ml cryotubes on ice. Add 5 ml of 20 % (v/v) DMSO in media mixture, i.e., the final concentration of DMSO is 10 % (v/v), and triturate gently.
10.3
Aliquot 1 ml each in 10 tubes. Freeze in -80°C overnight in a foam insulated box.
10.4
Transfer to liquid nitrogen for long term storage.
11. In vivo imaging of CNiFER clones
11.1
Perform a ‘thinned skull’ craniotomy as described9 or a conventional ‘open skull’ imaging window.
11.2
Pull capillary glass and break tip to inner diameter of 40 μm.
11.3
Harvest CNiFERs that were grown to 80 % confluency in a T75 flask with 10 ml ACSF (no trypsin) and triturate to remove clumps. Centrifuge and resuspend the pellet in 100 μl ACSF. Centrifuge for 30 seconds at 1400 rpm and remove the supernatant.
11.4
Load CNiFER pellet into glass pipette using a nanoinjector. Inject 2-5 nl of cells into neocortex through the thinned skull, ∼ 200 μm from the cortical surface.
11.5
After implantation in several adjacent sites (typically two injection sites per CNiFER variant), rinse with ACSF to remove cells that are outside of the injection site, seal craniotomy with a glass coverslip.
11.6
Attach a custom-built head-bar to the skull with dental cement.
11.7
To minimize potential immunological reaction to the human CNiFERs, inject mice daily with cyclosporine (20 μl/100 g, i.p.), starting the day before injection of CNiFERs.
11.8
Collect images of CNiFERs using in vivo two-photon microscopy and a 25× to 40 × dipping objective. Use 820 nm excitation light to excite the CFP in TN-XXL and split emission into two channels: 470 ± 20 nm for CFP and 535 ± 20 nm for YFP (Citrine).
12. Data analysis
12.1
Use Matlab or equivalent software for analyses. Draw regions of interest around CNiFER cells. Subtract background from CFP and YFP fluorescence intensities in these regions of interest and normalize the values to prestimulus baselines. Quantify responses as the fractional change in the FRET ratio ΔR/R, where ΔR is the change in the ratio the two emission channels, denoted FYFP and FCFP respectively, and R is the normalized baseline ratio, such that:
After low-pass filtering, determine the responses at the peak of AR/R.
For determining the sensitivity of CNiFERs plot the FRET ratio as a function of log agonist concentration. Fit with the Hill equation to determine the EC50 and Hill coefficient (n), using a scientific statistical software.
Hill Equation:
Representative Results
A CNiFER is derived from a human embryonic kidney (HEK293) cell that is engineered to stably express at least two proteins: (i) a specific G-protein coupled receptor (GPCR) and (ii) a genetically encoded [Ca2+] sensor, TN-XXL. TN-XXL undergoes fluorescence resonance energy transfer (FRET) between cyan and yellow fluorescent proteins (eCFP and YFP (citrine), respectively) in response to Ca2+ ions7,10. Activation of GPCRs that couple to endogenous Gq G-proteins trigger an increase in cytosolic [Ca2+] through the PLC/IP3 pathway, leading to an increase in FRET from the TN-XXL (Fig. 1).
Fig. 1. Scheme for developing CNiFERs.
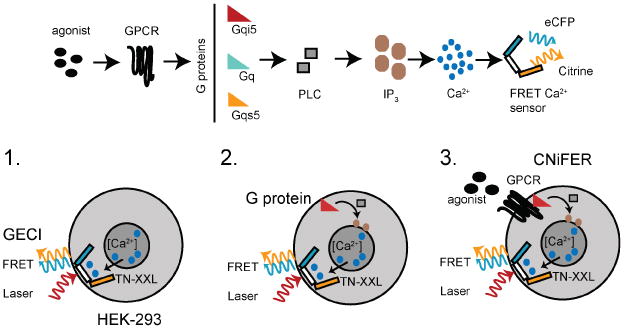
Top, signaling pathway for CNiFER cells. Bottom, 1. Ca2+-detector. 2. Gα G-protein chimera, if necessary. 3. Unique GPCR to create CNiFER.
The increase in FRET provides a rapid optical read-out of the change in neurotransmitter levels. To develop a CNiFER for a particular type of neurotransmitter, first determine the type of G protein that couples to the GPCR. For Gq-coupled GPCRs, the GPCR uses Gq proteins endogenously expressed in HEK293 cells. For Gi/o-coupled GPCRs, a clonal HEK293 line is first created that expresses a chimeric G protein that redirects the GPCR to the Gq-PLC/IP3 pathway. This is accomplished with a chimeric G-protein, Gqi5, which contains primarily Gαq sequence and five amino acids of the carboxyl terminus of Gαi – these five amino acids are sufficient for Gqi5 to communicate with Gi/o-coupled GPCRs but signal through the Gq pathway. For Gs-coupled GPCRs, a Gqs5 chimera is used11. The general strategy for producing a CNiFER is to: 1) create a clonal HEK293 cell that is stably expressing an optical Ca2+ detector (i.e. TN-XXL), using a lentivirus transduction of HEK cells, 2). stably express a G protein chimera, if necessary, in the HEK293 cell clone expressing TN-XXL, 3). create a stably expressing GPCR clone in the HEK293 cell clone expressing TN-XXL and the chimeric G protein. The clonal HEK293 line that lacks the GPCR but has the TN-XXL and chimeric G protein serves as the ‘control CNiFER’.
To generate lentivirus, a lentivector expression system is used (e.g. pCDH-CMV-MCS-EF1-Puro), which contains the genetic elements responsible for packaging, transduction, stable integration of the viral expression construct into genomic DNA, and expression of the target gene sequence. To produce a high titer of viral particles, expression and packaging vectors are transiently co-transfected into producer mammalian cells and virus is collected. There are several viral core facilities in the US that can generate high titer lentivirus. Following infection of HEK-293 cells, the Puro gene provides antibiotic selection for identifying transduced HEK293 cells (Fig. 2).
Fig. 2.
Plasmid map for Lentivector
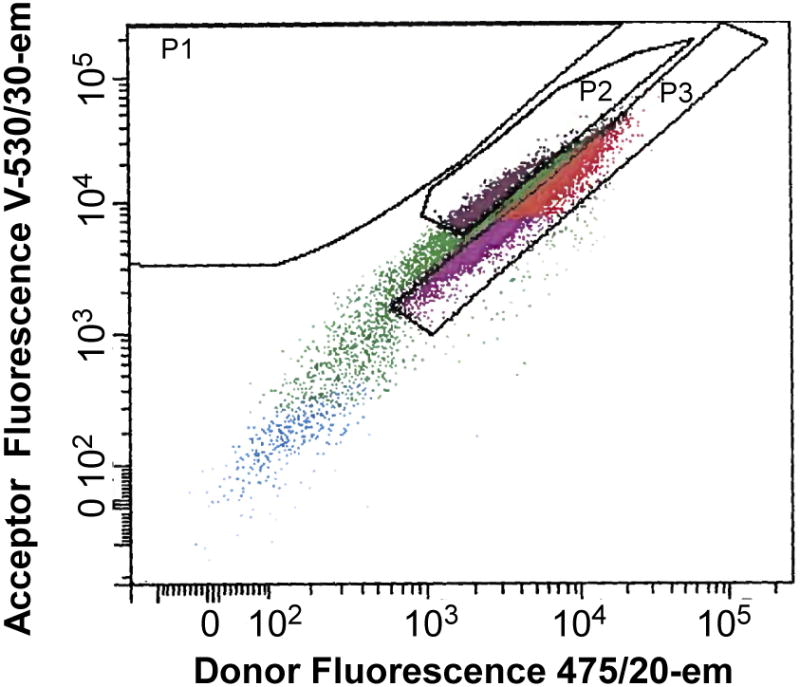
In order to identify specific clonal lines, transduced HEK293 cells are sorted using a fluorescence activated cell sorting (FACS) system. The objective is to isolate clones that contain a high expression level of FRET-based Ca2+ detector and the capability of undergoing FRET. In this example of FACS analysis, the fluorescence of the eCFP emission is plotted against FRET (eCFP excitation and YFP emission). The boxes mark regions (P2 and P3) that will be subsequently selected (“gated”) for sorting into 96 -well plates (Fig. 3). Generally, about four 96-well plates is sufficient to screen for successful creation of CNiFERs. From these 4 plates, approximately 100 clones are suitable for fluorometric plate reader analysis.
Fig. 3. Example of FACS analysis.
Graph plots the eCFP emission (“475/20-A”) as a function of YFP emission (citrine; “FRET V-530/30-A”), using eCFP excitation. Regions P2 and P3 show areas for selected for sorting.
Once the sorted cells have grown to sufficient density, the FRET response following agonist activation is determined using a 96-well fluorometric plate reader system equipped with solution handling and measuring FRET. To narrow down the number of clones to study, a “3-fold” agonists curve is generated for each clone. Approximately 10 clones are then analyzed further with a complete dose-response to the cognate agonist, and then examination of non-specific response with 10 other agonists. A 96-well drug plate is prepared as 3-fold concentration of drugs (e.g. agonists, antagonists, etc.) in artificial cerebral spinal fluid (ACSF). In this example, a drug plate is set up for testing a D2 CNiFER with its cognate agonist, dopamine, and potential non-specific response with a variety of other neurotransmitter and peptide agonists (Fig. 4).
Fig. 4. Example of layout for 96-well drug plate.
Drug plate is loaded with 3-fold concentrations of various neurotransmitters and peptides for fluorometric plate reader.
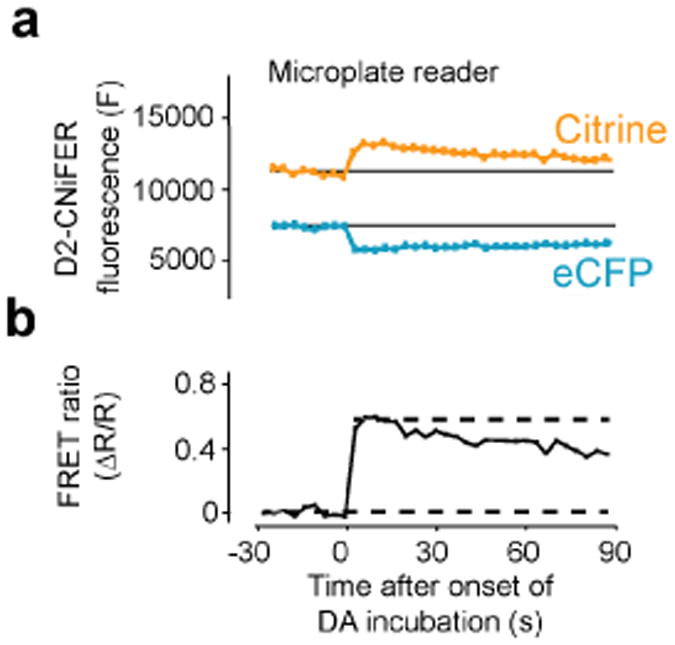
Stimulation of the GPCR is expected to increase the FRET response as a consequence of Ca2+ activation of TN-XXL. Under these conditions, FRET is produced by eCFP and YFP moving closer, so that excitation of CFP leads to smaller CFP emission and larger YFP emission. In this example, excitation is set to ∼ 435 nm and emission filters are set to 485 ± 7.5 nm for CFP (eCFP) and 527 ± 7.5 nm for YFP (Citrine) (Fig. 5). 30 seconds of baseline fluorescence is measured and then 50 μl of the “3-fold” agonists are delivered to each well containing 100 μl media (1:3 dilution). eCFP and YFP emission fluorescence are measured every 3.8 seconds for 180 seconds. Background measurements are taken from wells without cells and subtracted, if necessary. Fluorescence intensities are normalized to pre-stimulus baselines (ΔF/F), and peak responses are measured to calculate the FRET ratio (ΔR/R) using the ΔF/F of the 527 and 485 nm channels. A dose response curve is constructed by plotting the FRET ratio as a function of different agonist concentrations and fit with the Hill equation to determine EC50 and Hill coefficient (Fig. 6)
Fig. 5. Example of agonist-induced FRET response.
Left, plot of FRET (eCFP excitation and eCFP and YFP emission) response with application of dopamine with D2 CNiFERs. Right, plot of the FRET ratio. Taken from Muller et al., 20148.
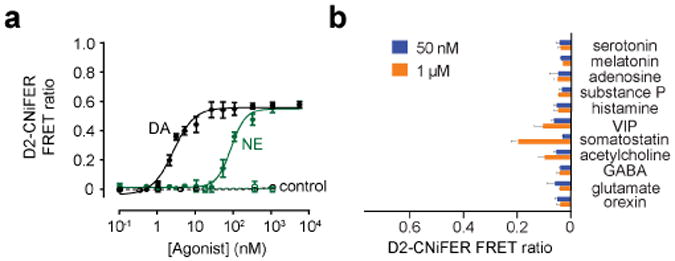
Fig. 6. Example of dose response curve for D2 CNiFER.
Left, dose response curve for dopamine (DA, black) and norepinephrine (NE, green) for D2 CNiFER and “control” CNiFERs lacking D2. Right, bar graph shows FRET ratio response for other neurotransmitters. Taken from Muller et al., 20148.
CNiFER clones are tested further for possible desensitization, temporal response and ultimately for detecting release in vivo.
Discussion
Creation of neuropeptide CNiFERs provides an innovative and unique strategy for measuring release of neurotransmitters in the brain. CNiFERs are ideally suited for measuring extrasynaptic release, i.e., volume conduction, for neurotransmitters. Importantly, each CNiFER possesses the properties of the native GPCR, providing a physiological optical measurement of changes in levels of neurotransmitters in the brain. To date, CNiFERs have been created for detecting acetylcholine (M1-CNiFER)7, dopamine (D2-CNiFER)8 and norepinephrine (α1a-CNiFER)8. In principal, a CNiFER can be created for any neurotransmitter that signals through a GPCR. For the case where the GPCR signals through Gq G proteins, no further modification is needed to the HEK293 cell. GPCRs that signal through Gi/o, however, require coexpression of a Gqi5 chimeric G protein to couple the GPCR to the Gq/PLC pathway8,11. Similarly, GPCRs that signal through Gs will require coexpression of a Gqs5 G protein11. Once completed, each CNiFER clone is screened and only those that have an affinity comparable to the native receptor, exhibit little or no desensitization and provide a signal-to-noise ratio that is adequate for measuring with in vivo two-photon microscopy, are selected for in vivo studies.
Two major issues to consider in constructing CNiFERs are sensitivity and desensitization. If the EC50 is too high, i.e. low affinity, relative to the native receptor, then the CNiFER may not have sufficient sensitivity to detect release of neurotransmitter in vivo. It may be possible to rescreen clones and find a different CNiFER clone that has higher affinity sensitivity. An alternative strategy would be to test other types of genetically encoded fluorescent Ca2+-detectors that may have a Ca2+ sensitivity that shifts the EC50 for the GPCR activation. Because the CNiFER design is modular, it is easily adapted to other types of genetically encoded Ca2+-detectors, such as GCaMP12. Isolating CNiFER clones with same receptor but different EC50s could be advantageous for extending the dynamic range of detecting release of endogenous receptors in vivo.
Desensitization of the CNiFER will also limit its use in vivo. If the peak response gradually decreases with each pulse of agonist, then the receptor may be desensitizing. In this case, examine other clones and determine if they respond the same way. Modifications to the receptor or use of another subtype of receptor may be necessary to address the agonist-dependent desensitization. The mechanism of desensitization must be determined for each receptor on a case-by-case basis. If there are known sites of phosphorylation or amino acids identified that associate with G protein receptor kinases (GRKs), it would be advisable to construct a non-desensitizing variant of the GPCR by mutating one or more sites.
Thus far, CNiFERs have been only implanted into superficial layers of cortex7,8, due to spectroscopic limitations with imaging fluorophores with two-photon microscopy13,14. In the future, it may be possible to adapt CNiFER technology with fiber-based measurements of fluorescence15 so that CNiFERs can be implanted in subcortical brain regions. Another consideration with implanting CNiFERs into the rodent brain is that CNiFERs are constructed with human cells. Thus, there is possibility of rejection or an immunological response with the CNiFER implants. It is therefore recommended to treat the mice with cyclosporine to minimize any potential immunological response. This was examined previously by immuonstaining for GFAP and MAC18. CNiFER implantation did not produce glial scars are generate any significant MAC1 staining8.
Table 1. Media and Buffers.
| HEK293 Growth Media | ||
|---|---|---|
| 500 ml | stock | |
| 445 ml | 1× | DMEM (high glucose) with Glutamax |
| 50 ml | 10% Fetal bovine serum | |
| 5 ml | 100× | 1× Pen/Strep antibiotics |
| Sterilize vacuum filter (0.2 um) | ||
| HEK293 Growth Media with Puro | ||
| 500 ml | stock | |
| 435 ml | 1× | DMEM (high glucose) with Glutamax |
| 50 ml | 10% Fetal bovine serum (heat inactivated) | |
| 5 ml | 100× | 1× Pen/Strep antibiotics |
| 10 ml | 100 ug/ml | 2 ug/ml Puromycin |
| Sterilize vacuum filter (0.2 um) | ||
| ACSF | ||
| Final | weigh | |
| 125 mM | 7.3 g | NaCl |
| 5 mM | 0.37 g | KCl |
| 10 mM | 1.8 g | D-glucose |
| 10 mM | 2.6 g | HEPES Na salt |
| Dissolve in 800ml of ddH20 or nanopure water | ||
| 3.1 mM | 3.1 ml | Stock of 1M CaCl2 |
| 1.3 mM | 1.3 ml | Stock of 1M MgCl2 |
| Adjust pH to 7.4 with NaOH | ||
| Bring final volume to 1L in graduated cylinder | ||
Table 2. Volumes for splitting cells.
| Dish | 0.05% trypsin/EDTA | Resuspend in HEK293 growth | Final Volume |
|---|---|---|---|
| 12-well plate | 200 μl | 0.5 ml | 1.2 ml |
| 6-well plate | 400 μl | 1.5 ml | 2 ml |
| T25 flask | 1 ml | 5 ml | 5 ml |
| T75 flask | 2.5 ml | 10 ml | 10 ml |
Acknowledgments
We thank B. Conklin (University of California, San Francisco) for providing the Gqi5 cDNA, A. Schweitzer for assistance with the electronics, N. Taylor for assistance with screening of clones, and Olivier Griesbeck for TN-XXL. This work was supported by research grants through the US National Institute on Drug Abuse (NIDA) (DA029706), the National Institute of Biomedical Imaging and Bioengineering (NIBIB) (EB003832), Hoffman-La Roche (88610A) and the “Neuroscience Related to Drugs of Abuse” training grant through NIDA (DA007315).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/53290.
Disclosures: The authors have nothing to disclose.
References
- 1.Day JC, Kornecook TJ, Quirion R. Application of in vivo microdialysis to the study of cholinergic systems. Methods. 2001;23:21–39. doi: 10.1006/meth.2000.1103. [DOI] [PubMed] [Google Scholar]
- 2.Robinson DL, Venton BJ, Heien ML, Wightman RM. Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clinical chemistry. 2003;49:1763–1773. doi: 10.1373/49.10.1763. [DOI] [PubMed] [Google Scholar]
- 3.Liang R, Broussard GJ, Tian L. Imaging Chemical Neurotransmission with Genetically Encoded Fluorescent Sensors. ACS chemical neuroscience. 2015 doi: 10.1021/cn500280k. [DOI] [PubMed] [Google Scholar]
- 4.Okubo Y, et al. Imaging extrasynaptic glutamate dynamics in the brain. Proc Natl Acad Sci USA. 2010;107:6526–6531. doi: 10.1073/pnas.0913154107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marvin JS, et al. An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods. 2013;10:162–170. doi: 10.1038/nmeth.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.IUPHAR/BPS Guide to PHARMACOLOGY. 2015 http://www.guidetopharmacology.org.
- 7.Nguyen QT, et al. An in vivo biosensor for neurotransmitter release and in situ receptor activity. Nat Neurosci. 2010;13:127–132. doi: 10.1038/nn.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller A, Joseph V, Slesinger PA, Kleinfeld D. Cell-based reporters reveal in vivo dynamics of dopamine and norepinephrine release in murine cortex. Nat Methods. 2014;11:1245–1252. doi: 10.1038/nmeth.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drew PJ, et al. Chronic optical access through a polished and reinforced thinned skull. Nature Methods. 2010;7:981–984. doi: 10.1038/nmeth.1530. Epub 2010 Oct 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamauchi JG, et al. Characterizing ligand-gated ion channel receptors with genetically encoded Ca2+ sensors. Public Library of Science ONE. 2011;6:e16519. doi: 10.1371/journal.pone.0016519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gqα to that of Gjα. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 12.Akerboom J, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 14.Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Cui G, et al. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature. 2013;494:238–242. doi: 10.1038/nature11846. [DOI] [PMC free article] [PubMed] [Google Scholar]